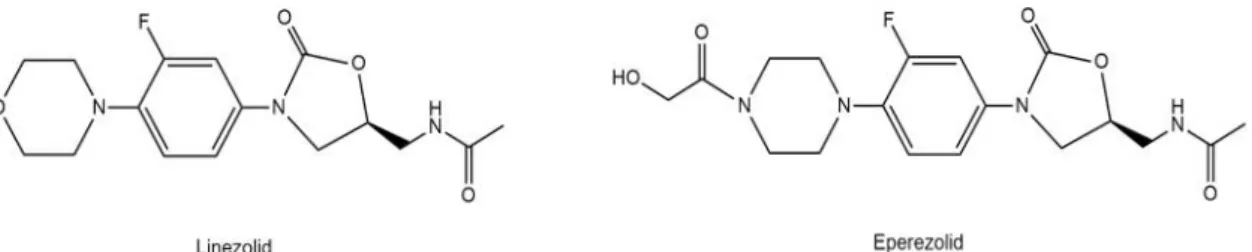
1. Introduzione
Uso di scaffold privilegiati per la progettazione dei farmaci
Piccole molecole organiche possono risultare di grande interesse sia a livello biologico che clinico, in quanto rappresentano utili strumenti sia terapeutici che diagnostici [1]. Nonostante i recenti progressi scientifici, quali la capacità dei chimici sintetici di preparare librerie comprendenti migliaia di composti, la scoperta di molecole efficaci da utilizzare nel trattamento di patologie specifiche rimane difficoltosa e fortuita [2]In gran parte questo è dovuto al fatto che non si conoscono in modo approfondito tutti gli elementi necessari alla progettazione di composti con attività biologica potente e specifica. Per esempio, le librerie di composti commerciali, sebbene facilmente reperibili, presentano alcuni svantaggi: proprietà chimico-fisiche non adatte e mostrano una bassa variabilità strutturale [3]. Al contrario, le librerie sviluppate da prodotti naturali bioattivi danno maggiori garanzie di successo, dal momento che il composto originario si è evoluto nei millenni per uno specifico scopo biologico [3]. Ad ogni modo, queste librerie di prodotti naturali portano raramente alla scoperta di molecole con una attività diversa da quella del composto “originario’’, in quanto si tratta di prodotti analoghi ottenuti per semplice modifica dei gruppi funzionali, piuttosto che da una progettazione razionale volta ad ottenere una nuova specificità di azione [1]. Di conseguenza, riuscire a creare librerie di piccole molecole altamente potenti e specifiche, potrebbe velocizzare l’identificazione di composti potenzialmente utili per il trattamento terapeutico, anche risolutivo, di alcune patologie.
In questo senso, una valida strategia è quella che prevede la creazione di librerie di composti sulla base di uno “scaffold privilegiato”.
Il termine “privileged scaffold” è stato coniato da Evans alla fine del 1980 per indicare elementi strutturali in grado di fornire ligandi per una vasta gamma di recettori [4]. Il lavoro svolto nel corso degli ultimi decenni da numerosi gruppi di ricerca, sia accademici che dell'industria, ha rivelato che svariate strutture chimiche possono essere identificate come privilegiate [5].
Effettuando uno studio approfondito della letteratura, risulta infatti che il termine “struttura privilegiata” viene utilizzato liberamente e in modo più ampio rispetto all'originaria concezione data da Evans, e cioè per indicare sia molecole capaci di legare bersagli multipli, sia molecole che, pur essendo caratterizzate da uno stesso scaffold, producono attività biologiche diverse. Al momento non esiste una lista esaustiva comprendente tutte le strutture privilegiate. Molte di queste sono comunque riportate nelle Tabelle 1-4; queste includono strutture privilegiate individuate sia da molecole “create de novo” dall'industria, e che sono ora farmaci, sia da composti di origine naturale che sono serviti da base strutturale per la sviluppo di composti di interesse farmaceutico. Come si può notare da queste tabelle, vi è una notevole sovrapposizione tra le strutture delle due classi; infatti, la stragrande maggioranza delle strutture si ritrovano come membri di entrambi i gruppi. Tutto ciò non è così sorprendente, nel senso che, una volta trovata la soluzione adatta per un particolare problema biochimico, la natura tende a ripetersi: le strutture macromolecolari dei sistemi viventi hanno infatti un alto livello di “patterning” non casuale. È interessante notare che ci sono pochi esempi di molecole per le quali non esistono analoghi in natura (Tabella 2).
Tabella 3.
Tabella 4.
considerazione lo scaffold purinico [6], probabilmente il più abbondante eterociclo azotato presente in natura [7]. La possibilità che il nucleo purinico abbia uno status privilegiato sembra intuitiva, dato il suo coinvolgimento in una vasta gamma di processi metabolici e cellulari [6,8]. L'obiettivo del gruppo di Schultz era quello di individuare composti contenenti lo scaffold purinico che potessero modulare l'attività delle chinasi ciclina dipendenti (CDK) e, di conseguenza, la crescita delle cellule leucemiche, visto il ruolo essenziale delle CDK nella regolazione del ciclo cellulare.
Un altro esempio di sviluppo di una libreria basata su strutture privilegiate proviene dal settore dell'industria, ovvero dagli scienziati della Merck. Questi hanno utilizzato lo
scaffold 2-arilindolico per la ricerca di nuovi ligandi per i recettori accoppiati a proteina G
(GPCR) [10]. Il fatto che il triptofano, contenente l’indolo, funga da precursore biosintetico della serotonina è una spiegazione plausibile per l'affinità della serotonina al recettore [11]. Questi ricercatori hanno scelto di preparare miscele combinatoriali nel tentativo di creare una vasta libreria di strutture indoliche che contenesse decine di migliaia di membri, in relativamente poche operazioni sintetiche. Al centro del loro progetto ritroviamo la classica sintesi di Fischer dell’indolo, che era stata descritta in precedenza in fase solida [12].
Come indicato nello Schema 1, prima un chetoacido arilalchilico viene bloccato su una resina solfonammidica, e successivamente si effettua una ciclizzazione con arilidrazina in modo da ottenere l'anello indolico. In totale, utilizzando diversi membri di ciascun componente, è possibile ottenere fino a 400 composti. La resina così prodotta è stata divisa equamente in 80 diversi pools, dove le sulfonammidi sono state alchilate nelle condizioni di Mitsunobu e sostituite con 80 ammine diverse; queste operazioni sono state utilizzate per la preparazione di 32.000 composti diversi. La resina è stata poi ricombinata e separata in due gruppi, portando in ultima analisi a 128.000 composti attraverso la
produzione separata di due nuove librerie, una costituita da ammine e l'altra costituita da ammidi.
Schema 1.
Il successivo screening biologico ha evidenziato alcuni composti quali potenti ligandi per specifici GPCR, fra cui i recettori di neurochinina, chemiochine, e serotonina. Uno di questi composti con elevata affinità di legame per la neurochinina umana-1 (kNK1), è stato usato come lead per un nuovo progetto della Merck, che ha portato all'ottenimento di un composto attualmente in fase di sperimentazione clinica [13].
Gli esempi appena descritti mostrano che il punto chiave per la costruzione di una libreria basata su una struttura privilegiata, non comporta solo lo sviluppo di nuove tecnologie, oppure di procedure sintetiche generali, ma una progettazione razionale che tenga in considerazione i parametri farmacocinetici e l'attività biologica degli scaffold.
La domanda che quindi ci si pone è: “come si fa a scoprire nuove strutture privilegiate?”
Recentemente Fesik e collaboratori hanno tentato d’identificare nuovi scaffold saggiando oltre 10.000 composti su 11 differenti bersagli proteici mediante test di binding basati sull’NMR [14]. Stranamente, la maggior parte delle strutture individuate erano elementi già presenti in composti biologicamente attivi, noti e considerati privilegiati [5].
Hu e collaboratori hanno recentemente condotto una valutazione sistematica del profilo di selettività del BindingDB database mediante metodi computazionali. Con questo studio su larga scala si sono identificati oltre 200 scaffold che hanno una selettività verso target simili; alcune di queste strutture potrebbero costituire nuovi potenziali scaffold [15]. A parte questi studi, dato l’enorme numero di modi con cui gli atomi possono essere combinati in strutture organiche, è ragionevole aspettarsi che le strutture attualmente esplorate siano estremamente poche rispetto all'intera gamma di complessità e proprietà strutturali che è possibile avere [16]. Sembra quindi molto probabile che ci siano decine di strutture privilegiate ancora da definire.
L'impiego della sintesi diversity-oriented, concetto introdotto da Stuart Schreiber, è un modo di affrontare questo problema. Le nuove molecole, sia in termini di struttura che di stereochimica, sono create in sequenze di reazioni relativamente brevi, in genere non più di 4 o 5 passaggi, utilizzando reazioni molto complesse (ad esempio cicloaddizioni Diels-Alder o reazioni multicomponenti di coupling di Ugi). Altre strategie per avere una maggiore varietà, includono percorsi di differenziazione basati sui reagenti [17], sui substrati [17], e su una strategia a tre fasi [18]. Questo approccio potrebbe facilitare la
scoperta di nuove strutture biologicamente utili e consentire l'individuazione di nuove strutture privilegiate (una volta noti i dati relativi ad un particolare scaffold).
Una seconda possibilità è valutare i motivi strutturali che hanno tradizionalmente dimostrato un difficile approccio sintetico, ma che sono presenti in decine di prodotti naturali. Tali esempi sono certamente più rari: la Tabella 3 illustra tre strutture onnipresenti in natura ma che non si trovano attualmente in farmaci in commercio. Un altro esempio è costituito dai prodotti naturali alogenati, come quelli mostrati nella Figura 1.
Figura 1.
Quello che può essere affermato con certezza è che la saturazione, in termini di numero di possibili strutture privilegiate che potrebbero modulare i sistemi biologici, non è stata ancora raggiunta.
