UNIVERSITÀ DEGLI STUDI DI GENOVA
Scuola di Scienze Mediche e Farmaceutiche
Tesi di Laurea
“L'ipermetabolismo del muscolo scheletrico predice la
sopravvivenza in pazienti con Sclerosi Laterale
Amiotrofica: un approccio computazionale alle
immagini FDG PET/TC”
Relatore: Prof. Gianmario Sambuceti
Correlatore: Dr. Matteo Bauckneht
Candidato: Emanuele Alexander Chiorino
Anno Accademico 2018/2019
Corso di laurea magistrale in
Medicina e Chirurgia
2 Indice
Capitolo 1. Considerazioni fisiopatologiche e cliniche sulla Sclerosi Laterale Amiotrofica (SLA) 1.1 Introduzione
1.2 Classificazione e spettro della SLA 1.3 Epidemiologia descrittiva
1.4 Eziopatogenesi della malattia
1.4.1 Fattori di rischio non genetici 1.4.2 Fattori di rischio genetici 1.5 Meccanismi fisiopatologici
1.5.1 Alterato metabolismo dell’RNA
1.5.2 Difetti del trasporto nucleocitoplasmatico 1.5.3 Omeostasi proteica compromessa
1.5.4 Riparazione DNA compromessa
1.5.5 Disfunzione mitocondriale e stress ossidativo 1.5.6 Difetti del trasporto assonale
1.5.7 Difetti del trasporto vescicolare 1.5.8 Neuroinfiammazione
1.5.9 Eccitotossicità
1.5.10 Disfunzione oligodendrocitaria 1.6 Clinica
1.6.1 Sintomi e segni 1.6.2 Segni di danno UMN 1.6.3 Segni di danno LMN 1.6.4 Segni bulbari
3 1.7 Scala di valutazione funzionale 1.8 Forme cliniche
1.8.1 Forma spinale classica
1.8.2 Sclerosi laterale primaria (SLP)
1.8.3 Atrofia muscolare progressivamente (AMP) 1.8.4 SLA a insorgenza bulbare
1.8.5 Flail Arm Sindrome 1.8.6 Flail leg sindrome 1.8.7 SLA emiplegica 1.8.8 Forma piramidale
1.8.9 Forma respiratoria assiale 1.8.10 Amiotrofia muscolare 1.8.11 SLA con FDT
Capitolo 2. Coinvolgimento muscolare nella SLA 2.1 Ruolo delle SC
2.2 Disfunzione mitocondriale del muscolo scheletrico 2.3 Disfunzione mitocondriale e giunzione neuromuscolare 2.4 MicrRNA del muscolo scheletrico
2.5 SOD1 mutato agisce indistintamente su tutti i muscoli? Capitolo 3. Generalità sulla FDG PET/TC in neurologia.
3.1 Radiofarmaci
3.2 Uso della FDG PET/TC nelle malattie neurodegenerative: implicazioni cliniche 3.2.1 FDG PET/TC nel Morbo di Alzheimer
4
3.2.3 FDG PET/TC nella Demenza a corpi di Lewy 3.2.4 FDG PET/TC in altre malattie neurodegenerative 3.3 Vantaggi e limitazioni
Capitolo 4. Uso della FDG PET/TC nella SLA
Capitolo 5. Analisi computazionale delle immagini FDG PET/TC nella SLA Capitolo 6. Introduzione dello studio sperimentale
Capitolo 7. Materiali e Metodi
7.1 Pazienti affetti da Sclerosi laterale amiotrofica 7.2 Gruppo di controllo
7.3 Immagini PET/TC
7.4 Analisi FDG PET/TC total body 7.5 Analisi cerebrale FDG PET/TC 7.6 Analisi statistiche
Capitolo 8. Risultati
8.1 Caratteristiche cliniche della coorte di pazienti
8.2 Descrizione dell’effetto della SLA sul sistema nervoso centrale all’imaging PET/TC
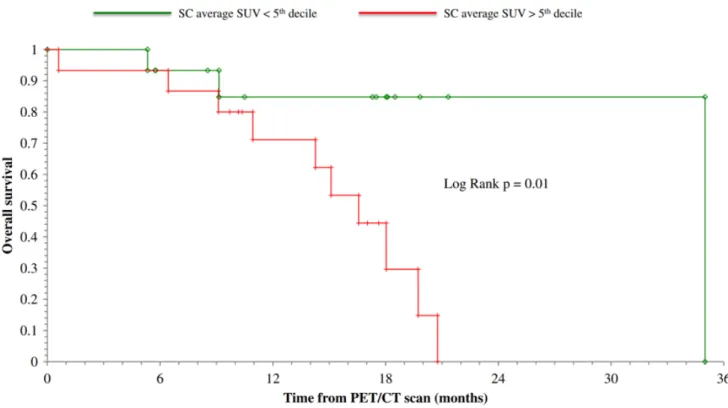
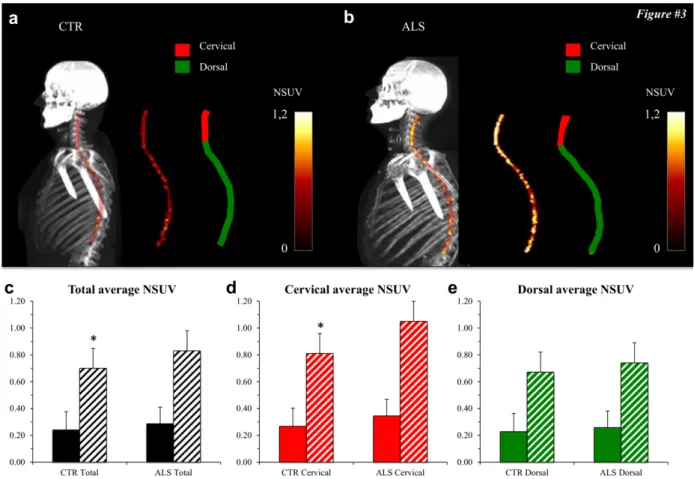
8.3 Descrizione dell’effetto della SLA sui muscoli scheletrici all’imaging PET/TC 8.4 Impatto sulla prognosi
Capitolo 9. Discussione Capitolo 10. Conclusioni Capitolo 11. Bibliografia
5
CAPITOLO 1
CONSIDERAZIONI FISIOPATOLOGICHE E CLINICHE SULLA SCLEROSI LATERALE AMIOTROFICA (SLA)
1.1 Introduzione
La sclerosi laterale amiotrofica (SLA) è una malattia neurodegenerativa ad esito fatale. È stata osservata per la prima volta da Charles Bell nel 1830 e descritta successivamente da Jean-Martin Charcot nel 1874. Egli descrisse la clinica della malattia con molta precisione, fatta eccezione per alcuni punti superati solo recentemente (Fazio et al., 2019; Mathis et al., 2017). La patologia colpisce nello specifico il motoneurone superiore, quello inferiore e il muscolo (Figura 1).
Figura 1. Rappresentazione schematica della via motrice con i sui rapporti sequenziali tra motoneurone superiore, motoneurone inferiore e muscolo effettore.
La distruzione costante di queste cellule nervose comporta un quadro clinico con decorso progressivo peggiorativo, in cui il paziente va lentamente incontro a paralisi amiotrofica generalizzata coinvolgente arti, tronco e i settori craniali (con particolare predilezione per quelli inferiori), andando a intaccare la deambulazione, la masticazione, la deglutizione, la comunicazione e la respirazione, rendendole via via più precarie fino a costringere all’immobilità con necessità di alimentazione e respirazione assistite (Fazio et al., 2019; Rowland et al., 2001).
La SLA divenne oggetto di attenzione mondiale quando il famoso giocatore di baseball Lou Gehrig dichiarò di esserne affetto. Recentemente la SLA è stata nuovamente oggetto di attenzione mediatica
6
a seguito della pubblicazione di uno studio che individuava una maggiore incidenza della malattia nei giocatori di calcio professionisti (Leigh et al., 1994; Vanacore et al., 2018).
Nella maggior parte dei pazienti l’eziologia della SLA è sconosciuta, in alcuni casi invece si è visto che la patologia è associata alla mutazione di alcuni geni che ne determinano la familiarità.
Recenti studi hanno inoltre rivelato che sebbene il coinvolgimento sia prevalentemente motorio, il 50% dei pazienti può sviluppare un danno cognitivo-comportamentale di entità variabile e che al 13% dei pazienti si associa una concomitante demenza frontotemporale (FTD).
Attualmente non esiste ancora alcun test definitivo per la diagnosi di SLA e il processo di indagine basato su criteri clinici e di esclusione di altre patologie risulta essere il metodo attualmente impiegato. L’elettromiografia (EMG) è oggi l’unico test sensibile abitualmente utilizzato nella pratica clinica. Il tempo tra l’insorgenza e la diagnosi è in media di 10-12 mesi, il che rende necessario ulteriori studi che portino al ridimensionamento di tale ritardo (Sabatelli et al., 2016; Hardiman et al., 2017; Mathis et al., 2017). Attualmente non esiste un trattamento curativo per questo tipo di disturbo neurodegenerativo. I trattamenti sono per lo più sintomatici (disfagia, insufficienza respiratoria, sintomi cognitivi e psichiatrici, dolore, spasticità affaticamento, disturbi del sonno ecc.) e palliativi (Mathis et al., 2017).
1.2 Classificazione e spettro della SLA
La SLA fa parte di un più grande gruppo di patologie note come malattie motoneuronali, di cui è la maggiore rappresentante. Tuttora emergono difficoltà ad arrivare ad una classificazione di questo gruppo di patologie che sia univoca e ubiquitariamente riconosciuta.
Un buon modo per classificarle è impostare tre sottogruppi a seconda del coinvolgimento dei motoneuroni:
- patologie che coinvolgono il motoneurone superiore; - patologie che coinvolgono il motoneurone inferiore;
- patologie a coinvolgimento misto (Fazio et al., 2019) (Tabella 1).
