INTRODUZIONE
TRODUZIONE
IN
1.1 Saccharomyces cerevisiae
Saccharomyces cerevisiae è l’organismo eucariote maggiormente
bientali ben definite e facilmente riproducibili. È
poco costosa;
etodologia simile espressione genica abbastanza semplice
ato sequenziato
dal “Centro Informazioni sulla Sequenza delle Proteine“ di Monaco.
studiato. È un ascomicete unicellulare di forma ovale o ellittica di diametro compreso tra i 5 e i 10 μm. Si riproduce per gemmazione multilaterale, ha un ciclo vitale aplodiplonte e il materiale genetico è suddiviso in 16 cromosomi. Nel ciclo cellulare alterna generazioni aploidi (16 cromosomi) e diploidi (16 coppie di cromosomi) con cellule capaci di riprodursi vegetativamente. È considerato un organismo modello in quanto soddisfa le richieste enunciate da Wills and Phelps (1975):
1. cresce a condizioni am
un organismo ubiquitario capace di effettuare il suo ciclo vitale sia in coltura liquida che solida, ha un tempo di duplicazione di 2-3 h e mostra buone capacità di resistenza al processo di conservazione criogenico che a 4° C;
2. la sua analisi genetica è
3. genera prodotti identificabili ed analizzabili con una m a quella usata per i batteri;
4. presenta uno schema di
nonostante la sua struttura interna sia riconducibile a quella complessa tipica degli organismi eucarioti superiori. Si stima che S. cerevisiae condivida circa il 23% del suo genoma con l'uomo.
E’ stato il primo genoma eucariotico ad essere st
completamente (Dujion, 1966). È composto da circa 13.000.000 coppie di basi e 6275 geni, sebbene soltanto 5800 di questi sono ritenuti essere i geni funzionali. Una delle più importanti banche dati sul S. cerevisiae è mantenuta
INTRODUZIONE
roduzione
altro utilizzo commerciale di lievito è la produzione di
al progresso del genere umano,
S. cerevisiae è un organismo che ha un ruolo essenziale negli studi
citologici e genetici;è inoltre l’agente microbiologico utilizzato nella p
di numerosi prodotti come il pane, il vino, la birra e altre bevande alcoliche. Il suo successo e la sua diffusione sono cresciuti di pari passo con lo sviluppo della civiltà. Esso è stato identificato nei residui di vino risalenti all’antico Egitto e databili nel 3150 a.c. (Cavalieri et al. 2003). I ceppi naturali di lievito vengono generalmente isolati da uve, da altri frutti e da insetti (Mortimer and Polsinelli 1999).
Sul mercato S. cerevisiae è presente sia come lievito fresco, sia come
lievito secco. Un
bioetanolo dalla fermentazione di zuccheri.
Alla luce di quanto detto si può affermare che nessun microrganismo è così strettamente legato al benessere e
INTRODUZIONE 1.1.1 Ciclo cellulare
Esso può essere diviso in quattro fasi (fig.1):
tempo Log n° c ell u le
Figura 1: curva di crescita di S. cerevisiae (molecularlab.it)
1. fase di latenza (A): periodo di incubazione nel quale il ceppo attiva i meccanismi biochimici necessari alla sopravvivenza nelle condizioni ambientali fornite.
2. fase replicativa o fase di crescita esponenziale (B): intervallo nel quale si ha il massimo livello di proliferazione delle cellule (Arava et al. 2002).
3. fase stazionaria (C): intermezzo temporale in cui i nutrienti presenti nel mezzo di coltura si esauriscono e la curva di crescita, raggiunge il
plateau.
4. senescenza (D): è la fase in cui le condizioni di coltura non sono più
favorevoli alla crescita a causa dell’esaurimento delle sostanze nutritive; le cellule si trovano ad uno stadio terminale e si osserva un decremento della loro concentrazione dovuto alla lisi cellulare.
INTRODUZIONE
La morte cellulare non è necessariamente una diretta conseguenza della mancanza di nutrienti ma può dipendere anche dal numero di gemmazioni compiute da ogni singola cellula madre; questo fenomeno prende il nome di invecchiamento cellulare (Mortimer and Johnston, 1959).