L'indolo quale struttura privilegiata
La struttura dell'indolo, rappresenta una delle più importanti subunità strutturali per la scoperta di nuovi farmaci. La dimostrazione che molti alcaloidi contengono il nucleo indolico, è riconosciuto dall'importanza del triptofano, aminoacido contenente la struttura indolica, essenziale nella nutrizione umana come costituente di molte proteine e precursore biosintetico del neurotrasmettitore serotonina, inoltre la scoperta di ormoni vegetali ha stimolato un'intensa ricerca sulla chimica indolo, dando origine a un vasto numero di derivati naturali e sintetici biologicamente attivi [21], saggiati farmacologicamente come agonisti e antagonisti dei recettori accoppiati a proteine G, bloccanti dei canali ionici e inibitori di enzimi. L'anello indolico è una subunità aromatica ricca di elettroni, dovuta alla frazione pirrolica. E' ampiamente distribuito nei sistemi biologici come importante costituente di biomolecole e prodotti naturali. Inoltre, la struttura indolica è presente in molti farmaci e in nuovi composti sintetizzati costituenti delle proteine; il triptofano ha la maggiore idrofobicità tra tutti gli altri amminoacidi e forma un ambiente idrofobico che potrebbe stabilizzare ulteriormente la struttura proteica [22]. È interessante notare che l'anello indolico del triptofano, nel sito attivo di alcuni metalloenzimi, ad esempio, il citocromo C e la perossidasi, è risultato coinvolto nel trasferimento di elettroni [23], indicando che la presenza di questi residui di aminoacidi nelle proteine non solo contribuiscono a fornire un ambiente idrofobico adeguato ma potrebbe anche prendere parte ad alcune reazioni enzimatiche.
Considerando tutte le sequenze proteiche , il triptofano è il più raro tra i comuni venti amminoacidi, (poco più dell'1%). Tra questi aminoacidi, solo quattro possiedono catene laterali aromatiche e solo tre di quelle presentano anelli ricchi di elettroni. Il nucleo imidazolico dell'istidina è povero di elettroni. Fenilalanina e tirosina, hanno una frequenza del 4 e 3%. Per quanto riguarda l'istidina (~ 2,5% occorrenza), la catena laterale presente
sulla struttura indolo del triptofano, è un eterociclo contenente azoto. Anche nella tirosina, l'indolo presenta una subunità donatore di legame idrogeno, cioè NH, e presenta un anello eterociclico ricco di elettroni, che possono essere coinvolte in molte interazioni supramolecolari. Queste caratteristiche rendono l'anello indolico del triptofano, un quadro chimico raro ma molto speciale di nostri peptidi e proteine [24].
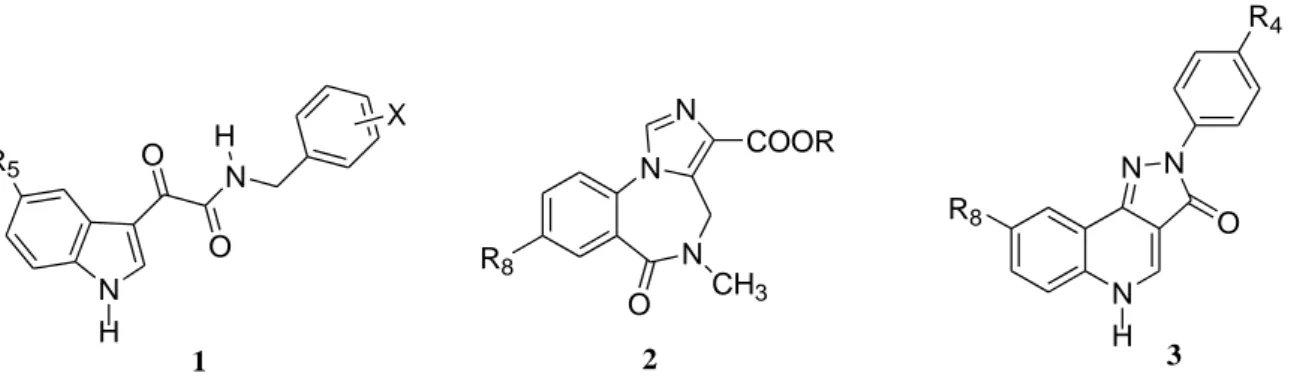
Alcuni ricercatori come Da Settimo e collaboratori, hanno messo in evidenza l'importanza della presenza del gruppo NH indolico nelle interazioni con il recettore, mediante sostituzione isosteriche del nucleo indolico con benzotiofene e benzofurano in una serie di derivati gliossilammidici, (composto 1, Figura 2) progettati come ligandi del recettore delle benzodiazepine, [25].
Questo lavoro ha dimostrato che, nonostante la letteratura indicasse che la presenza del gruppo NH come donatore di legami idrogeno non fosse necessaria per avere una risposta agonista, tutti derivati benzofuranici e benzotiofenici derivati, hanno mostrato una bassa affinità per il recettore, dimostrando che l'NH dell'anello indolico, svolge un ruolo chiave nell'interazione di questa classe di gliossilammidi. [26].
Il doppietto di elettroni dell'azoto dell'anello indolico, è coinvolto nel mantenimento del sistema aromatico, cosicchè il legame NH è acido (pKa ~ 17). L'anello indolico è in grado di avere legami non covalenti con cationi ed altre molecole, tramite il legame ad idrogeno della componente NH, attraverso la funzione aromatica [22].
Tutti gli esempi riportati in letteratura dei diversi composti e recettori, confermano l'importanza della struttura privilegiata. Non importa se l'ispirazione di questi composti sintetizzati è stata basata sulla natura (alcaloidi indolici, triptofano) o sulla chimica combinatoriale, questi approcci hanno dato luogo ad una serie diversificata di strutture biologicamente importanti, molte delle quali sono diventate farmaci, altre sonde per saggi farmacologici e la grande maggioranza composti lead. Bondensgaard e co, hanno
ligandi. Utilizzando frammenti di strutture privilegiate selezionate dalla letteratura, come il 2-fenil-indolo, hanno fatto il daking di tre paia di ligandi raggruppando i tipi recettoriali in vari modelli. Ad esempio, il recettore della serotonina 5-HT6 legato al composto 2 e il recettore melanocortina-4 (MC4) legata al composto 3 (Figura 1) [27].
Figura 2.
Dal confronto dei complessi recettore-ligando, gli autori hanno mostrato che la natura conservata della tasca di legame dove gli scaffold privilegiati si possono posizionare e le ulteriori interazioni con le parti non conservate della tasca, sono responsabili del diverso riconoscimento delle molecole da parte del recettore corrispondente. [27].
Inoltre Rad e colleghi, hanno riportato che la sottostruttura dell'indolo potrebbe essere convalidata come struttura privilegiata per bersagliare GPCR. Tra le 615 strutture della letteratura della chimica farmaceutica, 41 sottostrutture sono state classificate come "chimicamente privilegiate" e solo 6 di loro, tra cui l'indolo, sono state classificate come "biologicamente privilegiate". Questo concetto di "validazione biologica" è stato utilizzato per distinguere sottostrutture "chimicamente privilegiati" da quelle che hanno superato i test biologici e che forse hanno acquisito rilevanza clinica [28].
N H O H N O OCH3 1 N H N Br N H Br H3C N H 2 3
La capacità delle ammidi di formare legami a idrogeno, è stata ampiamente studiata e indagata in studi di ingegneria cristallografica [29]. Le ammidi acicliche secondarie formano legami a idrogeno tramite le loro catene laterali, mentre le ammidi primarie generalmente formano questi tipi di legami grazie alla presenza di un ulteriore donatore di legame a idrogeno. Le gliossilammidi hanno un maggior numero di caratteristiche rispetto alla semplice funzione ammidica.
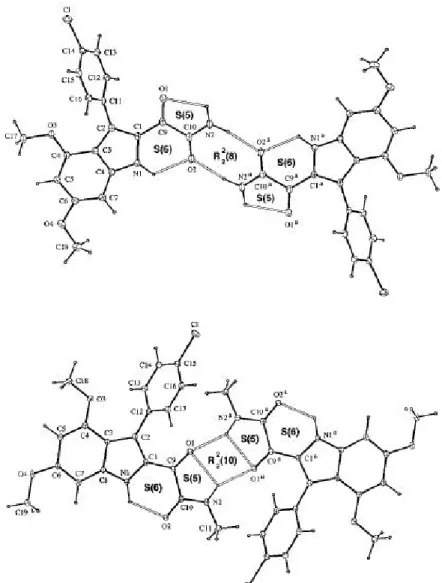
Le gliossilammidi sono dotate di una maggiore versatilità del sistema, grazie alla presenza di un legame idrogeno supplementare del gruppo chetonico e l'angolo di torsione variabile. La tendenza della funzione indolo-2-gliossilammidica, esemplificato nei composti 1a e 1b, a formare anelli con legami a idrogeno intramolecolari, conferisce rigidità al sistema ed elimina la necessità di sintetizzare anelli con legami covalenti. [30] I composti 1a e 1b, formano due anelli di legami a idrogeno intramolecolari, un anello a sei termini (S (6)), e un anello a cinque termini (S (5)) (Figura 3). [31]
Figura 3.Struttura cristallina a raggio X e rappresentazione del legame ad idrogeno della gliossilammide primaria 1a (in alto) e secondaria 1b (in basso).
Il sistema S(6) forma legami a idrogeno intramolecolari tra l'NH dell'indolo e l'ossigeno carbonilico dell'ammide ed è presente in entrambe le molecole 1a e 1b (Figura 2). Il sistema S(6) contiene la gliossilammide nella porzione dell'anello indolico, massimizzando l'interazione sterica con le molecole vicine.
Il legame a idrogeno intramolecolare nell' anello a 5 termini, dell'NH ammidico con il gruppo chetonico, è un interazione più debole ma altrettanto importante ed è presente in entrambe le strutture.
La gliossilammide primaria 1a, dimerizza preferenzialmente in direzione "down" (figura 4) in quanto coinvolge l'ossigeno carbonilico dell'ammide, che è un accettore di legame idrogeno più forte rispetto all'ossigeno carbonilico chetonico. [32]
1a: R = H 1b: R = CH3 Figura 4.