Si può pensare allo spettro fenotipico della SLA come un continuum compreso tra due poli. Al primo polo avremo la Sclerosi Laterale Primaria (espressione di un danno ai motoneuroni superiori), all’ estremo opposto invece l’Atrofia Muscolare Progressiva (espressione di un danno quasi esclusivo ai motoneuroni inferiori). Ragionando sempre per estremi entro cui si vanno poi ad inserire le forme borderline è possibile delineare un ulteriore spettro in funzione del deficit cognitivo, in cui da una
7
parte avremo la SLA accompagnata da demenza frontotemporale e dall’altra la SLA senza segni di decadimento cognitivo comportamentale (Fazio et al., 2019).
Tabella 1. Classificazione delle principali malattie motoneuronali in funzione delle classi neuronali lese.
8 1.3 Epidemiologia descrittiva
Dal punto di vista epidemiologico la SLA rientra tra le patologie rare. La prevalenza (numero delle persone affette che ci aspettiamo di trovare in ogni momento nella popolazione) si aggira sulle 8-10 su 100.000. L’incidenza (numero di nuovi casi per anno) si aggira intorno ai 2,5-3 su 100.000/anno. La maggior parte dei dati riguardo l’incidenza della SLA provengono principalmente dall’Europa occidentale, in misura minore dall’Europa orientale, dal Nord America, dall’Asia (soprattutto Cina e Giappone) e quasi nulla dai paesi in via di sviluppo (Tabella 2).
Tabella 2. Tasso di incidenza grezzo della SLA specifico per ogni paese.
Il picco di esordio va dai 55 anni ai 75 anni di età. Il fatto che vi sia un picco d’età preferenziale, abbastanza simile nei due sessi, indica che la malattia è definibile come malattia età correlata e allontana l’ipotesi che vi sia correlazione con l’invecchiamento come per esempio nella malattia di Alzheimer. Facendo il rapporto fra prevalenza e incidenza è possibile ottenere la durata media di malattia, che in questo caso è di 3 anni (Chio et al., 2013).
1.4 Eziopatogenesi della malattia
L’eziopatogenesi della malattia non è nota ma si distingue una forma sporadica (s-SLA) nel 90% dei casi e forme familiari (f-SLA) che rappresentano il restante 10%, caratterizzate prevalentemente da
9
trasmissione autosomica dominante, sono rare le forme autosomiche recessive o x-linked (Fazio et al., 2019). Inoltre, la SLA risulta essere influenzata sia da fattori genetici sia ambientali.
1.4.1 Fattori di rischio non genetici
Numerosi studi hanno permesso di analizzare molti fattori di rischio coinvolti nell’insorgenza della malattia, evidenziando peculiari caratteristiche.
I metalli pesanti, in particolare il piombo, sembrano essere in grado di portare all’insorgenza della SLA. Tuttavia, l’inquinamento da piombo ormai risulta essere estremamente diminuito, mettendo in seria discussione la presenza di questo tra i fattori di rischio nell’insorgenza della malattia (Nowicka et al., 2019).
Il trauma (distinto in trauma cranico e altri traumi) è stato analizzato in numerosi studi come fattore di rischio e i risultati emersi non hanno identificato alcuna correlazione tra il trauma non cranico e l’insorgenza di SLA, che invece potrebbe essere correlata al trauma cranico (Lian et al., 2019). È stato analizzato anche l’impatto dell’attività fisica sull’insorgenza della malattia, portando a risultati contrastanti (Lian et al., 2019; Nowicka et al., 2019).
I campi elettromagnetici e le radiazioni non ionizzanti sono stati studiati come possibile fattore di rischio; tuttavia la correlazione con l’insorgenza della malattia risulta essere incerta (Yu et al., 2014; Nowicka et al., 2019).
L’alcool è considerato un fattore di rischio. Uno studio cinese ha identificato la correlazione tra frequenza e quantità totale del consumo di alcool con l’insorgenza della SLA, mettendo in evidenza un minor rischio di sviluppo della malattia nei non bevitori (Lian et al., 2019).
Il fumo di sigaretta è sicuramente uno dei fattori di rischio più studiati sia nelle patologie in generale sia nella SLA. Già molti studi hanno messo in evidenza una maggior incidenza di SLA nei fumatori rispetto ai non fumatori anche se non è ancora noto in che modo vada a inserirsi nella genesi della patologia (Yu et al., 2014; Lian et al., 2019; Nowicka et al., 2019).
Studi basati sull’osservazione del fattore di rischio Body Mass Index (BMI) hanno evidenziato una differenza significativa nell’incidenza della malattia: in particolare è stato osservato che un BMI basso correla con un’aumentata insorgenza. Inoltre, si è evidenziato che un BMI maggiore al momento della diagnosi correli con una maggiore sopravvivenza (Lian et al., 2019; Nowicka et al., 2019).
10 1.4.2 Fattori di rischio genetici
I fattori di rischio genetici sembrano essere implicati sia nelle forme familiari sia nelle forme sporadiche della SLA. Sebbene siano stati identificati più di 50 geni causativi o modificanti la malattia le varianti più frequenti risultano essere SOD1, TARDBP, FUS e C9ORF72 (Boylan, 2015). - SOD1 (in precedenza denominato SLA1 “amyotrophic lateral sclerosis 1”) è stato il primo gene ad essere stato chiamato in causa nell’insorgenza della malattia (1993). La proteina superossido dismutasi (SOD1) usa come cofattori il rame e lo zinco e converte gli ioni superossido in acqua ossigenata riducendo il danno ossidativo della cellula (Figura 2) (Mejzini et al., 2019).
Figura 2. Struttura SOD1 umana.
Un recente studio ha individuato che le varianti patogene nella SOD1 rappresentano circa il 15-30% dei casi di f-SLA e meno del 2% dei casi di s-SLA (Zou et al., 2017). Inizialmente si ipotizzò che la malattia fosse dovuta ad una perdita dell’attività della dismutasi, tuttavia uno studio successivo dimostrò che l'attività dell’enzima non era correlata alla gravità della malattia, indicando anche un possibile guadagno funzionale della proteina. Inoltre un ripiegamento errato della SOD1 wild-type potrebbe essere implicato direttamente nell’insorgenza o far parte di un evento a valle comune nella progressione della SLA (Mejzini et al., 2019).
- TARDBP è un gene che codifica per la proteina TDP-43 la quale lega DNA/RNA regolando l’espressione genica. Da numerosi lavori si è osservato che una proteina TDP-43 aberrante potrebbe essere la causa della neurodegenerazione.
In particolare, analizzando gli esami istologici di campioni di midollo spinale di pazienti con SLA si sono riscontrate inclusioni ubiquitinate di citoplasma neuronale. Il componente principale degli
11
aggregati proteici ubiquitinati era la proteina 43 (TDP-43) la quale è ora considerata un segno patologico della SLA.
L'accumulo citoplasmatico della proteina TDP-43 e il suo depauperamento a livello nucleare ha portato a proporre meccanismi di sviluppo della malattia che comportano una perdita della normale funzione della proteina TDP-43 a livello nucleare, un guadagno di funzione probabilmente tossico o entrambi. Ciò evidenzia l'importanza di una regolazione finemente controllata di questa proteina la quale avviene attraverso un meccanismo di feedback.
Ad oggi, almeno 48 varianti di TARDBP sono state associate alla SLA (Mejzini et al., 2019). - FUS è un gene che codifica per una proteina legante l’RNA. Le varianti patogene di questo gene sono state identificate in un sottogruppo di pazienti con SLA. Queste varianti sono associate alla SLA precoce e alla SLA ad insorgenza giovanile. FUS-SLA è caratterizzato da aggregazione patologica della proteina FUS. L'aggregazione di TDP-43 non è comunemente osservata nei pazienti FUS-SLA, ciò identifica un percorso della malattia indipendente tra le due mutazioni (Vance et al., 2009). La proteina FUS normale è coinvolta nell’espressione genica, nei meccanismi di riparazione del DNA e di difesa cellulare contro vari tipi di stress. Sono state identificate oltre 50 varianti autosomiche dominanti della proteina FUS nei pazienti con SLA, tuttavia non si è ancora chiarito se sia la perdita o un guadagno del meccanismo funzionale a causare la malattia nella FUS-SLA. (Mejzini et al., 2019) - Nel 2011 è stata identificata la causa ereditaria più comune di SLA nelle popolazioni europee (34% dei casi di f-SLA e il 5% dei casi di s-SLA) in un'espansione ripetuta di sei nucleotidi (GGGGCC) nella regione non codificante del gene C9ORF72 (DeJesus-Hernandez et al., 2011). Nei pazienti con fenotipo normale il gene presenta 5-10 copie dell’espansione ripetuta di esanucleotidi ma i pazienti con SLA possono avere da centinaia a migliaia di copie di ripetizioni. La funzione del gene C9ORF72 è poco conosciuta, ma recenti studi hanno evidenziato un possibile ruolo nella regolazione del traffico endosomiale, nell'autofagia e nella regolazione del sistema immunitario. Secondo quanto studiato, i livelli di mRNA e proteina C9ORF72 sono diminuiti nei pazienti con SLA, portando all'ipotesi che una perdita della funzione della proteina possa essere implicata nella malattia. La perdita della sola funzione di C9ORF72, come dimostrato da alcuni studi, non è da considerarsi però sufficiente a causare la malattia dei motoneuroni. Al contrario il guadagno di funzione della proteina potrebbe essere alla base dello sviluppo della SLA.
Ulteriori varianti genetiche correlate alla patologia sono state recentemente scoperte grazie a nuove metodiche di sequenziamento del DNA. I geni coinvolti influenzano: l'elaborazione dell'RNA, l'omeostasi proteica e la dinamica citoscheletrica. (Mejzini et al., 2019)
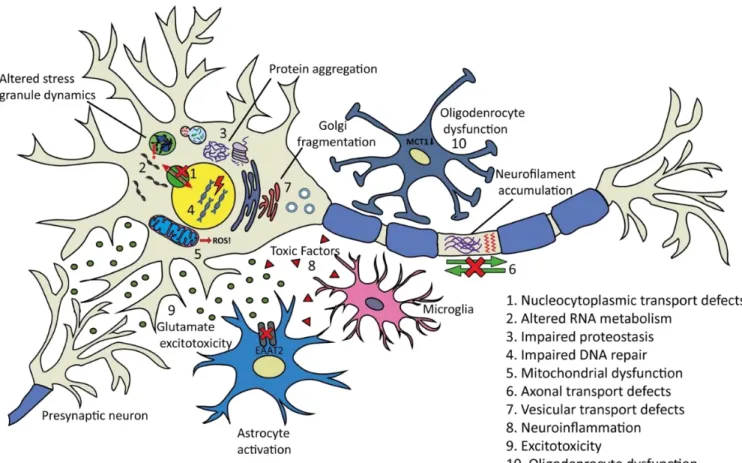
12 1.5 Meccanismi fisiopatologici
Figura 3. Schematizzazione grafica dei meccanismi fisiopatologici proposti per la SLA.