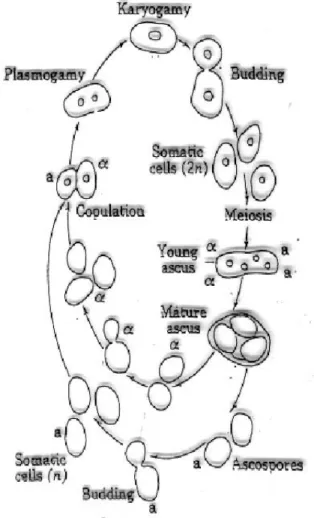
Figura 2: ciclo vitale di S. cerevisiae (Dikinson et al. 2004)
Le forme aploidi di S. cerevisiae sono di due tipi sessuali (mating type) rispettivamente a ed α. Ogni mating type produce un fattore specifico: dalle cellule aploidi MATa si genera il “fattore a”, mentre gli aploidi di tipo MATα producono il “fattore α”. La coniugazione può avvenire solo tra spore di tipo sessuale opposto (Polsinelli et al. 1993).
INTRODUZIONE
La cellula diploide va incontro a meiosi su fonti di carbonio povere come l’acetato. In queste condizioni le cellule di lievito attivano una serie di segnali biochimici, si ha la formazione di una struttura detta Asco, contenente una tetrade di spore aploidi, due di tipo sessuale a e due di tipo sessuale α (Dickinson and Schweizer 2004). Quando una spora trova le condizioni nutrizionali adatte germina: l’asco viene digerito da glucanasi e le spore, poste su di un mezzo di crescita fresco, si riproducono vegetativamente dando origine a una generazione aploide.
La sporulazione è un fenomeno tipico delle cellule diploidi eterozigoti (MATa/MATα): essa non avviene nelle forme petite e nelle specie diploidi omozigoti per il mating type.
INTRODUZIONE 1.1.2 Il flusso metabolico di Saccharomyces cerevisiae
Esso è composto dai seguenti processi:
Glicolisi.
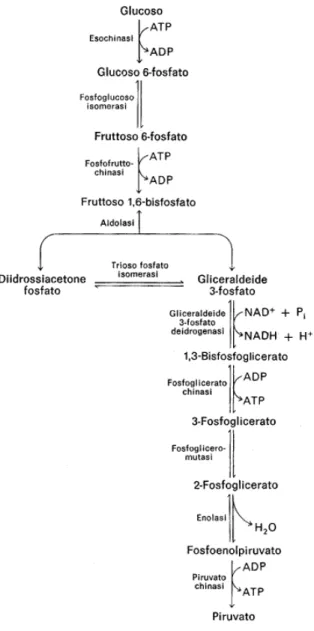
La glicolisi è la prima fase della catabolismo del glucosio e si realizza interamente nel citosol.
INTRODUZIONE
L’intero processo, che si compone di diversi step, è svolto da enzimi costitutivi che presentano un elevato tasso di conservazione per lieviti e altri organismi superiori mentre i composti intermedi ottenuti possono essere utilizzati in altre vie biosintetiche per la produzione di amminoacidi, nucleotidi e lipidi. Il glucosio, il mannosio e il fruttosio sono i substrati dell’esochinasi I e l’esochinasi II, codificate dai geni HXK1 e HXK2. Questi enzimi costitutivi presentano un’alta similarità di sequenza tra loro e sono inibiti da elevate concentrazioni di ATP e trealosio-6-fosfato. Il gene HXK1 è represso ad alte concentrazioni di glucosio mentre HXK2 ne è attivato e rappresenta un’importante punto di regolazione dell’intero processo glicolitico (Rodriguez et al. 2001).
Il bilancio energetico della glicolisi è:
1D-glucosio + 2 ADP + 2 Pi + 2 NAD+ → 2 Piruvato + 2ADP + 2 NADH + 4H+.