Il risultato è la formazione di un anello a 8 termini con un legame intermolecolare che coinvolge l' NH ammidico e l'ossigeno del carbonile ammidico (R (8)). Il sistema R (8), è costituito da legami ad idrogeno molto forti ed è quasi completamente coplanare con l'anello indolico. A differenza delle semplici ammidi primarie, la gliossilammide primaria 1a, non forma un motivo a nastro, a causa dell'ingombro sterico dovuta alla planarità del dimero.
È interessante notare che nella formazione dell'indolo-2-gliossammide 1a come dimero, presenta alcune somiglianze strutturali con al coppia di basi adenina timina di Watson-Crick, caratteristica importante da un punto di vista biologico. [33]
La semplicità di sintesi, la versatilità dei derivati indolici e la potenza attribuita alle gliossilammidi, hanno portato allo sviluppo di una serie di composti, contenente lo scaffold indolgliossilammidico, che hanno mostrato una serie di attività in Medicinal Chemistry. Le patologie in cui questi composti hanno mostrato di avere una certa attività sono
N H O OCH3 H3CO N O H R Cl up down
2. Attività antitumorale
Nei prossimi anni in tutto il mondo, è previsto un drammatico aumento di morti per casi oncologici e tumori. Nel 2001 in tutto il mondo, circa 10 milioni di persone soffrivano di cancro e oltre 6 milioni di persone sono morte di questa malattia. Dietro il termine cancro o tumore si nasconde un quadro clinico che comprende più di 200 malattie diverse. I tumori più importanti sono quelli del polmone, della mammella, dello stomaco, del collo dell'utero, della prostata, della testa e del collo, del grande e piccolo intestino, del fegato e del sistema sanguigno. Ci sono grandi differenze rispetto l'andamento, la prognosi e la reazione della terapia. Più del 90% dei casi conosciuti che riguardano i tumori solidi, in particolare quelli in fase avanzata o in metastasi, sono difficili da trattare o addirittura incurabili. I tre interventi nel controllo del cancro, sono ancora la rimozione chirurgica, l'irradiazione e la chemioterapia. Nonostante i grandi progressi, non è ancora stato possibile sviluppare farmaci che determinino un marcato prolungamento del tempo di sopravvivenza o addirittura una completa guarigione nei casi di tumori solidi [34].
Attualmente sono disponibili molti metodi utilizzabili nel trattamento del cancro. Nonostante i notevoli progressi, i trattamenti per molti tipi di cancro risultano inadeguati per una serie di motivi. Ci sono ancora dei tumori, che non rispondono o rispondono poco ai trattamenti attualmente disponibili. I pazienti con tumori curabili, spesso devono sottoporsi a chemioterapia con farmaci che provocano gravi effetti collaterali. Pochi di questi farmaci possono essere usati per via orale. Forse il problema più grave associato alla chemioterapia per il cancro, è lo sviluppo di multi-resistenza ai farmaci da parte di molti tumori. Ad esempio, molti tumori che inizialmente rispondono positivamente ad una terapia anti-cancro diminuendo in dimensioni o addirittura regredendo, spesso
sviluppano resistenza al farmaco. I tumori che hanno sviluppato una resistenza a più di un farmaco, sono detti " multi-farmaco resistenti ". Una volta che il cancro di un paziente è diventato resistente a più farmaci, purtroppo si può fare ben poco per fermare o ritardare ulteriormente la progressione della malattia.
C'è quindi una necessità di nuovi farmaci, che superino una o più carenze dei farmaci attualmente utilizzati nel trattamento del cancro. Le proprietà desiderabili dei nuovi farmaci anti-cancro includono l'efficacia contro i tumori che sono attualmente incurabili o poco curabili, l'efficacia contro i tumori resistenti a molti farmaci, la biodisponibilità orale e/o effetti collaterali ridotti [35].
Un agente antitumorale recentemente scoperto, il D-24851 (Indibulin) (Tabella 5), contiene lo scaffold indolgliossilammidico e possiede un attività antitumorale in vitro/in
vivo [36-38].
E 'stato visto che sostituzioni isosteriche, con eterocicli, portano a vantaggi di tipo farmacologico e farmacocinetico [39-42].
Altri studi, hanno dimostrato che sostituzioni eterocicliche possono causare cambiamenti nel grado di ionizzazione dei composti a pH fisiologico, modificare la basicità e lipofilia e portare a differenze sostanziali nelle proprietà farmacocinetiche [40].
Inoltre, sostituzioni eterocicliche del fenile, con anilina, o con una struttura isosterica, hanno portato ad una migliore efficacia farmacologica, migliori proprietà farmacocinetiche e minore tossicità [41].
Il cambiamento di orientamento (posizione di sostituzione) dell'anello isossazolo, modifica l'attività biologica [39].
Sarebbe interessante sapere se la sostituzione degli anelli benzenici con anelli eterociclici in posizione 1 e/o 3 dello scaffold indolgliossilammidico possono migliorare le loro attività biologiche.
Nel 2003 Li et al.[43], hanno riportato la sintesi di una serie di 1-15 indolgliossammidi N-eterocicliche e la valutazione della loro attività biologica come inibizione della crescita
in vitro di cellule tumorali, e come aumento della sopravvivenza degli animali malati di
cancro.
Figura 5.
Tutte le indolgliossilammidi sintetizzate, sono state esaminate inizialmente per l'attività antitumorale utilizzando cellule gastriche umane, le NUGC3. Le indolgliossilammidi attive selezionate, sono state poi ulteriormente valutate per ottenere i loro IC50, la concentrazione che provoca il 50% di inibizione della crescita delle cellule tumorali nei confronti di sei linee di cellule umane, cellule gastriche NUGC3, cellule epatocellulari HepG2, cellule del seno MCF7 ed il suo ceppo doxorubicina (adriamicina) MCF7-resistente/ADR, cellula dell'utero MES-SA ed il suo ceppo MES-SA doxorubicinresistente/Dx5, e una linea leucemica murina cellulare P388. L'attività antitumorale è stata valutata con un test colorimetrico [44], 3-(4,5 dimetiltiazol-2-il)-5-(3-carbossimetossifenil)-2-(4-sulfonil)-2H-tetrazolina (MTS) e fenazina metosolfato (PMS), misurando l'attività delle cellule tumorali residue dopo il trattamento con le indolgliossilammidi.
I cambiamenti apoptotici nella morfologia, nei nuclei di condensazione e nei corpi delle cellule tumorali al trattamento con le indlogliossilammidi, sono stati visualizzati con il metodo di fluorescenza Hoechst [45].
Come visualizzato con l'analisi della frammentazione del DNA nelle cellule del cancro gastrico NUGC3 umano, questi composti inducono apoptosi.
I composti 6 e 10 con attività antitumorale in vitro, sono stati ulteriormente valutati per la loro attività antitumorale in vivo su un modello di sopravvivenza leucemica al cancro P388, in un giovane topo femmina DBA/2J [46].
E' stato calcolato il coefficiente di ripartizione per le indolgliossilammidi tra acqua e n-ottanolo [47].
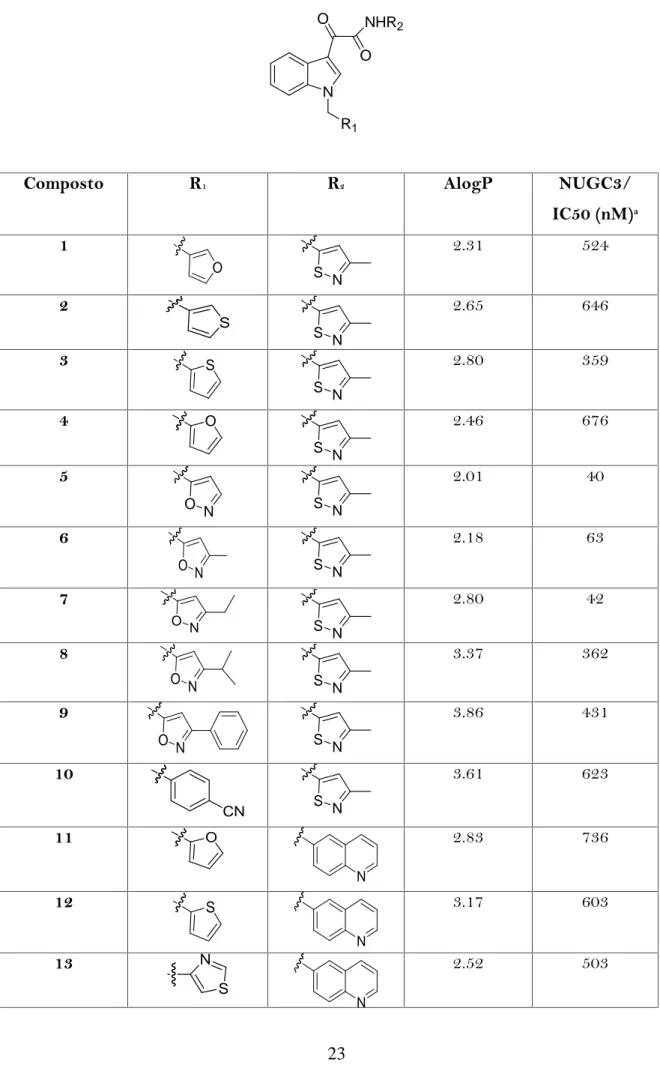
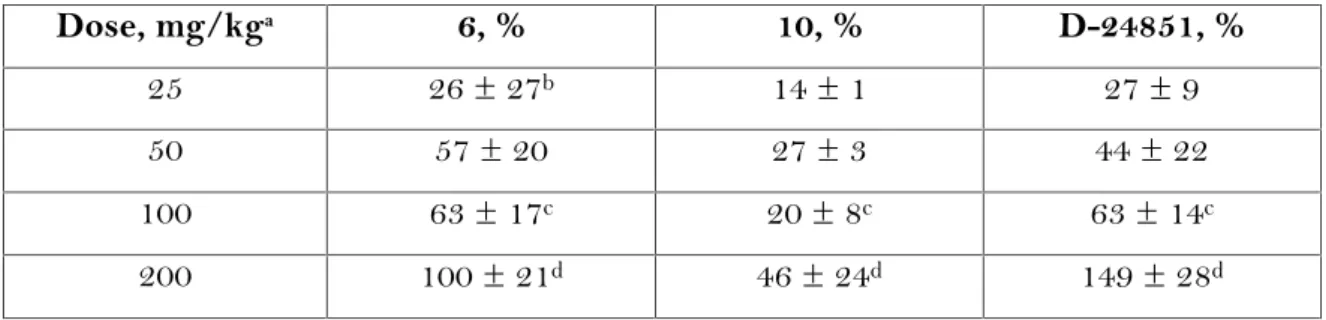
Il valore AlogP è un indice della capacità di una molecola di penetrare la membrana cellulare e tissutale. Il valore AlogP previsto per D-24851 è 3.30 mentre i valori per le altre indolgliossilammidi sintetizzate, sono elencati nella tabella 5.