Nonostante decenni di ricerca, i meccanismi patogeni causali nella SLA rimangono ancora poco chiari, specialmente nei casi sporadici. È probabile che molteplici fattori, anziché un singolo evento iniziale, contribuiscano allo sviluppo e alla progressione della malattia. Inoltre, la variazione genetica e fenotipica tra i pazienti rende difficile scoprire e trarre conclusioni sui meccanismi patogeni della SLA in generale.
L'elevato numero di geni e processi cellulari implicati nella SLA ha portato alla proposta di numerosi meccanismi operativi (Figura 3). La maggior parte delle conoscenze attuali sulla fisiopatologia sono state ottenute grazie a studi condotti su topi transgenici portatori di mutazioni a carico del gene della SOD1 (Mitchell et al., 2007). È necessario chiarire i tempi e la misura in cui ciascuno di questi meccanismi contribuisce alla patologia.
1.5.1 Alterato metabolismo dell'RNA
13
malattie neurodegenerative. Nella SLA sono state individuate variazioni nei geni delle proteine che legano e guidano il metabolismo dell’RNA come TARDBP e FUS. Inoltre, domini simili a prioni, coinvolti nella formazione di granuli di stress, sono stati identificati al loro interno (Protter e Parker, 2016).
1.5.2 Difetti del trasporto nucleocitoplasmatico
La SLA è caratterizzata dall'errata localizzazione nel citoplasma e dall'esaurimento a livello nucleare delle proteine leganti l'RNA come TDP-43 e FUS. Ciò suggerisce che alla base della patogenesi della malattia ci potrebbero essere difetti del trasporto nucleare.
1.5.3 Omeostasi proteica compromessa
L'accumulo di proteine danneggiate contribuisce a diverse malattie neurodegenerative tra cui il morbo di Alzheimer, la malattia di Huntington, il morbo di Parkinson ed è emerso come possibile caratteristica chiave anche nella SLA.
I geni associati alla SLA più comunemente mutati (SOD1, C9ORF72, TARDBP e FUS) danno origine ad aggregati proteici nei neuroni dei pazienti. Sebbene l'aggregazione proteica sia fondamentale per la patologia della SLA, rimangono dubbi riguardanti la formazione, il ruolo e la tossicità di questi elementi.
Alcuni studi hanno dimostrato inoltre che l'interruzione delle due principali vie di eliminazione delle proteine, l'autofagia e il sistema ubiquitin-proteasoma (UPS), potrebbero essere coinvolti nella patogenesi.
1.5.4 Riparazione del DNA compromessa
La compromissione della riparazione del DNA è un altro meccanismo suggerito che può contribuire alla patogenesi della SLA. Le proteine normali TDP-43 e in particolar modo FUS sembrano essere importanti nella prevenzione e nella riparazione del danno al DNA.
1.5.5 Disfunzione mitocondriale e stress ossidativo
Lo stress ossidativo è stato suggerito come possibile fattore iniziale nella patogenesi della SLA. Tale processo si verifica quando la capacità antiossidante di una cellula viene meno e non neutralizza più le specie reattive dell'ossigeno (ROS). I ROS inducono un danno importante alle macromolecole come DNA, fosfolipidi e proteine (determinando un’errata localizzazione e una maggiore tendenza all'aggregazione di TDP-43 e FUS).
14
I mitocondri producono la maggior parte delle specie reattive dell'ossigeno, ciò spingerebbe a ipotizzare l’esistenza di potenziali meccanismi patogenetici SLA-correlati a livello mitocondriale. Un’ulteriore ipotesi sulla patogenesi della malattia riguarda la correlazione che potrebbe esserci tra stress ossidativo indotto dai mitocondri e la disregolazione dell'RNA, sebbene ancora non sia noto quale sia la successione temporale tra i due.
1.5.6 Difetti del trasporto assonale
I difetti del trasporto assonale sono comunemente osservati nelle malattie neurodegenerative. Le prime prove neuropatologiche coerenti con i difetti del trasporto assonale nella SLA sono state ottenute da studi post mortem. Questi hanno rivelato accumuli anomali di neurofilamenti, microtubuli, mitocondri, lisosomi, nonché sferoidi contenenti vescicole a livello neuronale.
Ulteriori studi condotti su modelli murini hanno evidenziato difetti del trasporto assonale all'inizio della progressione della malattia, prima dell'insorgenza dei sintomi, suggerendo così un ruolo nei meccanismi di sviluppo.
1.5.7 Difetti del trasporto vescicolare
Diverse proteine coinvolte nel trasporto vescicolare sono state collegate alla SLA e alla Demenza fronto-temporale (FTD). Un trasporto vescicolare difettoso può portare all'accumulo di proteine e alla frammentazione dell’apparato di Golgi, la quale si verifica nelle prime fasi della cascata patologica della malattia. Questo evento potrebbe essere classificato come un fattore scatenante della neurodegenerazione.
1.5.8 Neuroinfiammazione
Vi sono prove crescenti che la neuroinfiammazione svolge un ruolo nella fisiopatologia della SLA. La neuroinfiammazione associata alla perdita neuronale è caratterizzata da attivazione di microglia, astrociti, sovrapproduzione di citochine infiammatorie e infiltrazione di linfociti T.
1.5.9 Eccitotossicità
L'eccitotossicità è un processo patologico di sovrastimolazione del recettore del glutammato con conseguente danno neuronale o degenerazione. Nel sistema nervoso centrale, i livelli di glutammato extracellulare sono mantenuti bassi con livelli intracellulari molto più alti.
15
stimolazione dei recettori glutammatergici. L'eccitotossicità è stata a lungo sospettata come mediatore della SLA.
L’assunzione orale di eccitotossine è responsabile di particolari forme di malattie dei motoneuroni. Tuttavia, gli attuali dati sull’aumento dei livelli di glutammato nei pazienti con SLA sono ancora contraddittori e resta da chiarire se la segnalazione glutammatergica alterata sia direttamente patogena o se sia guidata da altri fattori.
Mentre l’uso di Riluzolo ha mostrato benefici modesti per alcuni pazienti, numerosi altri farmaci che mirano alla riduzione della trasmissione glutammatergica non hanno avuto successo negli studi clinici (portando a sospettare un diverso meccanismo d’azione di questo farmaco).
1.5.10 Disfunzione oligodendrocitaria
Vi sono prove crescenti a sostegno del contributo delle cellule non neuronali, come gli oligodendrociti, alla patogenesi della SLA. Si sospetta che queste cellule siano coinvolte nella degenerazione assonale attraverso un’alterata metabolizzazione del lattato. (Mejzini et al., 2019)
1.6 Clinica
1.6.1 Sintomi e Segni
“Era molto intelligente, sembrava capire perfettamente tutte le domande che le facevo, tuttavia rispondeva con grande difficoltà e in modo del tutto incomprensibile. In particolare, la deglutizione della signora era problematica, spesso pezzetti di alimenti penetravano nella laringe, determinando pericolosi accessi di soffocazione. Madame Aubel presentava inoltre un’accentuazione dei riflessi osteotendinei e movimenti fibrillari assai rilevanti a livello della lingua. Osservai anche l’assenza di disturbi della sensibilità e delle funzioni sfinteriche.” Jean-Martin Charcot,1869
Venne così descritta per la prima volta la Sclerosi Laterale Amiotrofica.
Le manifestazioni cliniche della SLA (Tabella 3) sono il risultato della compromissione del primo e del secondo motoneurone. L’eterogeneità dei sintomi con cui fa il suo esordio rendono spesso difficoltosa una diagnosi di certezza.
16
Tabella 3. Sintomi e segni della SLA suddivisi a seconda del motoneurone coinvolto.
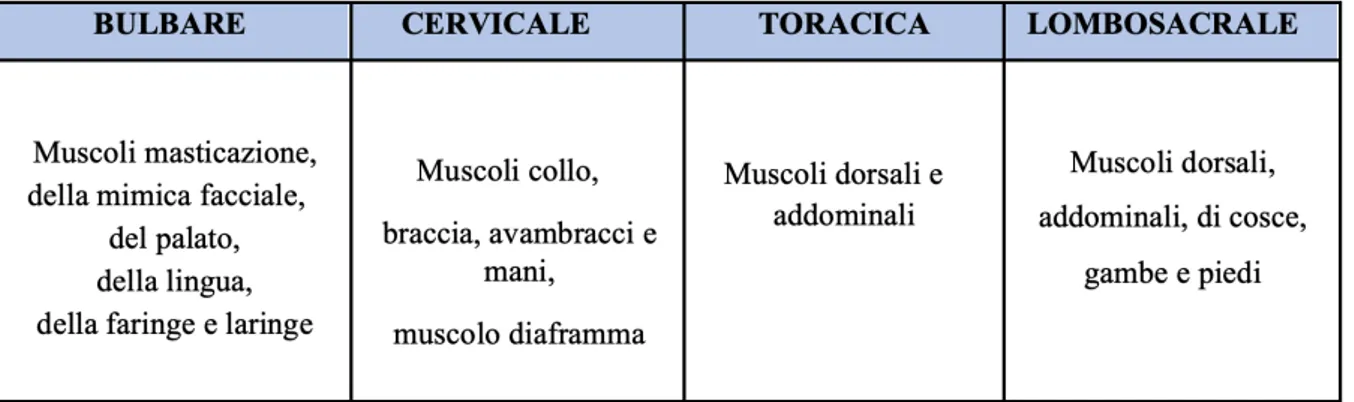
Il quadro clinico varia molto e si presenta con sintomi e segni in varie combinazioni e nelle differenti regioni anatomiche del corpo (tabella 4) a seconda della sede di esordio, dell’estensione del processo e dello stadio di patologia.