Il piruvato è una molecola centrale nel metabolismo di S. cerevisiae in quanto può seguire due diverse vie biochimiche:
• Respirazione: in presenza di ossigeno si ha la riduzione del composto a CO2 durante il TCA con conseguente liberazione di energia sottoforma di NADH e FADH2, i quali possono essere convertiti in ATP
attraverso il trasporto elettronico mitocondriale.
• Fermentazione: in carenza di ossigeno o durante le fasi iniziali della crescita si ha decarbossilazione e riduzione del composto in etanolo con conseguente riossidazione del NADH glicolitico.
INTRODUZIONE
Ciclo di Krebs (TCA) e Trasporto elettronico mitocondriale :
Entrambi i processi avvengono nei mitocondri.
Figura 4. Ciclo di Krebs (Taddei)
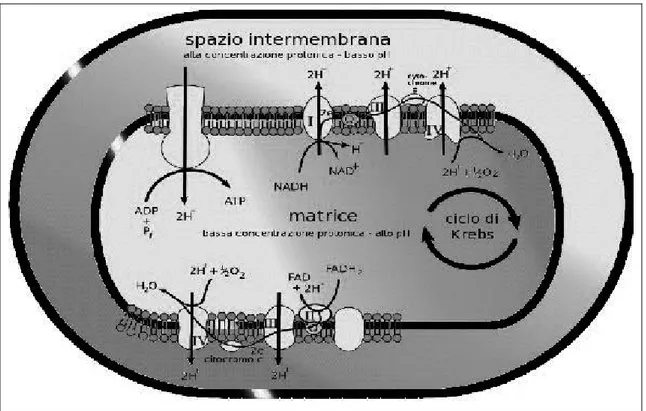
Nel trasporto elettronico mitocondriale il potenziale ossdoriduttivo immagazzinato in NADH e FADH2 viene convertito in un gradiente
protonico coinvolgendo una serie di complessi proteici posti sulla membrana interna del mitocondrio. Essi sono: complesso I (NADH deidrogenasi o coenzima Q reduttasi), complesso II, complesso III (Citocromo C reduttasi) e il complesso IV (Citocromo ossidasi). In particolare il complesso II partecipa sia al trasporto elettronico mitocondriale sia al TCA catalizzando l’ossidazione del succinato in fumarato; inoltre ha domini altamente conservati attraverso il processo evolutivo. Studi condotti su S. cerevisiae (Saliola et al. 2004) hanno messo in evidenza che l’espressione dei geni per la succinato deidrogenasi
INTRODUZIONE
(SDHs), codificati sia in condizioni fermentative sia respirative, sono repressi in presenza di eccesso di glucosio e la perdita della funzione SDH genera un’incapacità delle cellule a crescere su qualsiasi forma di carbonio utile per la respirazione. Successivamente il complesso ATP-sintetasi convoglia e trasforma l’energia trasportata in ATP. L’ossigeno è indispensabile al compimento del metabolismo respiratorio poiché funge da accettore finale di elettroni e rende possibile la generazione del gradiente protonico.
Figura 5. Trasporto elettronico mitocondriale (Nucci 2006) Il bilancio energetico dell’intero processo respirativo è: 1D-glucosio + 6 O2 + 32 ADP + 32 Pi → 6 H2O + 6 CO2 + 32 ATP
INTRODUZIONE
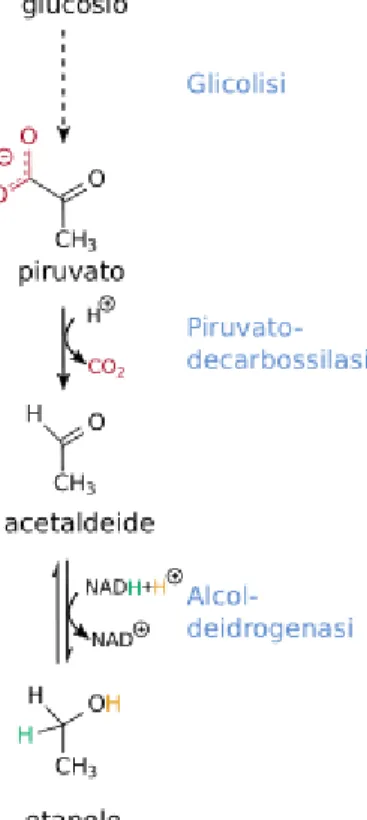
Fermentazione.