In generale, sostituzioni N-eterocicliche delle indolgliossilammidi, riducono AlogP e quindi si ha una maggiore solubilità in acqua. Si può notare tuttavia, che non vi è alcuna correlazione tra i valori AlogP e l'attività antitumorale in vitro. La concentrazione IC50dei composti 1-15 e D-24851 sono stati determinati e riportati in Tabella 6.
Tabella 5. N R1 NHR2 O O
Composto R1 R2 AlogP NUGC3/
IC50 (nM)a 1 O S N 2.31 524 2 S S N 2.65 646 3 S S N 2.80 359 4 O S N 2.46 676 5 O N S N 2.01 40 6 O N S N 2.18 63 7 O N S N 2.80 42 8 O N S N 3.37 362 9 O N S N 3.86 431 10 CN S N 3.61 623 11 O N 2.83 736 12 S N 3.17 603 13 S N N 2.52 503
14 S N 3.02 206 15 O N N 2.54 262
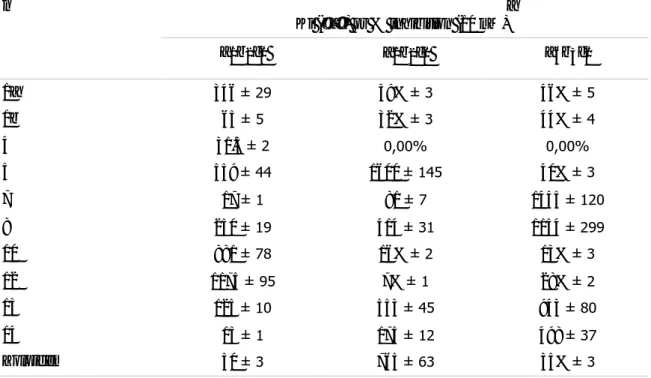
Tabella 6.Attività Anticancerogena delle Indologliossilammidi nella Serie Cellule Cancerogenea
Composto Gastric
NUGC3 HepG2Liver BreastMCF7 MCF7/ADRBreast MES-SAUterus Uterus MES-SA/DX5 P388 D-24851 524 ± 34 > 10000 1134 ± 430 5416 ± 784 7371 ±164 3018 ± 516 77 ±15 1 524 ± 26 5962 ± 116 957 ± 18 879 ± 30 652 ± 42 314 ± 17 10 ± 1 2 646 ± 31 2537 ± 162 958 ± 23 338 ± 37 151 ± 5 247 ± 31 167 ±22 3 359 ± 34 > 10000 6825 ± 316 1188 ± 130 729 ± 9 439 ± 36 7 ± 2 4 676 ± 53 7216 ± 723 3592 ±193 397 ± 24 85 ± 11 26 ± 6 5 ± 1 5 40 ± 6 > 10000 2608 ± 135 2333 ± 154 855 ±205 240 ± 57 6 ± 1 6 63 ± 1 1711 ± 301 73 ± 2 33 ± 7 19 ± 1 17 ± 7 18 ± 2 7 42 ± 5 1149 ± 79 627 ± 77 230 ± 57 32 ± 15 166 ± 37 5 ± 1 8 362 ± 50 > 10000 828 ± 72 2131 ± 59 673 ± 11 2132 ± 187 756 ± 138 9 431 ± 81 > 10000 946 ± 71 877 ± 46 820 ± 7 512 ± 13 54 ± 27 10 623 ± 126 3663 ± 1970 605 ±178 619 ± 122 385 ± 54 310 ± 96 12 ± 3 11 736 ± 74 > 10000 1260 ± 116 497 ± 12 349 ± 18 279 ± 7 5 ± 4 12 603 ± 23 5768 ± 692 1197 ±96 961 ± 55 576 ± 8 369 ± 28 62 ±22 13 503 ± 55 > 10000 3085 ± 301 486 ± 39 347 ± 45 280 ± 17 9 ± 1 14 206 ± 10 1889 ± 258 736 ±115 438 ± 44 378 ± 78 261 ± 27 112 ±15 15 262 ± 17 1837 ± 61 92 ± 9 95 ± 4 74 ± 20 32 ± 2 3 ± 2
aI dati sono stati espressi in IC50 nM come media ± deviazione standard di almeno tre esperimenti.
Queste indolgliossilammidi con anelli N-eterociclici in posizione R1- e R2-, mostrano un ampio spettro di attività antitumorale verso le cellule tumorali umane e quelle del topo. La maggior parte di questi composti sono risultati più attivi del D-24851. Tuttavia, è stato osservato che D-24851, è più potente verso P388 rispetto alle altre cellule tumorali
umane. Allo stesso modo, queste indolgliossilammidi con strutture chimiche diverse, hanno differenti attività in varie linee cellulari tumorali.
Le coppie isomere isosteriche, composti 1 e 2 o 11 e 12, hanno mostrato attività antitumorali comparabili. I composti 6-8, perdono gradualmente la loro efficacia, perchè hanno la catena laterale alchilica di grosse dimensioni, (R1= isossazolil). Queste osservazioni, suggeriscono che i fattori sterici ed elettronici possono svolgere un ruolo cruciale in posizione R1, influenzando l'affinità di legame tra il composto ed il suo target. Sono stati sintetizzati i composti con varie ammine eterocicliche in posizione R2, e sono stati valutati per la loro attività antitumorale come mostrato in tabella 5. La sostituzione dell'anello piridinico a sei membri di D-24851, con un anello eteroaromatico a cinque membri o con un anello chinolinico in posizione R2, non determina un significativo cambiamento dell'attività antitumorale. Come suggerito dai dati del composto isotiazolilaminico 2, tale gruppo sembra essere una funzione importante in posizione R2. Osservazioni simili sono state fatte per i composti 5-7, che contengono il gruppo isotiazolilaminico in posizione R2.
È stata anche valutata la possibilità di resistenza crociata di questi composti con altri farmaci antitumorali, come la doxorubicina, utilizzata frequentemente in clinica. La maggior parte di queste indolgliossilammidi N-eterocicliche, hanno mostrato un'analoga efficacia o addirittura maggiore verso due sottolinee resistenti doxorubicina, MCF7 del seno/ADR e dell'utero MESSA/Dx5 rispetto alle cellule del cancro al seno MCF7 native e dell'utero MESSA, indicando che non c'è una resistenza crociata ai multi -farmaco resistenti alla doxorubicina.
La mancanza di resistenza crociata a più farmaci, può essere clinicamente importante. Dall'altro lato, i composti 6-8, perdono gradualmente la loro efficacia con l'aumentare della dimensione della catena laterale alchilica isossazolica in R1. L'aumento delle dimensioni della catena laterale alchilica su R1, in qualche modo, rende queste molecole
substrati migliori per la multi-resistenza, portando ad una graduale perdita di attività come suggerito dai dati in Tabella 6.
In accordo con i risultati precedentemente mostrati, D-24851 ha mostrato la sua efficacia in modo analogo sia verso MCF-7 che per la sua linea multi-resistente. I differenti valori di IC50 osservati, possono essere dovuti all'utilizzo di diversi metodi di analisi (MTS vs 2,3-bis (2-metossi-4-nitro-5-solfonil)-5[(fenilamino)carbonil]-2H-tetrazolina idrossido XTT, o solforodamina B, SRB) e ai periodi di trattamento. I risultati hanno dimostrato che l'inserimento di eteroatomi in posizione 1 e 3 nelle indolgliossilammidi, in generale, aumenta l'attività antitumorale in vitro. Cellule umane di cancro gastrico, le NUGC3, sono state trattate con le indolgliossilammidi per 16 ore, e l'induzione di apoptosi è stata osservata dal cambiamento morfologico nei nuclei di condensazione e nella formazione di corpi apoptotici come dimostrato dall'analisi della frammentazione del DNA. Come mostrato in Figura 6, i corpi apoptotici delle cellule tumorali trattate con il composto 10 a 100 nM, sono stati osservati dopo colorazione di Hoechst con un microscopio a fluorescenza.
Come mostrato in Figura 7, diverse indolgliossilammidi a 100 µM, hanno indotto frammentazione del DNA dopo un trattamento di 24 ore nelle cellule tumorali. I risultati indicano che l'induzione di apoptosi può essere un meccanismo attraverso il quale queste indolgliossilammidi N-eterocicliche, uccidono le cellule tumorali. Il bersaglio molecolare di queste nuove indolgliossilammidi è ricercato anche se l'attività antitumorale di D-24851 può essere spiegato da una interazione con la Tubulina .
Figura 7.
Topi femmina DBA/2J di un gruppo di controllo negativo, sopravvivono per 7-8 giorni dopo una singola iniezione intravenosa di un milione di cellule di leucemia murina P388. Nell'esperimento di controllo, la doxorubicina ha mostrato un'attività consistente nel prolungare il periodo di sopravvivenza nei topi DBA/2J inoculati con P388. L'aumento della durata della vita mediante trattamento con doxorubicina, alla sua dose massima tollerata era 119 ± 18% (media ± deviazione standard), è stato usato come controllo sperimentale di qualità per ogni esperimento. I composti 6 e 10 hanno mostrato un'attività dose-dipendente correlata all'aumento della durata della vita degli animali malati di cancro come mostrato in Figura 8.