Tabella 4. Sintomi e segni della SLA suddivisi a seconda del distretto anatomico colpito.
I sintomi di esordio sono vari, generalmente si manifesta agli arti in modo asimmetrico e focale con ipostenia, ridotta resistenza all’esercizio, fascicolazioni e ipotrofia muscolare.
17
L’ipostenia risulta essere il sintomo cardine, valutabile con perdita di forza, faticabilità, riduzione di ampiezza e velocità dei movimenti, alterata coordinazione, determinando insuccesso nel portare a termine i movimenti.
La debolezza muscolare è spesso ciò che spinge il paziente a rivolgersi al medico (presente già fin dall’esordio nel 58-63% dei casi). (Traynor et al., 2000)
Le due presentazioni cliniche iniziali più frequenti sono: - SLA ad insorgenza agli arti (circa il 70% dei casi); - SLA ad insorgenza bulbare (circa il 25%).
Leggermente più frequente sembra essere l’insorgenza agli arti superiori rispetto agli arti inferiori. Raramente si ha generalizzazione della sintomatologia già negli stadi iniziali (7%). Poco comune è anche l’esordio combinato ad arti superiori e inferiori (11%). L'esordio respiratorio è raro (< 3% dei casi di SLA) e sembra essere collegato alla mutazione di SOD-1. (Chiò et al., 2011)
Successivamente la malattia si diffonde in altre regioni. In una proporzione molto più piccola di pazienti, il solo coinvolgimento del secondo motoneurone (LMN,lower motor neuron) provoca Atrofia Muscolare Progressiva (PMA), mentre il solo coinvolgimento del motoneurone superiore (UMN,upper motor neuron) porta a Sclerosi Laterale Primaria (PLS) delineando due patologie “pure” che si inseriscono agli estremi dello spettro della SLA classica. (Fazio et al., 2019)
1.6.2 Segni di danno UMN
Il danno al UMN è rilevato dalla presenza di alcuni o tutti i seguenti segni: iperreflessia, spasticità e clono. Nella pratica clinica vengono generalmente testati: il riflesso masseterino, il segno di Hoffmann e il segno di Babinski (gli altri riflessi primitivi non sono informativi). Se i segni al UMN sono ancora isolati dopo 4 anni dall'insorgenza dei sintomi, la diagnosi è di PLS clinicamente puro. Questa sindrome è caratterizzata da progressione lenta, insufficienza funzionale bassa e deperimento dell'arto inferiore. Le caratteristiche cliniche al momento della prima valutazione sono utili per prevedere la sua probabile progressione verso PLS, SLA a prevalente coinvolgimento dell'UMN o SLA tipica. (Grad et al., 2017)
1.6.3 Segni di danno LMN
Le caratteristiche cliniche del danno al motoneurone inferiore comprendono: fascicolazione, ipo-atrofia, ipo-areflessia osteo-tendinea, crampi e ipotonia/flaccidità. (Fazio et al., 2019)
18
rapide, spontanee, aritmiche, parcellari, di una sola o poche unità motorie. Non comportano spostamenti di segmenti corporei. Possono essere documentate sia con la clinica che con l’Elettromiografia EMG. Se prima si pensava che potessero essere un fenomeno ad esclusivo appannaggio del motoneurone inferiore, recenti studi dimostrano la compartecipazione del motoneurone superiore (de Carvalho et al., 2017).
1.6.4 Segni bulbari
I muscoli che consentono fonazione, masticazione e deglutizione sono innervati dai nervi cranici: V (nervo trigemino), VII (nervo facciale), IX (nervo glossofaringeo), X (nervo vago) e XII (nervo ipoglosso), con nuclei somato-motori situati nel Bulbo. Quando si ha un coinvolgimento bulbare sono generalmente colpiti il nucleo motore dell'ipoglosso e il nucleo ambiguo, mentre più raro è l'interessamento di quelli del V e del VII nervi cranici.
Nella SLA si assiste ad una paralisi bulbare a commistione flaccida-spastica. Nella forma flaccida pura i muscoli mimici sono ipotonici, la lingua è atrofica e presenta fascicolazioni, il riflesso masseterino è assente. Nella forma spastica, invece, non c'è debolezza e/o flaccidità a carico dei muscoli facciali, la lingua presenta normale trofismo, le fascicolazioni linguali sono assenti, mentre il riflesso masseterino è vivace. I segni e sintomi bulbari, oltre all'atrofia e alle fascicolazioni linguali, comprendono disartria, disfagia e scialorrea.
1.6.5 Segni respiratori
L’affaticamento respiratorio e la dispnea non sono di solito sintomi precoci. Tuttavia, l’insufficienza respiratoria dovuta alla debolezza del diaframma e dei muscoli intercostali insieme alle complicanze infettive polmonari, soprattutto ab ingestis, rappresentano le cause più comuni di morte nei pazienti affetti da SLA.
1.7 Scala di valutazione funzionale
L’esame neurologico quantifica in modo approssimativo il deficit funzionale del paziente ma questa valutazione può essere efficacemente implementata tramite opportune scale preformate e riconosciute, di cui la più utilizzata a livello clinico è la Amyotrophic Lateral Sclerosis Functional Rating Scale Revised (ALSFRS-R). (Figura 4-a; Figura 4-b; Figura 4-c)
19
20
21
Figura 4-c. Amyotrophic Lateral Sclerosis Functional Rating Scale Revised
1.8 Forme cliniche
I fenotipi della SLA sono così differenti l’uno dall’altro che spesso si pone la questione se la SLA sia un’unica malattia o un gruppo di patologie. Dal punto di vista del fenotipo f-SLA e s-SLA sono indistinguibili.
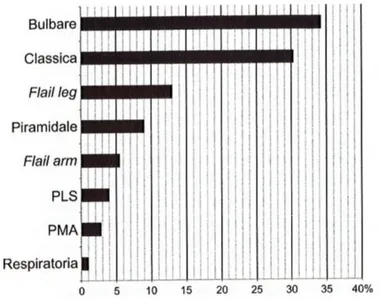
I fenotipi clinici della SLA possono essere classificati in modo grossolano in base al livello e all'area anatomica del coinvolgimento dei motoneuroni e al modello di insorgenza (Figura 5).
1.8.1 Forma Spinale Classica
La SLA tipica o "classica" comporta segni simultanei di UMN e LMN ed è generalmente fatale entro 4 anni dall'esordio. La debolezza muscolare inizia in una regione del corpo ristretta (solitamente viso, braccio o gamba) e avanza costantemente nel tempo e nello spazio. Nel 40-50% dei casi si ha iniziale compromissione degli arti superiori, soprattutto alla mano e una crescente ipostenia viene riferita dal paziente come una difficoltà a manipolare gli oggetti (abbottonarsi, girare le chiavi per aprire la porta, usare la penna e le posate). Sono per lo più colpiti i muscoli dell’eminenza tenar innervati da C8-T1 (Nervo Mediano: opponente del pollice, abduttore breve del pollice; Nervo Ulnare: 1°interosseo dorsale) con relativo risparmio di quelli dell’eminenza ipotenar. Questo tipo di differente coinvolgimento regionale determina quella che viene definita come “Split Hand”, assai specifica della SLA. Con l’avanzare della patologia vengono coinvolti anche i muscoli dell’eminenza ipotenar determinando una conformazione della mano scimmiesca. L’estensione alla muscolatura bulbare e
22
respiratoria è generalmente più tardiva e ciò spiega una maggior sopravvivenza rispetto a forme che invece ne hanno un coinvolgimento precoce.
1.8.2 Sclerosi laterale primaria (SLP)
Si riferisce a una sindrome che coinvolge prevalentemente l’UMN; non è chiaro se questo fenotipo sia un disturbo puro o una variante della SLA. Nella maggior parte dei pazienti con SLP, i sintomi iniziano nelle gambe e ascendono in modo simmetrico alle braccia e ai muscoli bulbari. I pazienti con sclerosi laterale primaria e assenza di anomalie all’EMG quattro anni dopo l'insorgenza dei sintomi, possono avere sopravvivenza sopra i 10 anni. I pazienti con lievi cambiamenti all’EMG o un coinvolgimento LMN possono avere una sopravvivenza e una prognosi sfavorevole, che è coerente con la SLA classica.
1.8.3 Atrofia muscolare progressiva (AMP)
Si riferisce a una sindrome con coinvolgimento prevalente del LMN, anche in questo caso non è chiaro se sia un disturbo puro o una variante della SLA. L'insorgenza può verificarsi in qualsiasi regione del corpo, ha una maggiore incidenza nei maschi. Circa il 30% dei pazienti con AMP sviluppa sintomi dell’UMN entro 18 mesi dall'esordio della malattia. Le designazioni di fenotipo clinico SLP e AMP si basano sul livello del coinvolgimento dell’UMN o LMN della patologia sottostante. Altre designazioni fenotipiche per la SLA si basano sulla regione del corpo interessata per la prima volta all'insorgenza della malattia.
Figura 5. Incidenza percentuale delle varie forme di Sclerosi laterale amiotrofica (SLA), Atrofia muscolare progressiva (PMA)
23 1.8.4 SLA a insorgenza bulbare
Fenotipo ad esordio nei muscoli del linguaggio, della masticazione e della deglutizione. Tradizionalmente indicata con il coinvolgimento prevalente del LMN, mentre la variante pseudobulbare è indicativa dell’interessamento prevalente del UMN. Entrambe le forme, bulbare e pseudobulbare, hanno una progressione simile.
1.8.5 Flail Arm Sindrome
La variante regionale agli arti superiori, conosciuta anche come sindrome delle braccia cadenti o sindrome di Vulpian-Bernhart, è caratterizzata bilateralmente da debolezza e atrofia inizialmente confinata alla parte prossimale degli arti superiori senza significativo coinvolgimento degli arti inferiori. Il deficit motorio può estendersi alla muscolatura del collo determinando “dropped head” in cui si ha caduta della testa, costringendo il paziente all’utilizzo di collari. Non vi è alcuna differenza nell'età di esordio tra questa variante e la SLA tipica, sebbene la prima sia più comune nei maschi, il rapporto M:F si è visto essere di 9:1. Nella Flail Arm Sindrome la progressione di malattia è più lenta e la sopravvivenza media è di 53 mesi (quasi il doppio della SLA tipica).