Indipendentemente dalla presenza di ossigeno, la maggior parte del piruvato in uscita dalla glicolisi segue il processo fermentativo che si avvale dell’attività di due enzimi (van Dijken et al.1993):
1. Piruvato decarbossilasi. È codificata dai geni PDC1, PDC5 e PDC6 e converte irreversibilmente il piruvato in acetaldeide e CO2. Questo
enzima è attivato allostericamente dal substrato. La proteina Pdc1 è l’isoenzima prevalente nei ceppi wild type (tra l’80 e il 90%) e insieme a Pdc5 sono altamente espresse nei mezzi di coltura con glucosio e etanolo; la loro induzione è relazionata all’attività dei fattori di trascrizione Rap1 e Gcr1. Pdc6 è generalmente silente. L’acetaldeide formata viene utilizzata per la sintesi dell’acetil-CoA citosolico mediante un processo chiamato pituvato deidrogenasi by-pass (Flikweert et al. 1997).
2. Etanolo deidrogenasi o alcol deidrogenasi. È codificata dai geni ADH e genera etanolo e CO2 ossidando il NADH. L’etanolo prodotto diffonde
attraverso la membrana plasmatica nel mezzo di crescita e il NAD+ che si forma verrà utilizzato nel processo glicolitico.
INTRODUZIONE
Figura 6. Fermentazione alcolica (Anne-Kathrin Sauer) Il bilancio energetico della fermentazione
1D-glucosio + 2 ADP + 2 Pi → 2 Etanolo + 2 CO2 + 2 ATP
Esso evidenzia che vengono prodotte solo 2 moli di ATP per mole di glucosio, mentre nella respirazione il rapporto è di 32:1. Nonostante questa notevole differenza nella resa energetica, in condizioni aerobiche, le cellule di S.
cerevisiae possono operare contemporaneamente entrambi i processi,
soprattutto se la concentrazione dei etanolo nel terreno è bassa. Questo avviene perché la fermentazione consente di produrre energia molto velocemente (Voet 2005).
Altre fonti di carbonio come etanolo e il glicerolo incrementano invece i processi respirativi.
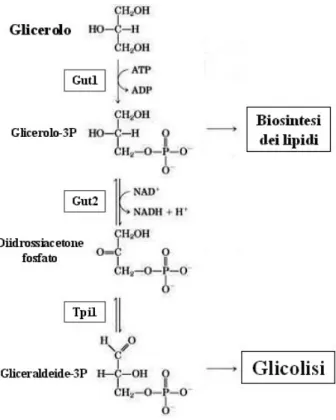
Metabolismo del glicerolo
INTRODUZIONE
fonte di carbonio solo in assenza di glucosio. Esso entra all’interno della membrana plasmatica di S. cerevisiae sia per trasporto passivo attraverso il canale specifico (Fps1) codificato dalle proteine MIP (Major Intrinsic Protein) in risposta a stress osmotici (Tamas et al.1999), che per simporto protonico a carico di Gup1 e Gup2 in risposta agli stress salini (Sutherland et al.1997). Questa molecola gioca un ruolo cruciale nel mantenimento del bilancio redox in condizioni anaerobiche poiché è l’unica via per riossidare il NADH citosolico e produrre biomassa (van Dijken and Scheffers 1986).
Figura 7. Metabolismo del glicerolo (Nucci, 2006)
Il glicerolo, una volta convertito in glicerolo-3fofato dalla glicerolo chinasi (Gut1), può essere avviato alla biosintesi dei lipidi oppure alla glicolisi. Per entrare nella glicolisi, il glicerolo-3P viene prima ossidato in diidrossiacetone fosfato (DHAP) dalla glicerolo-3P deidrogenasi (Gut2), e poi convertito in gliceraldeide-3P dalla triosofosfato isomerasi (Tpi1). L’enzima
INTRODUZIONE
Gut2 è una glicerolo-3P FAD dipendente localizzata sulla membrana interna del mitocondrio dove concorre al trasferimento di potenziale redox tra mitocondrio e citosol (Gomes et al. 2005).