Figura 8.
La relazione dose-risposta in vivo (p <0.05, ANOVA) dei composti 7, 13 e D-24851 nel prolungare la sopravvivenza al cancro negli animali, è stata riassunta nella tabella 7.
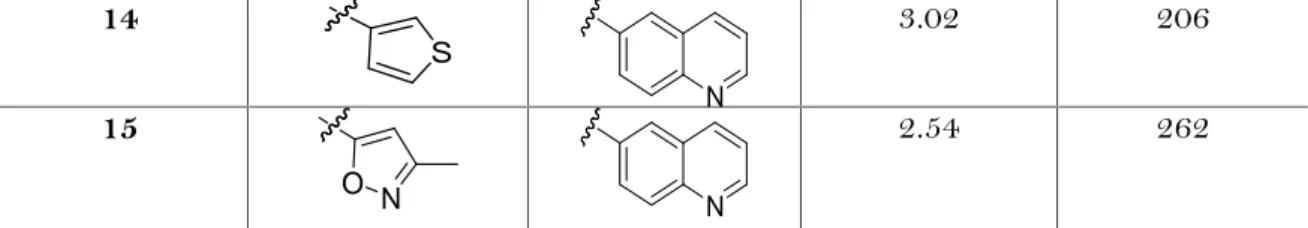
Tabella 7.Prolungamento della sopravvivenza al cancro in P388 inoculato nei topi leucemici trattati con
6, 10, e D-24851 Dose, mg/kga 6, % 10, % D-24851, % 25 26 ± 27b 14 ± 1 27 ± 9 50 57 ± 20 27 ± 3 44 ± 22 100 63 ± 17c 20 ± 8c 63 ± 14c 200 100 ± 21d 46 ± 24d 149 ± 28d
È stata osservata un'attività orale dose-dipendente in vivo tra 7 e D-24851. I composti 7 e D-24851, hanno mostrato attività analoga nel prolungamento della sopravvivenza al cancro a dosi orali fino a 100 mg/kg, mentre D-24851 è risultato più attivo a dose orale più elevata, 200 mg/kg. Ulteriore prolungamento della sopravvivenza al cancro, è stato osservato dopo continue somministrazioni orali giornaliere fino a 9 giorni con i composti 7 e 13, senza perdita evidente del peso corporeo (dati non riportati). Come mostrato nella Tabella 6, la potenza in vitro dei composti 7 e 13 era di circa 4 e 6 volte maggiore rispetto a D-24851, tuttavia quest'ultimo ha mostrato uguale o migliore attività orale in vivo rispetto a 7 e 13 nel prolungare la sopravvivenza di animali con P388 inoculato. La differenza di attività orale in vivo dose-dipendente tra 7 e il D-24851, implica un possibile coinvolgimento dei processi di farmacocinetica, assorbimento, distribuzione, metabolismo, ed escrezione. Di solito le caratteristiche farmacocinetiche tra attività in vivo ed in vitro, spesso non sono correlate. Ad esempio, la glicoproteina-P intestinale limita la biodisponibilità orale di paclitaxel e quindi il suo uso per via orale [48].
Inibitori della glicoproteina P sono stati sviluppati per aumentare la biodisponibilità orale di farmaci [49,90].
Queste osservazioni giustificano gli studi farmacocinetici delle indolgliossilammidi N-eterociclici. Tuttavia, i risultati del modello di sopravvivenza alla leucemia P388, hanno dimostrato che la sostituzione dell'N-eterociclico mantiene l'attività antitumorale in vivo, ed inoltre ha dimostrato un'attività antitumorale orale di queste indolgliossilammidi N-eterocicliche.
È stato osservato che il D-24851 è selettivamente più potente verso P388 del topo, rispetto alle cellule tumorali umane. La differenza tra la IC50 di cellule tumorali umane e del topo P388 per i composti studiati, era generalmente inferiore a quella per D-24851, implicando una possibile attività orale del composto 7 verso la crescita in vivo delle cellule tumorali umane. Dal momento che per via orale è attivo contro il cancro leucemico,
questa osservazione incoraggia la valutazione dello spettro dell'attività orale verso la crescita di xenotrapianti di tumori solidi umani, negli animali ed il loro ulteriore sviluppo.
N Cl O R H N O 1 2: R = 3-metil-5-isotiazolil 3: R = 4-carbossammidefenil 4: 6-quinolinil
Sulla base di questi risultati, i nuovi derivati 1 3-indolgliossilammidici, sono stati riportati in questo brevetto del 2003, per il trattamento dei soggetti con cancro. Una nuova serie di composti innovativi, è rappresentata dai composti di formula 5.
N X R3 Z2 NR1R2 Z1 A R4 5
L'anello A è sostituito o non sostituito e può essere fuso con un gruppo arilico. Z1e Z2possono essere O, S, N-OR9o NR9.
R1può essere H, un gruppo alifatico, un gruppo alifatico sostituito, un gruppo arilico non sostituito o un gruppo arilico sostituito.
N S N H N N N H N O B C D E F I II III IV N X1 O N N N N H N N X1 G H J I V VI VII VIII K S N L IX M X O P
Preferibilmente R2è un gruppo arilico sostituito con–C(O)-NR5R6, o è rappresentato da una struttura I, IV, VI o X.
In questo brevetto è riportata anche la composizione farmaceutica che può includere un carrier o un diluente per i composti della serie 1 o 5. Preferibilmente, la composizione farmaceutica comprende la somministrazione al soggetto di una quantità efficace del composto. Inoltre è riportato un metodo di trattamento di un soggetto con cancro: il metodo consiste nella somministrazione al soggetto di una quantità efficace di composto 1 o 5. I composti 3-indolgliossilammidici hanno molti vantaggi quando vengono utilizzati contro i tumori. Ancor più significativo è il fatto che sono citotossiche per molte linee di cellule multi-resistenti e quindi possono essere utilizzati quando le terapie tradizionali non hanno funzionato. Inoltre, essi mostrano effetti collaterali minimi e sono attivi somministrati per via orale.
Nel 2005 [34] sono state descritte nuove indol-3-gliossilammidi N-sostituite di formula generale 6, per il trattamento di tumori benigni e maligni nei mammiferi, compreso l'uomo. N Y X R6 R3 R4 R5 R2 N R7 Het R1 6
Se questi composti hanno gruppi sufficientemente basici (ammine secondarie o terziarie) possono essere convertiti nel loro sale corrispondente, con acidi inorganici o organici; alternativamente, se hanno gruppi sufficientemente acidi (carbossile) possono essere convertiti nei loro sali con basi inorganiche ed organiche. Le 6 indol-3-gliossilammidi, sono utili per il trattamento dei mammiferi, compresi gli esseri umani.
Un ulteriore aspetto riportato nel brevetto è il trattamento di tumori nei mammiferi, compresi gli umani, che consiste nella somministrazione di una 6 indol-3-gliossilammide, ad una dose efficace per il trattamento dei tumori. La dose terapeuticamente efficace da somministrare nel trattamento della indol-3-gliossilammide, dipende dalla natura e dallo stadio della malattia tumorale, l'età e il sesso del paziente, la modalità di somministrazione e la durata trattamento. I farmaci possono essere somministrati in forme farmaceutiche liquide, semisolide e solide.
I composti di formula generale 6, possono essere utilizzati come una singola sostanza o in combinazione con altre sostanze citotossiche, come il cisplatino, la doxorubicina, la ciclofosfamide, il metotressato e in particolare in combinazione con gli inibitori di trasduzione del segnale, quali Herceptin, Glivec o Iressa.
Tra le nuove indol-3-gliossilammidi, l'Indibulin (ZIO-301; D-24851; (N-(piridina-4-il)-[1-(4- clorbenzil)-indol-3-il]-gliossil-ammide)) è una nuova piccola molecola sintetica somministrata per via orale, con attività antitumorale dovuta alla destabilizzazione dei microtubuli [51]. N Cl O H N O N Indibulin
L'indibulin induce l'accumulo di cellule con nuclei condensati e di fusi mitotici anormali, arrestando le cellule in metafase. Questo farmaco antimitotico, è attivo contro una vasta gamma di linee cellulari tumorali umane e di xenotrapianti, comprese le cellule tumorali multi-resistenti e tumori refrattari [52, 53].
Studi pre-clinici hanno mostrato la mancanza di neurotossicictà nell'indibulin, tipicamente associata ad altri farmaci come i taxani e gli alcaloidi della vinca [54–57].
Inoltre è stata messa in evidenza la buona biodisponibilità dopo somministrazione orale e la terapia a dosi quasi non tossiche [52, 53].
Così indibulin, potrebbe avere un indice terapeutico notevolmente migliorato rispetto agli altri composti che inibiscono i microtubuli, come il paclitaxel e la vincristina [58, 59].
In precedenza è stata selezionata una soluzione orale di indibulin al 10% di acido lattico da una serie di formulazioni per ulteriori test clinici, in uno studio di fase I [53, 60].
Durante lo studio di fase 1 [60], è stato scelto un programma di somministrazione giornaliera per 14 giorni consecutivi, in cui si è riscontrata una maggiore efficacia dopo
una prolungata somministrazione giornaliera orale. Inoltre è stato notato che la somministrazione di indibulin in pazienti a digiuno o a stomaco pieno, non ha portato differenze di efficacia o di tollerabilità.
Tuttavia in questo studio di fase 1 [60], è risultato evidente che con il proseguimento del trattamento di indibulin, si sono manifestate in modo crescente fenomeni di nausea e vomito. Il cattivo gusto della formulazione lamentato dai pazienti, probabilmente è legato alla presenza dell' acido lattico. Pertanto è stata preparata una nuova formulazione in capsule, per migliorare la tollerabilità della formulazione.