1.8.6 Flail leg sindrome
La variante regionale degli arti inferiori è confinata alle gambe e può essere alternativamente identificata come variante di Patrikios o pseudopolinevritica. L’insorgenza è tipicamente asimmetrica con assenza dei riflessi tendinei all’arto inferiore, lenta progressione e lievi o assenti segni di coinvolgimento del primo motoneurone. È relativamente rara, prevalente negli uomini e con una sopravvivenza media compresa tra 76 e 96 mesi. Il fenotipo flail leg si è rilevato essere particolarmente correlato con la demenza frontotemporale (Chiò et al., 2011).
1.8.7 SLA emiplegica
La SLA emiplegica o variante di Mill è un raro tipo di variante di SLA caratterizzata da emiplegia progressiva che sale dalla gamba o scende dal braccio. Esiste una scarsa letteratura sulla variante di Mill, anche se uno studio di tomografia ad emissione di positroni (PET) su un paziente ha mostrato una chiara lateralizzazione dell'attivazione microgliale nell'emi-sfera controlaterale all'emiplegia (Turner et al., 2005).
24 1.8.8 Forma piramidale
Può manifestarsi inizialmente agli arti superiori o inferiori ma tende a generalizzarsi configurando un quadro di tetraparesi spastica, iper-reflessica per il maggior coinvolgimento del motoneurone superiore, a cui possono associarsi sintomi bulbari e pseudobulbari.
1.8.9 Forma respiratoria assiale
È caratterizzata dalla predominante compromissione già in fase iniziale dei muscoli respiratori. È considerata una delle forme più aggressive e ha un tasso di sopravvivenza molto basso tra i vari fenotipi della SLA.
1.8.10 Amiotrofia monomelica
Forma sporadica (eccezionalmente familiare) di SLA con compromissione del motoneurone inferiore limitata ad un arto. Sebbene sia considerata una forma di amiotrofia benigna sono stati descritte rare eccezioni con evoluzione sfavorevole, in cui, anche dopo intervalli stazionari di patologia intorno ai 10 anni, il quadro amiotrofico si è aggravato diffondendosi bilateralmente fino a diventare generalizzato.
1.8.11 SLA con FDT
I fenotipi clinici della SLA possono coinvolgere anche funzioni non motorie. La sovrapposizione tra FTD e SLA è ben documentata. Fino al 50% dei pazienti può sviluppare un danno cognitivo-comportamentale di entità variabile e che al 13% dei pazienti si associa una concomitante demenza frontotemporale. Al momento non è chiaro se la SLA con FDT abbia meccanismi patogenetici nettamente diversi dalla SLA tipica o sia semplicemente un'estensione di un singolo spettro di malattie. (Fazio et al., 2019; Grad et al., 2017)
25 CAPITOLO 2
Coinvolgimento muscolare nella SLA
Il coinvolgimento del muscolo scheletrico è stato oggetto di numerosi studi condotti su pazienti e su modelli animali (Loeffler et al., 2016). Il muscolo scheletrico è ad oggi considerato come un tessuto importante nella patogenesi della SLA e sembra in grado di attivare una cascata di segnalazione retrograda che degrada i motoneuroni (Tsitkanou et al., 2016). Tuttavia, le miocellule non sono colpite in modo uniforme ma esistono differenze in relazione alle loro caratteristiche contrattili e del loro metabolismo. Come per il motoneurone, la maggior parte delle informazioni disponibili sul coinvolgimento del muscolo nella SLA si devono a modelli murini con forme mutanti di SOD1. È comunemente accettato che la distruzione delle giunzioni neuromuscolari si verifichi prima della degenerazione dei motoneuroni. Inoltre, è stato ipotizzato che il miocita scheletrico potrebbe svolgere un ruolo attivo, invece di risentire passivamente della perdita dei motoneuroni (Figura 6.). Nella SLA risultano attivati diversi meccanismi per mantenere la massa e la funzione dei muscoli scheletrici sani tra cui la proliferazione delle cellule satelliti (SC), l’attività mitocondriale e la regolazione dei miRNA. È interessante notare che questi fattori possono agire in sinergia per mantenere il muscolo scheletrico sano e possono influenzare il numero e l'attività delle giunzioni neuromuscolari. (Loeffler et al., 2016)
Figura 6.Potenziale crosstalk tra SC, mitocondri e miRNA per influenzare
la giunzione neuromuscolare e la salute dei muscoli scheletrici nella SLA. Le perturbazioni nei miRNA, come il miR-206, possono influire
26 2.1 Ruolo delle SC
I meccanismi all'interno del muscolo che contribuiscono alla perdita iniziale della massa muscolare e all'eventuale degenerazione della giunzione neuromuscolare e dei motoneuroni non sono noti. Il muscolo scheletrico si ripara attraverso il processo ben coordinato che si basa sull'attivazione, proliferazione e differenziazione delle SC.
In condizioni basali le SC sono inattive, ma in risposta a lesioni acute, denervazione muscolare o altri stimoli come l’esercizio fisico, vengono attivate, si differenziano e proliferano con conseguente riparazione e / o crescita delle fibre muscolari.
Alcuni studi evidenziano che le SC derivate da pazienti con SLA non sono in grado di differenziare in modo del tutto efficiente e completo. Di conseguenza, il muscolo scheletrico non è in grado di ripararsi e rigenerarsi efficacemente, causando grave atrofia muscolare e debolezza.
Un possibile target terapeutico potrebbe quindi mirare ad aumentare la capacità proliferativa delle SC.
2.2 Disfunzione mitocondriale del muscolo scheletrico
In modelli murini SOD1G93A la disfunzione mitocondriale precede la paralisi clinica dei muscoli. Pertanto, il mitocondrio potrebbe giocare un ruolo fondamentale. La mutazione SOD1 del muscolo scheletrico è sufficiente a causare una patologia muscolare simile alla SLA e supporta ulteriormente il coinvolgimento di questo sito nella patogenesi della malattia. Inoltre, la somministrazione orale di un antiossidante mitocondriale nei topi SOD1G93A ha portato dei lievi miglioramenti nell’energia muscolare a 90 giorni, rallentando il declino mitocondriale e stabilizzando le giunzioni neuromuscolari.
Tra i possibili bersagli molecolari che possono regolare la disfunzione mitocondriale nella SLA abbiamo i mRNA (sensibilmente ridotti) e PGC-1alfa. La regolazione di PGC-1alfa sembra migliorare infatti i livelli di atrofia.
2.3 Disfunzione mitocondriale e giunzione neuromuscolare
Le fibre muscolari scheletriche dei topi SOD1G93A pre-sintomatici di 37 giorni hanno una riduzione del potenziale della membrana interna mitocondriale nei segmenti prossimali alla giunzione neuromuscolare. Questi segmenti di fibre hanno anche una maggiore attività di rilascio di Ca 2+
indotta da stress osmotico. È interessante indagare se ciò può contribuire alla degenerazione della giunzione neuromuscolare.
27
Da notare che la riduzione del potenziale di membrana mitocondriale e la disregolazione del rilascio di calcio sono eventi che precedono temporalmente molti dei difetti presenti a livello motoneuronale, deponendo ulteriormente per un precoce interessamento muscolare nella patologia. Effettuando trattamenti per stabilizzare la membrana mitocondriale e la regolazione del rilascio di calcio, si è diminuito il deterioramento della giunzione neuromuscolare, deponendo quindi per una correlazione tra i due compartimenti.
2.4 MicroRNA (miRNA) del muscolo scheletrico nella SLA
I miRNA sono piccoli RNA non codificanti che regolano post-trascrizionalmente l'espressione genica e inibiscono la traduzione proteica. Per ora pochi studi hanno avuto come principale interesse il ruolo dei miRNA nel muscolo scheletrico. Sembra che questi elementi abbiano un ruolo regolatorio: nella proliferazione delle SC, nell’innervazione delle giunzioni neuromuscolari e nella stabilità della membrana mitocondriale.
All’interno di campioni muscolari di pazienti con SLA, rispetto ai controlli sani, sono stati osservati livelli elevati di alcuni sottotipi di miRNA a funzioni specifiche (aumento attivazione, proliferazione e differenziazione delle SC) (Figura 7). Tuttavia, sono necessari ulteriori studi per estendere e validare queste osservazioni.
Figura 7.Segnalazione miR-206 nei muscoli a contrazione lenta e veloce. I livelli di miR-206 risultano più alti nei muscoli a contrazione lenta.
28
Non è attualmente noto se vi sia prima una disregolazione dei miRNA che porti poi ad un’alterazione nella miogenesi oppure se la loro alterazione sia solo un tentativo di attivazione per far fronte alla patologia. (Tsitkanou et al., 2016)
2.5 SOD1 mutato agisce indistintamente su tutti i muscoli?
Il muscolo scheletrico è un tessuto eterogeneo composto da diversi tipi di fibre con caratteristiche ultrastrutturali, contrattili e metaboliche distinte che consente ai muscoli di adattarsi alle mutevoli esigenze funzionali. I primi esperimenti condotti su topi SOD1 mutanti hanno mostrato una riduzione del consumo massimo di ossigeno nei mitocondri del muscolo Soleus a metabolismo ossidativo e contrazione lenta, rispetto a quello osservato negli estensori lunghi delle dita EDL a metabolismo glicolitico, suggerendo che la malattia influenzerebbe in particolare i muscoli che mostrano un metabolismo ossidativo.
Inoltre, gli studi di spettrometria di massa del modello murino di Wobbler sulla SLA hanno rivelato un aumento della quantità dell'enzima glicolitico G3PDH, suggerendo quindi un passaggio dal metabolismo ossidativo a quello glicolitico nel tentativo di far fronte alla malattia.
Tuttavia, studi contrastanti hanno stabilito che, nei SOD1 mutanti, le fibre a contrazione rapida isolate dai topi transgenici hanno sviluppato meno forza delle fibre a contrazione lenta (stimolazione con calcio), rispetto alle fibre di controllo isolate da topi in cui non era stata indotta la mutazione. La quantificazione del numero delle unità motorie si è visto calare già in uno stadio presintomatico nei muscoli a contrazione rapida mentre il calo delle unità ha colpito le unità a contrazione lenta in uno stadio sintomatico e quindi più tardivo. Le ragioni della diversa vulnerabilità tra fibre muscolari sono ancora oscure.