Per la sua importanza biochimica come marker per il vino, all’inizio del XX secolo, sono stati condotti una serie di esperimenti su S. cerevisiae per la produzione massiva di glicerolo: Neuberg e Reinfurth nel 1918 hanno determinato che l’aggiunta di solfito nel mezzo di coltura si accompagna a una diminuzione dell’acetaldeide che è substrato coinvolto nella riossidazione del NADH glicolitico poiché vengono bloccati gli enzimi fermentativi. Hanoi nel 2004 ha creato ceppi di lievito deificienti per le alcol deidrogenasi (ADHs), per il gene triofosfato isomerasi (TPI1 che genera un accumulo di DHAP) e per la piruvato carbossilasi (PDC) minimizzando il consumo di NADH nella via biochimica di produzione dell’etanolo e rendendo la produzione di glicerolo 4,7 volte maggiore rispetto ai ceppi wild type (Hanoi 2004).
1.1.3 Bilancio energetico cellulare
Il bilancio degli equivalenti Redox gioca un ruolo cruciale nell’omeostasi cellulare. In S. cerevisiae le reazioni di ossidoriduzione avvengono principalmente nel citoplasma e nei mitocondri, ma NADH e NADPH sono cofattori coinvolti anche in processi biochimici localizzati nei gliossisomi e nei perossisomi (Veenhius et al. 1987). I diversi comparti cellulari non sono permeabili ai coenzimi pirimidinici e, in linea di massima, il metabolismo di queste molecole è regolato autonomamente dall’organello che le produce.
NADH e NADPH occupano ruoli differenti nel metabolismo cellulare: • Il NADH è il principale cofattore che partecipa ai processi
ossidativi (catabolici) quali la glicolisi, il ciclo di Krebs e la fosforilazione ossidativa. In generale per 1g di biomassa fresca di lievito vengono prodotti con l’ausilio di glucosio e ammonio 10 mmol di NAD+ (Verduyn et al.1990).
INTRODUZIONE
• Il NADPH è il cofattore usato nei processi anabolici ed è coinvolto sia nell’aumento della biomassa in lievito che nelle sintesi riduttive come la formazione degli acidi grassi, degli acidi nucleici, degli amminoacidi e nella via dei pentosi fosfati (PPP) (Albers et al. 1998). Esso si trova per il 60-80% nel citosol mentre il 20-40% viene prodotto nella matrice mitocondriale. Nei perossisomi il NADPH è il coenzima dell’isocitrato deidrogenasi NADP+-dipendente (IPDs) che converte l’isocitrato a
2-ossoglucarato; questo enzima è presente anche nei mitocondri (Van Roermund et al. 1998). Il turnover del NADPH avviene essenzialmente nel citosol.
Nonostante il NADPH sia preferenzialmente utilizzato nelle vie biochimiche assimilatorie, molte reazioni biosintetiche utilizzano come riduttante per la produzione di monomeri cellulari destinati alla biosintesi degli amminoacidi, il NADH formato nelle riduzioni di metaboliti centrali come il piruvato, l’ossalacetato e l’acetil-CoA.
Il NADH può essere convertito NADPH attraverso una trans-deidrogenasi mitocondriale detta NADH-chinasi scoperta da Griffiths e Bernofcky (1970). Questo sistema richede una molecola di ATP per ogni NADH. La reazione è la seguente :
NADH + ATP → NADPH + ADP.
In generale valori pari a 1000 nel rapporto NAD+/NADH favoriscono reazioni ossidative mentre valori di 0,1 stimolano processi riduttivi (Voet e Voet 1995).