L'indibulin formulato in capsule, è stato sottoposto ad uno studio dove veniva somministrato a dosi sempre maggiori, con l'obiettivo primario di determinare la dose massima tollerata (MTD) per via orale, somministrata una volta o due volte al giorno per 14 giorni ogni 3 settimane, in soggetti con diagnosi di tumori solidi avanzati. Gli obiettivi secondari erano la determinazione della tossicità dose-limitante (DLT), la caratterizzazione del profilo delle reazioni avverse, la farmacocinetica dopo somministrazioni singole e multiple a diversi livelli di dose, e l'attività antitumorale preliminare.
Lo studio di fase I, in cui si hanno somministrazioni sempre più elevate, mostra che indibulin in capsule somministrato per via orale, è stato ben tollerato alle dosi saggiate in pazienti con tumori solidi avanzati. Tuttavia, prima di raggiungere la MTD, è stata osservata una fase di plateau a partire da livelli superiori a 250 mg QD. Non vi era alcuna differenza significativa nella AUC di indibulin dopo dosi multiple (nei giorni 1-14) rispetto alla singola somministrazione (giorno 4). Vi è un'alta variabilità individuale nella AUC . Non si è osservata nessuna neurotossicità con le capsule di indibulin.
Indibulin è stato selezionato per la sperimentazione in pazienti con tumori solidi avanzati sulla base di dati preclinici, che ne hanno mostrato l'elevata attività ed un buon profilo di
Una soluzione di indibulin al 10% di acido lattico più glucosio, è stata selezionata per lo studio di fase 1 [53, 60]. Tuttavia l'aumento dell'incidenza e della gravità della nausea e del vomito, nei pazienti ai quali è somministrato indibulin continuamente , è correlato all'acido lattico presente nella soluzione.
Pertanto nel 2010 [61] è stata testata la nuova formulazione di indibulin in capsule. La formulazione è costituita da una miscela di tensioattivi, amido di mais, cellulosa microcristallina, aerosil, polisorbato e stearato di magnesio, che solubilizzano indubulin e funziona da agente di riempimento delle capsule di gelatina dura. Le capsule di indibulin sono state ben tollerate dai pazienti. Con le capsule orali dosati fino a 600 mg BID (1.200 mg totale/giorno), non è stato raggiunto in questo studio la MTD. Tuttavia, ogni caso è stato discusso e non vi era alcuna relazione apparente tra la quantità di dose e la frequenza o l'intensità del trattamento che produce tossicità. Prima di raggiungere la MTD, è stato osservato un plateau in esposizione al farmaco per via orale con la formulazione in capsule di indibulin, con un calo dei valori di AUC con la dose iniziale di 250 mg-dose (Tabella 8, Figura 9.).
Tabella 8.
Il protocollo è stato modificato ad una somministrazione di due volte al giorno, nel tentativo di aumentare ulteriormente la distribuzione sistemica di indibulin. Tuttavia questa strategia, non ha avuto alcun effetto benefico sulla distribuzione sistemica di indibulin. Inoltre, c'è stata un alta variabilità tra paziente e l'altro nell'AUC, ed un assorbimento relativamente lento e variabile delle capsule indibulin. Continui aumenti di dosi di indibulin, non hanno prodotto un aumento dell'assorbimento sistemico di indibulin, quindi si è deciso di interrompere questo studio.
Una spiegazione plausibile per il basso assorbimento sistemico, è la bassa solubilità dell'indibulin nella sua attuale formulazione orale.
Questa formulazione non può essere adatta per evitare che indibulin precipiti nel tratto gastro-intestinale.
La formulazione di indibulin dovrebbe essere ottimizzata per aumentare l'assorbimento sistemico dopo la somministrazione orale. Questo è un problema comune per lo sviluppo
Sono stati descritti diversi studi clinici con nuove formulazioni di paclitaxel per via orale, tuttavia finora le formulazione di paclitaxel per via orale, non sono clinicamente applicabili [62–64].
È stata osservata una stabilizzazione della malattia in quattro pazienti affetti da carcinoma progressivo , NSCLC e il cancro del colon, che hanno ricevuto un trattamento a dosi che variano da 100 mg/die a 900 mg/die per tre o quattro cicli di trattamento. In conclusione l'indibulin è stato ben tollerato alle dosi testate. E' stato osservato un plateau in esposizione al farmaco prima di osservare l'MTD. Pertanto, si è deciso di interrompere questo studio. Continuando ad aumentare la dose di indibulin, è risultato improbabile avere un ulteriore aumento dell'assorbimento sistemico. Una spiegazione plausibile per il basso assorbimento sistemico, è dovuta ala bassa solubilità del indibulin nella sua formulazione attuale. La formulazione necessita un'ottimizzazione nella formulazione per aumentare l'assorbimento sistemica dopo somministrazione orale.
3. Attività Ansiolitica
Negli anni '80 iniziò uno studio sulle indol-3-gliossilammidi 1 (Tabella 9), che ha mostrato di legarsi al Recettore centrale delle Benzodiazepine (CBR), spostando [3H] flunitrazepam dalle membrane cerebrali dei bovini, e un certo numero di questi composti ha mostrato avere un profilo di agonista/antagonista parziale tipico di ansiolitici. [65-71].
Gli studi sullo scaffold indolgliossililamidico e una profonda analisi della letteratura sulla proteina Translocator (TSPO), precedentemente denominato "Recettore Periferico delle Benzodiazepine" (PBR), hanno portato allo sviluppo di una nuova classe di ligandi ad alta affinità, che sono i derivati N, N-dialchil-2-fenilindol-3-gliossammidi (IGA) 2 (Figura 10) [72,73] tra i quali alcuni composti hanno mostrato attività ansiolitica anche in vivo.[73] I ligandi ad alta affinità per TSPO, come Ro5-4864, PK 11195, [74] alpidem, [75] e FGIN-1-27 [76,77] (Tabella 9), aumentano i livelli dei cosiddetti neurosteroidi, come pregnenolone e allopregnenolone, agendo come modulatori allosterici positivi della neurotrasmissione del GABA. Così ligandi al TSPO neurosteroidogenici, possono rappresentare un significativo passo avanti nello sviluppo di nuovi agenti farmacologici che mostrano un profilo ansiolitico, senza i tipici gli effetti collaterali indesiderati delle classiche benzodiazepine. [78]
Figura 10.
La promettente attività in vitro di questa classe di indolgliossilammidi 2 sostituite, ha portato a saggiare un gran numero di questi ligandi ad una concentrazione di 40 µM, per la loro capacità di aumentare la produzione di pregnenolone nelle cellule di glioma C6 di ratto. Il motivo per cui gli effetti dei ligandi del TSPO si abbiano a concentrazioni più elevate rispetto a quelle previste dalle loro affinità nell'ordine del nanomolare, non è stato ancora chiarito. Tuttavia, è stato riportato che la velocità del ligando di stimolare i siti di legame delle proteine intracellulari, può superare quelli necessari per saturare la proteina stessa [72,73,79-85]. La misurazione della produzione di pregnenolone è stata effettuata mediante il test radioimmunologico [72,82] o tramite metodo immunoenzimatico (ELISA) [73,83] La Tabella 9 elenca i composti più performanti, che hanno indotto un tasso di produzione di pregnenolone superiore al 100%.
I composti più efficaci erano 3 e 4 con un aumento della produzione di pregnenolone > 150% , seguiti da 5 e 7. N H O R2 N O R1 R3 R4 R5 N H O R2 N R1 R3 2 N N Cl O CH3 Cl Ro 5-4864 N N Cl N O Alpidem FGIN 1-27 N N Cl O PK 11195 N H O H N O R1 R4 1
I più performanti ligandi in termini di affinità per il TSPO e di produzione di pregnenolone, sono stati testati in un modello di ansia nel ratto, per verificare se la loro capacità di aumentare la sintesi dei neurosteroidi in vitro era associato alla produzione degli effetti ansiolitici in vivo.
Tabella 9. Composto R1 R2 R3 R4 Ki (µM) o inibizione (%)a Aumento produzione di pregnenolone production vs controllo (%)b 3 (CH2)5CH3 (CH2)5CH3 Cl Cl 5.8 ±0.6 109 ± 9 4 CH2CH3 CH2C6H5 Cl Cl 3.33 ±0.3 171 ± 14 5 (CH2)3CH3 (CH2)3CH3 Cl Cl 1.9 ±0.2 147 ± 13 6 (CH2)5CH3 (CH2)5CH3 F Cl 7.75 ±1.55 135 ± 4 7 (CH2)2CH3 (CH2)2CH3 CH3 H 5.50 ±0.38 148 ± 12 PK 11195 9.3 ±0.5 48 ± 5 Ro 5-4864 23 ±3.1 41 ± 4 Alpidem 0.5-7
aLa concentrazione dei composti testati che inibisce il legame nelle membrane mitocondriali del ratto, [3H]
PK11195 (IC50) del 50%, è stato determinato con sei concentrazioni degli dislocatori, ciascuno triplicato. I
valori Ki sono la media ± SEM di tre determinazioni. Le cellule di glioma BC6 sono state incubate per 2 ore a 37°C in presenza di ciascun composto. bIl pregnenolone è stato quantificato mediante il test
radioimmunologico come descritto nel testo. Sono inclusi anche gli effetti della PK 11195 e Ro5-4864 sulla produzione di pregnenolone.