Inoltre, è stato osservato che le unità motorie a contrazione rapida diventano iperattive in risposta a lieve denervazione. Sembra plausibile che la conversione di fibre a rapido affaticamento in fibre ad alta resistenza alla fatica le renda più resistenti alla SLA. Questo fenomeno è stato osservato nei topi SOD1 mutanti nel corso della malattia. Tuttavia, il passaggio dal metabolismo glicolitico a quello ossidativo non è privo di rischi ma è accompagnato da disfunzione mitocondriale e stress ossidativo, fornendo ulteriori prove della vulnerabilità selettiva dei muscoli nella SLA. (Loeffler et al., 2016).
29 CAPITOLO 3
Generalità sulla FDG PET/TC in neurologia
La tomografia a emissione di positroni (Positron Emission Tomography, PET) è una tecnica di medicina nucleare che valuta la concentrazione locale di radiofarmaci emettitori di positroni (Salomon et al., 2015).
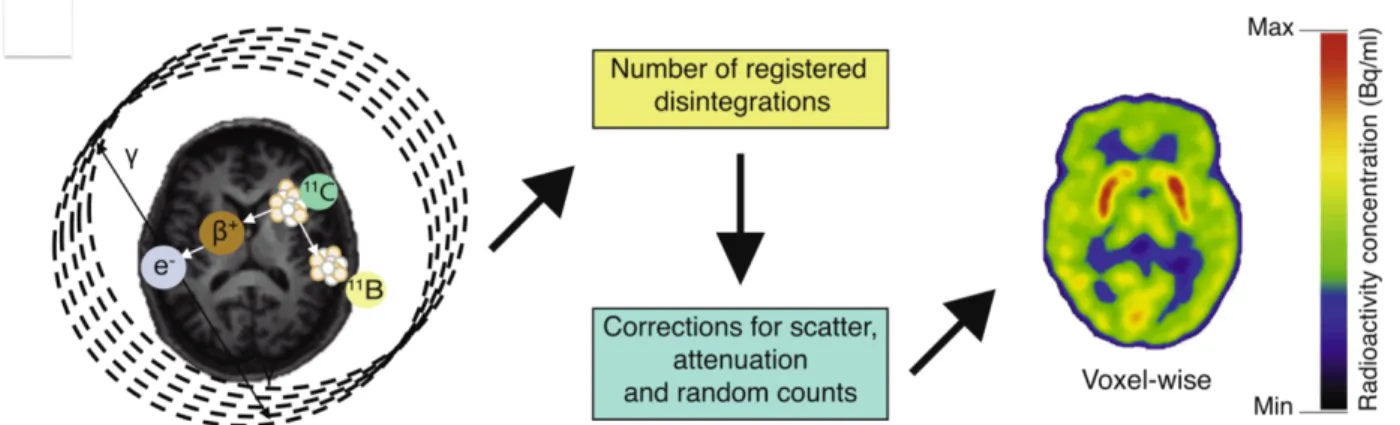
La PET si basa quindi sull’utilizzo di traccianti radiomarcati con isotopi radioattivi che decadono attraverso l’emissione di positroni, particelle con la stessa massa dell’elettrone ma con carica opposta. Una volta emesso, il positrone interagisce con un elettrone del tessuto circostante dando origine al fenomeno dell’annichilazione in cui, a seguito dell’impatto, vengono emessi due fotoni gamma ad alta energia (511 KeV), aventi stessa direzione ma verso opposto (Figura 8). I due fotoni possono essere rilevati in coincidenza temporale tramite un rilevatore circolare incluso nel tomografo PET, che è in grado di attribuire a tale segnale di coincidenza delle coordinate spaziali. Il rilevamento coincidente produce i cosiddetti conteggi che vengono successivamente ricostruiti e corretti per dare origine all’immagine. Le immagini PET vengono comunemente prodotte utilizzando scale di colori come la scala arcobaleno o la scala di colori Sokoloff. Convenzionalmente i colori caldi indicano le sedi di maggior concentrazione del tracciante ed i colori freddi ne indicano un accumulo minore. Un altro approccio è quello di mostrare le immagini in scala di grigi dove il nero rappresenta l’assenza di radioattività e il bianco la radioattività più elevata.
Figura 8. Fondamenti di tomografia ad emissione di positroni (PET). Un isotopo di breve durata, qui illustrato da un atomo di carbonio-11, si disintegra ed emette una particella beta carica positivamente. Questo si scontra con un elettrone che provoca il rilascio di due particelle gamma in direzioni opposte. I rivelatori nello scanner PET registrano simultaneamente particelle gamma registrate come un'unica disintegrazione. Le registrazioni di un'intera scansione PET, chiamate conteggi, vengono quindi corrette per perdite dovute a dispersione e attenuazione nei tessuti nonché conteggi casuali, e i dati vengono ricostruiti matematicamente dando come risultato un volume di immagine con i dati assegnati nella distribuzione spaziale di cui è stata stimata l’origine. Le immagini PET sono generalmente mostrate in scala di colori, con l'intensità del voxel (pixel 3-D) che rappresenta una diversa concentrazione di radioattività (Becquerel / ml di tessuto).
30
L’utilizzo di tecniche ibride combinando la tecnologia PET con la TC o la RM ha consentito di integrare informazioni di tipo funzionale, metabolico e biochimico della PET con informazioni di tipo anatomostrutturale, derivanti da TC o RM.
3.1 Radiofarmaci
Molte molecole di interesse biologico quali ossigeno, acqua, anidride carbonica, glucosio e vari farmaci, sono formati da atomi di cui esistono isotopi radioattivi che decadendo emettono positroni. I principali radionuclidi utilizzati in campo neurologico hanno la caratteristica di avere una breve emivita: 18F (110 min), 11C (20 min), 15O (2 min). Tutti i radiofarmaci utilizzati in PET sono
esclusivamente dei traccianti e non hanno azione farmaceutica vera e propria, questo è anche reso possibile dal fatto che vi sia un eccezionale sensibilità nella rilevazione dei fotoni emessi per cui la quantità di radiofarmaco iniettata è talmente bassa da non esplicare gli effetti del normale farmaco. (Heurling et al., 2017)
[18F] FLUORO-2-DESOSSIGLUCOSIO ([18F]-FDG)
Si tratta dell’analogo non metabolizzabile del glucosio. Come quest’ultimo, viene fosforilato dall’esochinasi ma a differenza del glucosio non subisce ulteriori processi della via di metabolizzazione per la mancanza del gruppo
ossidrile in posizione 2. Perciò tutto il [18F]-FDG si accumula
all’interno del neurone in maniera proporzionale all’attività neuronale. (Fazio et al., 2019)
È noto che il sistema nervoso utilizzi principalmente glucosio per la produzione di energia. Il glucosio entra nell'unità funzionale
neurone-astrocita dove viene poi metabolizzato, sia seguendo una via aerobia lenta sia una anaerobia rapida, per produrre energia. È stato inoltre dimostrato che il metabolismo del glucosio è strettamente associato alla funzione neuronale sia quando vige uno stato di riposo sia quando vi sia stato di attivazione. (Sokoloff et al., 1977; Magistretti et al., 1999)
Associando il [18F] con il glucosio, è possibile misurare in modo quantitativo (o semiquantitativo) il
metabolismo locale dei terminali sinaptici, poiché la maggior parte dell'utilizzo del glucosio avviene proprio a questo livello. Il tasso metabolico cerebrale di riferimento della sostanza grigia per i valori di glucosio (CMRglc) varia da circa 40 a 60 mmol di glucosio / 100 g di tessuto cerebrale / 1 min. a riposo in un adulto normale; nella sostanza bianca varia circa da 1/3 a 1/4 rispetto alla materia grigia. I valori più alti si trovano nei gangli della base, nel talamo e nella corteccia occipitale, mentre la
31
corteccia temporale mediale e il cervelletto hanno i valori più bassi. Il ridotto assorbimento di glucosio rappresenta essenzialmente una riduzione del numero di sinapsi o una ridotta attività metabolica sinaptica. (Rocher et al., 2003)
Nel soggetto sano, sia a livello corticale che sottocorticale il metabolismo glucidico cerebrale risulta simmetrico e omogeneo, con accentuata captazione della sostanza grigia rispetto a quella bianca (Fazio et al., 2019).
Dato che le immagini rappresentano la concentrazione di radioattività misurata nel tessuto, la PET è di per sé una tecnica quantitativa; la concentrazione di radioattività in ciascun voxel (“pixel tridimensionale”) determina l'intensità dell'immagine. Il segnale misurato fornisce una stima della concentrazione totale di radioattività, comprendente diversi stati del tracciante nel tessuto. Il segnale PET misurato è costituito da radioattività nel sangue, tracciante nello spazio extracellulare libero, tracciante intracellulare, tracciante legato al bersaglio e tracciante temporaneamente e vagamente legato ad altre entità all'interno del tessuto. (Heurling et al., 2017; Fazio et al., 2019)
3.2 Uso della FDG PET/TC nelle malattie neurodegenerative: implicazioni cliniche
Le malattie neurodegenerative sono caratterizzate dalla progressiva degenerazione e morte neuronale. Rappresentano un gruppo eterogeno dal punto di vista eziopatologico ma condividono alla base una comune deposizione anomala di proteine patologiche, ed è proprio sulla base di tali proteine che generalmente vengono classificate (Kovacs, 2016).