I tre composti selezionati 4, 6, e 7 sono stati valutati nel ratto per i loro potenziali effetti
N H O R2 N O R1 R3 R4
costituito da quattro braccia di cui due aperte e due chiuse, e misura la preferenza dell'animale per i luoghi chiusi e bui (strade chiuse) o luoghi esposti e più luminosi (strade aperte) [86-88]. E' stato osservato un aumento sia del numero di entrate che del tempo speso dai topi nelle strade aperte, dopo la somministrazione di 30 mg / kg, i.p dei ligandi 4 o 7, rispetto a ratti trattati con il veicolo. Sia 4 che 7 non hanno mostrato di influenzare il sistema d'esplorazione spontanea dei topi, come il numero totale delle vie di entrata (sia aperta che chiusa) scelte dai ratti trattati non differivano significativamente da quelle scelte da ratti di controllo. Pertanto, le modifiche notate nell'EPM sembravano essere attribuibili a un effetto ansiolitico in vivo dei composti in esame. Un aumento delle esplorazioni delle vie aperte nell'EPM, è infatti, un effetto distintivo suscitato dagli agenti ansiolitici [86]. Probabilmente gli effetti osservati dipendono dall'abilità di 4 e 7 ad aumentare in vivo i livelli di neurosteroidi, come suggerisce il loro effetto sulla produzione di pregnenolone in cellule di glioma C6 e loro mancanza di affinità per il CBR.
Nonostante l'efficacia di 6 nel promuovere la sintesi di pregnenolone nelle cellule di glioma C6, la somministrazione di questo composto ai ratti, ha prodotto risultati diversi rispetto a quelli osservati in seguito al trattamento con i ligandi 4 e 7. La mancanza di effetti ansiolitici osservati in vivo dal composto 6 potrebbe essere attribuito all'influenza di diversi fattori, quali la possibilità che i ligandi vengono metabolizzati in modo diverso,così che i risultati possono differire da quelli attesi sulla base dei saggi in vitro.
Presi insieme questi risultati, ci suggeriscono che le N,N-dialchil-2-fenilindol-3-gliossilammidi, possono rappresentare dei promettenti strumenti farmacologici adatti al trattamento dell'ansia.
4. Attività Ipnotica
Alcuni anni fa, le N-(benzil) indol-3-gliossilammidi 1 sono state descritte come una nuova classe di ligandi per il recettore delle benzodiazepine (BzR) (Figura 11) [89,90].
Figura 11.Struttura delle N-(benzil)indol-3-ilgliossilamidi 1, Imidazobenzodiazepine 2 e Pirazolochinoline 3
Questi composti mostrano un'affinità maggiore per il sottotipo delle BzR, rispetto alle isoformee. La SARs dei derivati indolici ha rivelato che gli effetti della sostituzione dei sostituenti R5 e X sull'affinità di 1 sono indipendenti. In particolare, l'affinità è
favorita con X = idrossi / metossi o alogeni, a seconda che la posizione 5 del nucleo indolico sia sostituito (R5 = Cl / NO2) o meno (R5 = H). Così mentre nella serie 5-Cl/NO2 l'affinità è ottimizzata da X= 3',4'-(OH)2 o 3',4'-(OMe)2, nella serie 5-H l'affinità più alta è stata ottenuta con gruppi elettronattrattori X= 4'-Cl. Questi dati sono stati interpretati assumendo che i ligandi nella loro interazione con il sito recettoriale, possono adottare due modalità di legame alternativi, chiamati A e B (Figura 12) [90].
N O R5 H O N H X 1 N N N R8 COOR O CH3 2 N N N R4 R8 O H 3
Figura 12. Ipotetici modi di legame dei modelli indolici A e B per BzR [90] nel modello topico/farmacologico di Cook
Nel tentativo di identificare nuovi ligandi che possiedono selettività per i vari sottotipi BzR, l'attenzione è stata rivolta alle indol-3-gliossilammidi N-sostituite, che si inseriscono nelle regioni lipofile LDI e L2 in misura diversa. L'approccio si è sviluppato, concentrandosi sulle differenze nelle proprietà di riconoscimento dei sottotipi BzR. In particolare, è stato suggerito che la topologia di base di questi sottotipi è altamente conservata, ad eccezione delle tasche lipofile L2 e LDI, le cui diverse dimensioni potrebbero giocare un ruolo nella determinazione della selettività [87]. In particolare, è stato proposto che le regioni LDi e L2 sono più larghe nei siti di legame 1e, rispetto
agli altri sottotipi. Di conseguenza, la piena occupazione di LDi e L2 può spiegare la selettività 1 e [91,92]. Un ottimo esempio di selettività è data dalle
imidazobenzodiazepine 2 (Figura 11) dotate di sostituenti ingombranti in posizione 8 come etinil e (trimetil)-etinil, che si inseriscono nella regione L2 [91,92]. Yu et al. hanno proposto che la simultanea occupazione di L2 e LDi possa promuovere la selettività , grazie ai dati di legame forniti da alcuni pirazolodinoline 3 (Figura 11) sostituite in entrambe le posizioni 4 e 8, con gruppi ingombranti e lipofili. Tuttavia, i fattori che determinano la selettività , non sono ben definiti, infatti lo stesso gruppo di ricerca
afferma che i ligandi BzR si legano fortemente al sito 1 della regione LDi, nonostante
questi si leghino anche ad altre aree lipofile del recettore [91,92]
Prendendo le benzilindolgliossilammidi 1a e 1b come composti di riferimento (Tabella 10), [89] per misurare le proprietà di riconoscimento delle tasche LDi e L2, sono stati sintetizzati i composti 4-11 (ottenuti variando il sostituente sulla benzilammide dell'anello fenilico) e i composti 12-14 (ottenuti sostituendo la funzione benzilica con gruppi alchilici) [94]. Tabella 10. n R5 R Ki (µM) or % inhibition (10M) 1a H CH2-C6H5 120 ± 11 1b NO2 CH2-C6H5 117 ± 12 4 NO2 CH2-C6H4-4'-CH3 88 ± 6 5 H CH2-C6H4-4'-CH2CH3 2160 ± 180 6 NO2 CH2-C6H4-4'-CH2CH3 42 ± 3 7 H CH2-C6H4-4'-Br 93 ± 8 8 H CH2-C6H4-4'-C≡CH 164 ± 10 9 NO2 CH2-C6H4-4'-C≡CH 28% ± 1 10 H CH2-C6H4-4'-C≡C-CH3 3275 ± 299 11 NO2 CH2-C6H4-4'-C≡C-CH3 0% ± 3 12 H (CH2)3CH3 3720 ± 30 13 NO2 (CH2)3CH3 110 ± 9 14 NO2 CH(CH3)2 57.8 ± 4 Diazepam 120 ± 11 Flumazenil 120 ± 11 N COCONHR R5 H
L'affinità di legame dei derivati indolgliossilammidici sintetizzati 4-14 al recettore BzR nelle membrane di cervello, è stata determinata mediante esperimenti di competizione con il suo agonista radiomarcato [3H]flumazenil [95] ed è stata espressa come valore di Ki solo per quei composti che inibivano il radioligando oltre l'80% ad una concentrazione fissa di 10 iM (Tabella 10).
La Tabella 10 elenca i valori di affinità dei nuovi derivati indolici 4-14 e quelle degli indoli di riferimento 1a e 1b. Questi dati mostrano che la stessa catena laterale dell'ammide produce diversi effetti di affinità, a seconda che la posizione 5 del nucleo indolico sia sostituito (R5 = NO2) o meno (R5 = H). Questo andamento di affinità conferma che la 5-NO2 indolgliossilammide si lega al BzR in modo diverso dal corrispondente derivato 5-H (rispettivamente secondo modalità A e B) (Figure 12).
Secondo le SARs finora delineate, la regione LDi sembra essere più lunga della L2, perchè la prima può ospitare i sostituenti lunghi in posizione 4 dei 5-H indoli 5, 8 e 10 (orientati secondo il modo B), mentre l'altro non può ospitare gli stessi sostituenti in posizione 4 dagli analoghi 5-NO2 6, 9, e 11 (orientati secondo il modo A).
Allo stesso modo come osservato per composti 4-11 N-benzilici, l'impedimento sterico è un fattore che determina l'affinità anche per i derivati N-alchilici 12-14. Tuttavia, nel secondo sottoinsieme, l'affinità sembra dipendere non solo dalla lunghezza e larghezza totale, ma anche dalla forma della catena laterale. Le stesse N-sostituzioni sono ancora tollerate, quando vengono introdotte nell'analogo 5-H 12, (con affinità micromolare secondo modalità B), confermando che la regione LDi è più lunga della L2, che dovrebbe avere una lunghezza precisa, con il risultato che l'affinità è nulla quando la catena laterale dell'ammide aumenta al di sopra di un certo valore soglia. La ramificazione della catena R è tollerata all'interno della tasca L1/L2, effettuata nel 5-NO2 derivato 14 (con affinità elevata o moderata secondo la modalità A). Questi dati possono essere correlati alle
diverse forme e volumi delle regioni L1/L2 e LDi. Per quanto riguarda questi indoli, la tasca L1/L2 potrebbe ospitare sia catene N-aromatiche o N-alchiliche laterali dei derivati 5-NO2 (orientati in modalità A), così come l'anello benzenico fuso dei derivati 5-H (orientati verso il modello B). Per contro la tasca LDi, è in grado di accogliere l'anello benzenico fuso dei 5-NO2 indoli e le catene laterali N-benziliche dei ligandi 5-H, ma non è stericamente adatto per diverse catene N-alchiliche degli indoli 5-H. In altre parole, nonostante sia più lunga (rispetto alla L2), la regione LDi sembra essere essenzialmente ampia e può essere approssimativamente descritta come una cavità "tipo benzile". In realtà, qualsiasi deviazione da questo sostituente "migliore" nei 5-H indoli, ha dato effetti sfavorevoli sull'affinità.
In accordo con quanto descritto, molti studi di modellistica molecolare 17,18 hanno dimostrato che la maggior parte dei ligandi del BzR occupa la regione LDi con le funzioni aromatiche (ad esempio, un anello di benzenico fuso B-carboline, composti CGS, diidropiridoindolici) o piccoli gruppi alchilici (ad esempio, un metile dello zolpidem e della triazolopirimidina CL 218872).
Sono stati saggiati alcuni rappresentativi derivati indolici (4, 5, 7, 8, 10, 12, 13 e 14), insieme ai composti di riferimento 1a e 1b, per la loro affinità verso i sottotipi 1,e
del BzR.
Tabella 11. n Ki (µM) or % inhibition (10M) a 122 2 1a 346 ± 29 39% ± 3 46% ± 5 1b 65 ± 5 32% ± 3 44% ± 4 4 31.3 ± 2 0,00% 0,00% 5 559 ± 44 1600 ± 145 40% ± 3 7 17 ± 1 81 ± 7 1445 ± 120 8 250 ± 19 414 ± 31 1154 ± 299 10 881 ± 78 16% ± 2 13% ± 3 12 1175 ± 95 7% ± 1 28% ± 2 13 125 ± 10 553 ± 45 943 ± 80 14 15 ± 1 175 ± 12 498 ± 37 Zolpidem 50 ± 3 765 ± 63 35% ± 3
Esiste una buona correlazione tra le affinità wild-type e quelle sottotipo recettoriale1. Il
miglior risultato in termini di affinità e selettività, è rappresentato dal composto 4, che mostra una Ki di 31.3 µM verso il sottotipo1 e di non legarsi alle isoformeo.
Questi dati indicano che le indolgliossilammidi, a causa della loro forte interazione con la regione lipofila LDi (in una delle modalità di legame A o B), mantengono la selettività 1
indipendentemente dal grado di interazione con le tasche lipofile L1 / L2. Ciò è in accordo con quanto riportato in letteratura, dove è stato osservato lo stesso effetto in diverse serie di ligandi come le β-carboline [96] e i pirazolo[4,3-c]idrossi-3-oni
[92,93,97]. Il composto 4 è stato selezionato per essere sottoposto a screening per l'efficacia funzionale, e come illustrato in figura 2, la sua efficacia su co-applicazione con GABA al sottotipo BzR, era simile a quella dello standard diazepam (72% ± 2% e 78% ± 3% per il composto 4 e diazepam, rispettivamente), mostrando che la caratteristica principale del composto 4 è quello di essere un agonista di questo sottotipo recettoriale.
Grazie al suo profilo di efficacia, il composto 4 dovrebbe esercitare effetti sedativi in vivo. Questa ipotesi è stata inizialmente valutata con un modello comportamentale basato sulla rilevazione dell'attività motoria spontanea dei topi,[98] impiegando lo zolpidem 1
-selettivo [99] come farmaco di riferimento e i risultati hanno mostrato che il composto 4, anche se meno attivo dello zolpidem, è un agente sedativo-ipnotico. In conclusione, nessuna delle modifiche strutturali finora realizzate sulle indolgliossilammidi ha dato alcun spostamento di selettività dal sottotipo 1 verso il sottotipo o , confermando
così che un ligando che occupa favorevolmente la regione LDi mostra selettività verso 1,
nonostante le sue interazioni con altre aree lipofile nella tasca di legame del recettore. Il composto 4 è stato identificato come un ligando selettivo1 mostrando così un profilo di
5. Attività antinfiammatoria
Inibitori della fosfodiesterasi PDE 4
Finora, sono state identificate 11 famiglie di fosfodiesterasi (PDE). Questi enzimi sono coinvolti nella modulazione della trasduzione del segnale e agiscono mediante degradazione dei nucleotidi ciclici (cAMP e/o cGMP). Tra le PDE, la PDE4, la PDE7 e la PDE8, mostrano specificità per cAMP [100]. Nella ricerca di un trattamento anti-infiammatorio alternativo ai corticosteroidi, hanno avuto notevole importanza gli inibitori selettivi di PDE4, perché l'isoenzima PDE4 è il principale enzima che metabolizza cAMP nelle cellule immunitarie e infiammatorie [101]. L'inibizione selettiva della PDE4, ha effetti anti-infiammatori in pazienti con malattie infiammatorie come l'asma o la broncopneumopatia cronica ostruttiva (BPCO) [102]. Attualmente due composti, roflumilast e cilomilast, sono in fase di sviluppo clinico (fase III) per il trattamento della BPCO e nel caso del roflumilast, anche per l'asma.
Roflumilast Cilomilast
Gli inibitori di PDE4 sono potenzialmente migliori dei corticosteroidi in uso clinico, perché non solo inducono effetti anti-infiammatori, ma influenzano anche diversi altri tipi cellulari coinvolti in queste malattie; questi tipi di cellule sono cellule epiteliali delle vie
dati indicano che gli inibitori della PDE4 di nuova generazione, non hanno gli effetti collaterali spesso associati alla terapia con corticosteroidi, tra cui gli effetti negativi sull'asse ipofisario e sulla densità ossea [104]. Tuttavia ulteriori effetti collaterali come nausea, vomito, aumento della secrezione acida gastrica e mal di testa, hanno finora limitato l'uso terapeutico di inibitori della PDE4 [105]. Il composto AWD 12-281, è un nuovo e altamente selettivo inibitore della PDE4 [106-108]. Durante lo sviluppo clinico, questo composto ha dimostrato di avere un profilo di sicurezza migliore rispetto ad altri inibitori della PDE4.
AWD 12-281
Inoltre, è stato progettato per il trattamento topico. Ha una bassa biodisponibilità orale, una bassa solubilità ed esercita un forte e duraturo effetto farmacologico dopo somministrazione intra-tracheale in vari modelli animali, indicando la sua persistenza nel tessuto polmonare. Un alto valore di legame plasma-proteina ed un efficace metabolismo epatico della glucuronidazione, sono ulteriori fattori che contribuiscono al basso assorbimento sistemico dopo somministrazione endotracheale. Questi fattori contribuiscono ad una differenza tra livelli di dosi emetiche ed anti-infiammatorie [108]. In uno studio del 2004 [109], è stato valutato l'effetto di AWD 12-281 nelle cellule infiammatorie umane. Le preparazioni di cellule umane, PBMC, e cellule umane derivate
del tessuto bersaglio che possono essere utilizzati per prevedere effetti anti-infiammatori. Il rilascio del fattore di necrosi tumorale alfa (TNFè stato scelto come marcatore per i processi infiammatori in tutte le preparazioni cellulari testate. Inoltre è stato valutato il rilascio di citochine IL-2, IL-4 e IL-5, nel PBMC. Il TNF viene rilasciato da monociti attivati, macrofagi e linfociti, e l'attività di TNF è mediata attraverso il legame ai recettori del TNF di membrana.
IL-4 è importante per lo sviluppo dell'infiammazione allergica. Essa induce la produzione di IgE dei linfociti B [110] e provoca l'attivazione dei mastociti. IL-4 è essenziale per l'infiammazione allergica perché è in grado di regolare la differenziazione dei linfociti Th0 in linfociti Th2 [111]. IL-5 ha un ruolo chiave nello sviluppo e l'attivazione degli eosinofili e quindi anche nell'infiammazione eosinofila [112]. I linfociti Th2 sono la fonte principale di IL-5. IL-2 è una citochina derivata dai linfociti Th1 ed è coinvolta nella crescita e differenziazione delle cellule T e anche nell'infiammazione eosinofila in vivo. AWD 12-281 era in grado di sopprimere la produzione di citochine in PBMC indotti: interleuchina-2 (IL-2, stimolazione con fitoemoagglutinina ), IL-5 (stimolazione con concanavalina A ), IL-5 e IL-4 (costimolazione di anti-CD3 e anti-CD28), ed il rilascio di TNF stimolata da lipopolisaccaridi. I valori corrispondenti per l'inibizione EC50 per AWD 12-281, erano un intervallo ristretto (46-121 µM). Confrontando l'effetto di AWD 12-281 con roflumilast, cilomilast (SB 207.499), rolipram (RPR-73401), e 1-(3-nitrofenil)-3-(4-piridilmetil)pirido[2,3-d]/pirimidin-2,4/(1H,3H)-dione (RS-25.344-000), è stato possibile dimostrare che l'attività inibitoria sulla PDE4 era strettamente correlata con potenziale di inibizione misurato dai test sopra descritti. AWD 12-281 sopprimere il rilascio di TNF in polipi nasali dispersi (EC50 = 111 µM) e nel sangue (EC50 = 934 µM). L'attività ridotta nel sangue umano può essere correlato al legame con le proteine plasmatiche. Attualmente sono in corso, studi clinici di fase II, sono in corso per valutare
il potenziale terapeutico di AWD 12-281 nell'asma, nel BPCO, e nella rinite allergica. In sintesi AWD 12-281 ha dimostrato di sopprimere del tutto il rilascio di mediatori infiammatori in tre diverse preparazioni di cellule umane. La capacità di AWD 12-281 di inibire la PDE4 è risultata elevata come quella di altri inibitori di PDE4. Questi risultati, correlati alla potente attività di AWD 12-281 in modelli animali e la buona separazione tra attività emetica e anti-infiammatoria, specialmente dopo somministrazione topica, rendono AWD 12-281 un candidato interessante per il trattamento topico di malattie delle vie aeree come l'asma, BPCO, e rinite allergica. Attualmente sono in corso studi clinici di fase II, per valutare il potenziale terapeutico di AWD 12-281 in queste malattie.
Inibitori della Fosfolipasi A2
Gli enzimi della fosfolipasi secretoria A2 (sPLA2), prodotti e secreti nei vasi sanguigni umani e negli epatociti, contribuiscono allo sviluppo di aterosclerosi attraverso meccanismi che sono sia dipendenti che indipendenti delle lipoproteine. A-002 (1-H-indolo-3-gliossamide, metil varespladib), è un nuovo inibitore selettivo della sPLA2 umana con specificità verso sPLA2-IIA (/IC50/ 9-14 nmol / L), sPLA2 -V (IC50 77 nmol / L), e sPLA2-X (IC50 15 nmol / L) enzimi. Sono stati esaminati gli effetti di A-002 sulla concentrazione e l'attività di sPLA2 e sulle lipoproteine plasmatiche biomarcate, in pazienti con insufficienza coronarica.
. NH
O O NH2
![Figura 12 . Ipotetici modi di legame dei modelli indolici A e B per BzR [90] nel modello topico/farmacologico di Cook](https://thumb-eu.123doks.com/thumbv2/123dokorg/8014458.121671/43.892.208.739.130.373/figura-ipotetici-legame-modelli-indolici-modello-topico-farmacologico.webp)