Studi neuropatologici hanno dimostrato che la degenerazione sinaptica precede la morte neuronale per un considerevole periodo di tempo ed è a questo livello che la FDG PET/TC si inserisce come elemento prezioso (Price et al., 1999; Rocher et al., 2003). In vivo l’assorbimento di FDG è fortemente correlato alla densità e all’attività sinaptica cerebrale. Il modello ipo/iper-metabolico cerebrale fornisce informazioni sull’entità, sulla sede e sull’endofenotipo della disfunzione neuronale. La tecnica, ormai consolidata negli studi clinici e di ricerca del morbo di Alzheimer (AD, Alzheimer disease), sta esprimendo al meglio tutte le sue potenzialità anche nelle altre patologie neurodegeneretive. Le linee guida cliniche e procedurali orientate al neuroimaging citano il ruolo clinico dell’FDG-PET ma ancora mancano raccomandazioni specifiche basate sull'evidenza per l’uso dell'FDG-PET nelle malattie neurodegenerative (Sorbi et al., 2012; Varrone et al., 2009). Ad oggi, il ruolo principale dell’FDG-PET si ha nella diagnosi precoce dell’AD a seguito del riscontro di mild cognitive impairment (MCI) e nella diagnosi differenziale dell'AD da altri disturbi, come la demenza a corpi di Lewy (DLB) e le malattie appartenenti allo spettro della degenerazione frontotemporale
32
(Herholz et al., 2002; Mosconi et al., 2008). Possono quindi essere messi in evidenza eventuali schemi patologici già in fase pre-demenza. Infatti, grazie alla sua sensibilità e alla sua significatività come valore predittivo negativo, un imaging PET-TC non alterato nella MCI indica una bassa probabilità di progressione verso AD, anche se sono presenti deficit di memoria clinicamente importanti (Dubois et al., 2007). Inoltre, nei Parkinsonismi, FDG-PET è stato anche dimostrato essere superiore per la diagnosi differenziale rispetto all’imaging dopaminergico (Hellwig et al., 2012).
3.2.1 FDG PET/TC nel Morbo di Alzheimer
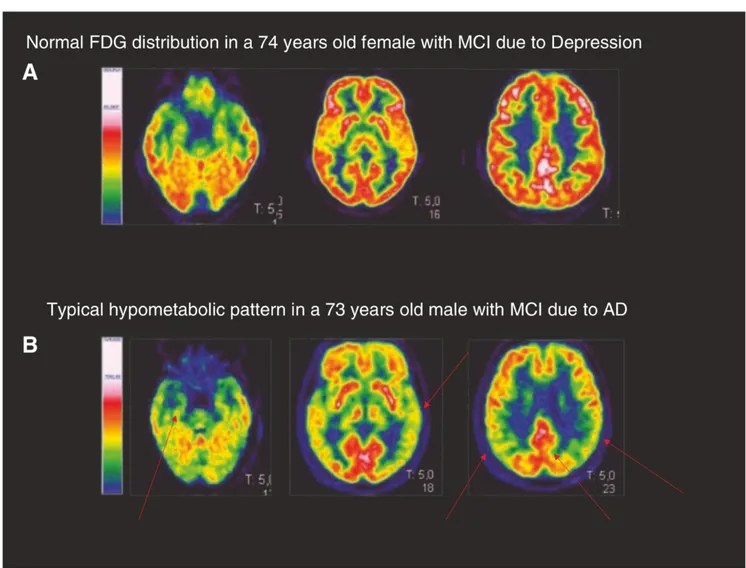
Lo schema tipico dell’AD all’ FDG PET/TC presenta ipometabolismo nei lobi temporali mediali (MLT), area di associazione parieto-temporale, cortecce cingolate posteriori e precuneus (Minoshima et al., 1995). Al contrario, le cortecce visive primarie, lo striato, il talamo, gli emisferi cerebellari e le cortecce motorio-sensoriali primarie registrano un assorbimento di glucosio conservato, distinguendosi quindi dalle aree di ipometabolismo (Figura 9).
Figura 9. Distribuzione normale FDG-PET e pattern tipico di AD in due pazienti con compromissione cognitiva lieve (MCI). Il paziente A, mostra una scansione normale, è stato confermato, al follow-up clinico, avere MCI a causa della depressione, mentre il paziente B ha un MCI a causa dell’ AD. Le frecce rosse evidenziano il modello tipico di AD nel paziente B (ipometabolismo nella corteccia parietale temporo-mediale, temporo-laterale e posteriore, nonché nel precuneo sinistro).
33
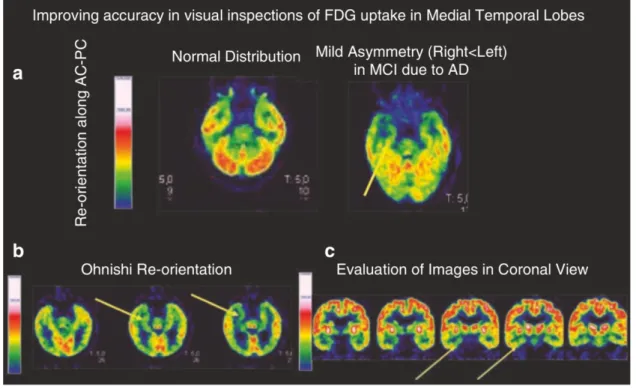
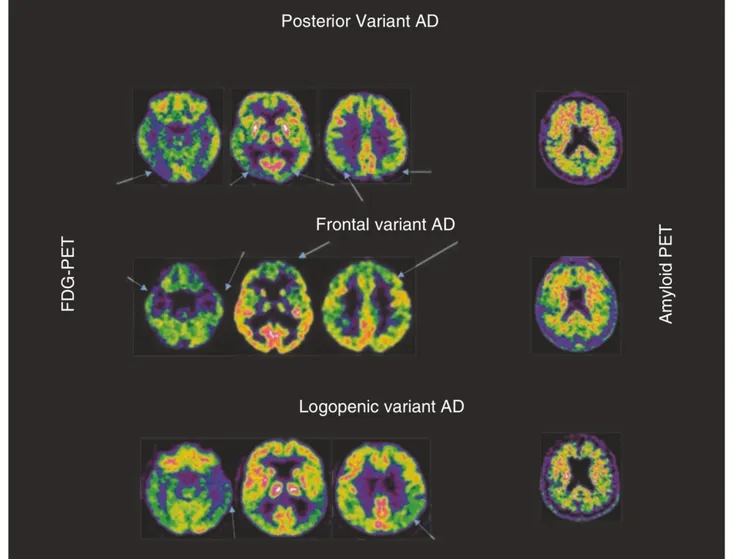
In stadi avanzati anche la corteccia associativa frontale potrebbe risultare ipometabolica (Herholz et al., 2007; Gallucci et al., 2008) con una relativa conservazione del giro cingolato anteriore. La corteccia entorinale e l’ippocampo sono tra le prime strutture colpite dall’AD. Inoltre, la ridotta attività del giro del cingolo posteriore risulta essere uno degli indicatori FDG-PET più sensibili e precoci di AD (Minoshima et al., 1997; Morbelli et al., 2010). La valutazione MTL non è sempre facile, spesso l’ipometabolismo non è chiaramente apprezzabile, per agevolare il suo rilevamento è stato proposto un diverso orientamento delle immagini PET (ri-orientamento di Ohnishi a 30–40° con il naso verso l'alto rispetto al piano bicommissurale) al fine di migliorare la valutazione dell'asimmetria nell'assorbimento di FDG in MTL (Figura 10-B) (Ohnishi et al., 1995). Specialmente nelle prime fasi, indipendentemente dalle regioni corticali interessate, una caratteristica molto frequente delle scansioni FDG-PET è il coinvolgimento asimmetrico dei due emisferi cerebrali (Figura 10-A). Di conseguenza, l’analisi visiva delle scansioni per il riconoscimento dello stato di patologia si basa fortemente su eventuali asimmetrie presenti. A parità di compromissione cognitiva i pazienti con AD a esordio precoce tendono a mostrare un ipometabolismo più esteso e con maggior coinvolgimento parieto-frontale rispetto ai pazienti con AD a esordio tardivo che al contrario sono caratterizzati da un coinvolgimento meno esteso e più evidente alla MTL (Figura 11) (Ohnishi et al., 1995; Kim et al., 2005). Anche le varianti atipiche di AD sembrano avere loro peculiarità (Figura
12). Nella variante logopenica, l’ipometabolismo sembra coinvolgere prettamente i lobi
temporo-parietali e posterolaterali di sinistra (Madhavan et al., 2013). L’atrofia corticale posteriore caratterizzata da difetti visuospaziali riporta una compromissione predominante nelle cortecce di associazione visiva parieto-occipitale coerente con il danno dorsale per l’elaborazione del flusso visivo (Laforce et al., 2014; Rosenbloom et al., 2011). In questo modo tutte queste varianti atipiche possono essere distinte tra loro e dalla demenza frontotemporale. Infine, mentre per supportare la diagnosi di demenza vascolare generalmente ci si avvale della clinica e dell’imaging RM, in alcuni casi particolarmente difficili di diagnosi differenziale potrebbe essere dirimente l’FDG PET/TC (Kerrouche et al., 2006).
34
Figura 10. Valutazione del lobo temporale mediale su FDG-PET. Pannello a: immagini FDG-PET di un controllo sano (lato sinistro, scansione normale) e di un paziente con AD (lato destro: lieve asimmetria in MTL con destra <sinistra). I pannelli b e c mostrano le immagini FDG-PET dello stesso paziente con AD visualizzate rispettivamente in base al riorientamento di Ohnishi e in vista coronale. L'asimmetria tra MTL destra e sinistra (frecce gialle) è evidente sia nei pannelli b e c che nel pannello a.
Figura 11.Modello ipometabolico tipico di AD a esordio tardivo (a) e AD a esordio precoce (b), allo stesso livello di MCI. In
esordio tardivo l'ipometabolismo è meno esteso e principalmente limitato ai lobi temporali mediali (frecce rosse) con. assorbimento solo lievemente ridotto nella corteccia parietale posteriore (frecce blu chiaro). In esordio precoce il modello ipometabolico è significativamente più esteso e coinvolge diverse regioni neocorticali tra cui la corteccia frontale bilaterale (frecce gialle).
35
Figura 12.Risultati di FDG-PET e PET amiloide nelle varianti atipiche di AD. Ogni variante atipica di AD è caratterizzata da un modello specifico di ipometabolismo (frecce blu). Tutte queste varianti sono caratterizzate da un'elevata deposizione di amiloide e sono quindi positive all'amyloid PET (il pannello di destra mostra la mancanza di contrasto tra assorbimento di materia grigia e bianca nelle scansioni amyloid PET di tutti i pazienti).
3.2.2 FDG PET/TC nella Demenza Frontotemporale (FTD)
La FTD non è una singola patologia ma rappresenta un gruppo di patologie eterogeneo dal punto di vista neuropatologico, genetico, clinico e radiologico di cui la variante comportamentale FTD (bvFTD) ne rappresenta la forma più frequente (Seltman et al., 2012). La bvFTD è caratterizzata da deterioramento della personalità, del comportamento sociale e della cognizione, può essere difficile da riconosce clinicamente in quanto i disturbi possono mimare patologie psichiatriche e l’MCI può essere lieve. Gli attuali criteri diagnostici per bvFTD richiedono conferma del sospetto clinico al neuroimaging (Rascovsky et al., 2011). La PET permette quindi di fare diagnosi differenziale (DD) tra FTD e AD oltre che a differenziare tra le varianti di FTD: variante comportamentale frontale, variante temporale/ demenza semantica (SD) e afasia progressiva non fluorescente (PNFA) (Whitwell et al., 2012; Morbelli et al., 2016) (Figura 13).
36
Figura 13.Pattern FDG-PET di ipometabolismo nelle varianti cliniche della demenza frontotemporale.
Risulta molto importante riconoscere una FTD anche sul piano terapeutico in modo da evitare la somministrazione di inibitori della colinesterasi che possono peggiorare ulteriormente lo stato clinico (Mendez et al., 2007; Chow et al., 2002). La bvFTD si caratterizza per ampio ipometabolismo nell’area temporale frontale e anteriore, giro del cingolo, uncus, insula e aree subcorticali includendo gangli della base e regioni talamiche (Jeong et al., 2005). Le cortecce temporali anteriori e cingolate sono le sedi in cui meglio si riesce a fare DD con AD (Womack et al., 2011).
La SD si caratterizza per un ipometabolismo fortemente asimmetrico ai lobi temporali prettamente nella porzione anteriore e a dominanza sinistra (Matias-Gulu et al., 2015; Rabinovici et al., 2008). La PNFA presenta invece ipometabolismo asimmetrico più evidente nell’area corticale perisilviana anteriore sinistra e nella corteccia fronto-insulare.
3.2.3 FDG PET/TC nella Demenza a corpi di Lewy
Malattia caratterizzata da sintomi simil malattia di parkinson a cui si accompagna declino cognitivo. Frequentemente esordisce con allucinazioni. L’FDG-PET è elencato tra i criteri di supporto per la sua identificazione ed è una tecnica sempre più utilizzata in questo tipo di patologia.
37
Lo schema ipometabolico si localizza nella corteccia associativa posteriore, nella corteccia visiva primaria e nella corteccia occipitale con un relativo risparmio delle strutture subcorticale e della corteccia somatomotoria primaria (Bauckneht et al., 2018).
Il precuneus si presenta ipometabolico, risulta conservato invece il giro del cingolo posteriore (segno dell’isola cingolata) (Graff-Radford et al., 2014).
3.2.4 FDG PET/TC in altre malattie neurodegenerative
Negli ultimi anni si sta cercando di trovare schemi anche per le altre patologie neurodegenerative. Il nostro studio cerca di fornire una caratterizzazione specifica e riconoscibile per la SLA che in questo senso differisce da quelli appena trattati perché non si limita alla mappatura del cervello ma cerca di creare uno schema integrato di tutti i sistemi colpiti dalla patologia.
3.3 Vantaggi e limitazioni
Come detto prima la FDG PET è in grado di rilevare la disfunzione sinaptica in uno stadio che precede la morte neuronale. La PET quindi è in grado di rilevare con l’ipometabolismo quelle aree che in un secondo momento saranno poi colpite da atrofia alla risonanza magnetica. Tuttavia, alcuni autori, ritengono che l’FDG PET debba essere strumento per seguire la progressione della malattia piuttosto che un criterio di diagnosi (Dubois et al., 2014; Morbelli et al., 2017). Considerando l'attuale disponibilità clinica dei biomarcatori di amiloidosi (CSF e PET amiloide) andrà valutata la costruzione di un algoritmo favorevole all’AD che dia la giusta collocazione diagnostica alle varie tecniche di imaging (Morbelli et al., 2017). Da considerare anche che gli approcci di mappatura statistica negli ultimi decenni sono riusciti a migliorare l’accuratezza diagnostica dell’FDG PET anche in fase precoce di malattia (Perani et al., 2014).
In conclusione, la disponibilità dell’FDG PET (e di altri biomarcatori) fornisce progressi imprescindibili nella valutazione clinica e nella comprensione dell’endofenotipo delle malattie neurodegenerative.
38 CAPITOLO 4
Uso della FDG PET/TC nella SLA
La SLA è una malattia neurodegenerativa di considerevole variabilità per quanto riguarda eziologia, età di insorgenza, fenotipo e progressione di malattia. Una prognosi attendibile riguardo ad un avanzamento lento o veloce è difficile nella pratica clinica e la diagnosi è spesso ritardata di almeno un anno dalla comparsa dei sintomi (Chiò, 1999), a volte anche per la difficoltà di una diagnosi differenziale con disordini che mimano la SLA e che non presentano anomalie rilevabili alla tomografia assiale computerizzata o alla risonanza magnetica nucleare (RMN). Quanto detto sottolinea come ci sia per la SLA un urgente bisogno di un biomarcatore affidabile, facilmente accessibile, con alta sensitività e specificità, con un alto valore prognostico e che vari con la progressione di malattia rendendolo adatto per il monitoraggio dei trattamenti. Una diagnosi precoce permetterebbe un pronto inizio della terapia oltre che una corretta comunicazione della diagnosi a pazienti e caregiver ed il possibile inserimento dei pazienti in trial clinici. La diagnosi di SLA necessita l’implicazione nella malattia sia del primo motoneurone (UMN) sia del secondo (LMN). I segni di quest’ultimo sono spesso apprezzabili alla valutazione clinica e all’elettromiografia, esame assai sensibile nel confermarne un suo coinvolgimento. Al contrario l’evidenza clinica dell’implicazione dell’UMN è più difficile dal momento che i segni clinici per determinarla sono poco sensibili e allo stato dell’arte non esistono test per valutarne il grado.
Studi che hanno utilizzato voxel-based RMN, pur con qualche inconsistenza, hanno dimostrato come nella SLA ci siano alterazioni del segnale nella corteccia motoria il cui spessore, a livello di gruppo, è diminuito (Cosottini et al., 2016). Tuttavia, per il paziente singolo un’atrofia che non si sovrapponga a quella trovata nei controlli sani della stessa età è riscontrabile solamente nel 50% dei casi. Inoltre, una bassa accuratezza diagnostica è stata riscontrata per le ipointensità a T2 che probabilmente riflettono gliosi e attivazione della microglia (Cosottini et al., 2016). D’altra parte, iperintensità a T2 nel fascio cortico-spinale sono state rilevate nella capsula interna nel 50% dei casi di SLA ma solo nelle fasi avanzate di malattia rendendole non adatte per una diagnosi precoce.
Al contrario, una significativa riduzione dello spessore corticale è stata messa in evidenza dalla morfometria surface-based che ha anche permesso di correlare quest’ultimo alla severità dei
sintomi nei pazienti con SLA e demenza frontotemporale (ALS-FTD); (Agosta et al 2012; Mezzapesa et al 2013).
39
La Diffusion Tensor Imaging (DTI) ha invece dimostrato come nella SLA sia presente una riduzione della anisotropia frazionaria nei tratti cortico-spinali, nel corpo calloso e nelle aree frontotemporali ed un aumento della diffusività media nei tratti cortico-spinali e nelle aree frontotemporali entrambe le condizioni proporzionali sia alla severità clinica sia alla rapidità della progressione di malattia (Agosta et al., 2014). Tuttavia, la DTI dei fasci cortico-spinali benchè sia un promettente biomarcatore non raggiunge livelli di accuratezza accettabili come discordanti sono i dati di stimolazione magnetica transcranica e potenziali evocati della corteccia motoria che riportano sia ipoeccitabilità sia ipereccitabilità. Studi di spettroscopia di risonanza magnetica (MRS) hanno messo in evidenza ridotte concentrazioni di N-acetilaspartato (NAA) nella corteccia motoria primaria, peraltro proporzionali alla gravità ed alla progressione della SLA, nelle regioni premotorie e frontali ed in alcune regioni sottocorticali (Stagg et al., 2013). Inoltre, sono stati descritti ridotti rapporti tra il NAA, la creatina e la colina nei fasci cortico-spinali intracerebrali e bulbari. Dal momento che le neuroimmagini strutturali convenzionali (RMN) e di recente applicazione (DTI, MRS) pur rilevando atrofia della corteccia motoria e alterazioni dei fasci cortico-spinali hanno una scarsa accuratezza diagnostica e una scarsa capacità di identificare i processi patologici fini che sottendono la SLA, un tentativo di approfondire la conoscenza a livello di gruppo e di singolo paziente dello stato neuroinfiammatorio e metabolico è stato fatto con la PET.
Nel passato alcuni traccianti sono stati usati per identificare nella SLA la perdita selettiva o la disfunzione neuronale. L’ 11C-Flumazenil si lega ai recettori GABA-A espressi dai neuroni ed il suo uptake è considerato proporzionale alla presenza dei neuroni piramidali motori e degli interneuroni. L’uso di questo tracciante ha rivelato recentemente un coinvolgimento selettivo delle cortecce motorie primarie e associativa correlato con lo score clinico di deficit del primo motoneurone (Turner et al., 2005). Un secondo tracciante, l’ 11C-WAY100635, affine ai recettori 5-HT1A della serotonina espressa nei neuroni piramidali, ha mostrato una diminuzione pronunciata degli stessi nella corteccia motoria e nelle regioni frontotemporali (Turner et al., 2005).
Altri studi PET sono stati intrapresi per evidenziare la presenza di neuroinfiammazione e gliosi, precedentemente dimostrate essere una delle caratteristiche patologiche più frequentemente descritte nella SLA. La presenza di neuroinfiammazione è stata evidenziata da 11C-PK11195, 18F-DPA-174 e 11C-PBR28, marcatori della translocator protein (TSPO) presente nelle membrane mitocondriali della microglia attivata. Il risultato comune a questi studi è stato un aumento della concentrazione di questi marcatori nella corteccia motoria (Turner et al., 2004; Corcia et al., 2012; Lavisse et al., 2012), positivamente correlato con gli score di UMN e negativamente con l’ALS-FRS, indice di declino