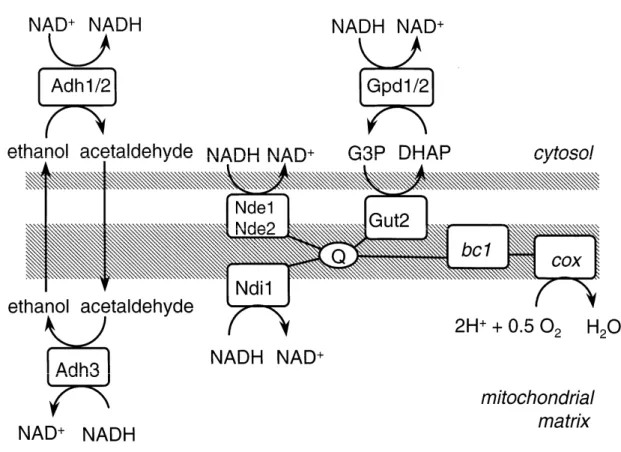
I principali meccanismi di ossidazione e trasporto del potenziale ossido-riduttivo sono:
• NADH-deidrogenasi interna (NDI): opera in condizioni aerobiche, si trova nella membrana interna del mitocondrio ed agisce durante il
INTRODUZIONE
trasporto elettronico. Questo enzima, detto NADH ubichinone ossido-reduttasi, è composto da un’unica proteina ed ha il sito attivo rivolto verso la matrice mitocondriale; esso riossida il NADH formato nel ciclo di Krebs e trasferisce due elettroni al pool dell’ubiquinone (De Vries and Marres 1997). È codificato dal gene NDI1, pesa 57 kDa e riconosce le sequenze target del coenzima attraverso la sua coda N-terminale. I suoi accettori di elettroni sono: 2, l’ubichinone-6, il diclorofenolo e la ferricianide. È represso in presenza dei tipici inibitori del complesso I come la pericidina e il rotenone, ed è trascritto maggiormente in presenza di fonti di carbonio non fermentabili (De Vries and Grivell 1988). Mutanti ndi1∆ non possono ossidare il NADH mitocondriale quindi S. cerevisiae utilizza meccanismi alternativi per trasportare gli equivalenti dalla matrice mitocondriale al citosol come i sistemi shuttle di cui parleremo più avanti.
• NADH-deidrogenasi esterna (NDE): opera in condizioni aerobiche e si trova nella membrana interna del mitocondrio ma, differentemente da NDI, volge il suo sito attivo verso lo spazio compreso tra la membrana interna e quella esterna del mitocondrio.
• Alcol deidrogenasi (ADHs): Differentemente da quanto avviene in altri lieviti, come Candida albicans e Kluveromyces lactis, S. cerevisiae non ossida il NADH esclusivamente nella catena respiratoria mitocondriale ma utilizza anche enzimi NADH-dipendenti, come le alcol deidrogenasi (ADHs), che catalizzano la conversione di NADH in NAD+
(Camourgrand et al. 1988). Il turnover del coenzima dipende inoltre dalla fonte di carbonio utilizzata come substrato di crescita del lievito. In mancanza di ossigeno e in presenza di fonti di carbonio limitate il meccanismo di riossidazione del NADH è affidato a Adh1 che catalizza la conversione dell’acetaldeide in etanolo nel citosol. Questa reazione
INTRODUZIONE
non è però sufficiente a riossidare tutto il NADH prodotto con la glicolisi e S. cerevisiae si serve dell’aiuto della glicerolo-3-P deidrogenasi con la produzione di glicerolo e dello shuttle etanolo-acetaldeide (Bakker et al. 2000). In presenza di etanolo come fonte di carbonio Ciriacy et al. (1997) hanno dimostrato che la conversione di NAD+ in NADH non è esclusivamente effettuata da Adh2, enzima citosolico responsabile della dissimilazione dell’etanolo, ma anche da Adh3, deidrogenasi presente nei mitocondri (Ciracy 1979).
• Glicerolo-3-fosfato deidrogenasi (GPD): è l’enzima citoplasmatico maggiormente coinvolto nella biosintesi del glicerolo e catalizza la riduzione del diirossiacetonfosfato DHAP in L-glicerolfosfato precursore del glicerolo. Esso occupa un ruolo determinante per il mantenimento del bilancio redox nel citoplasma in condizioni prettamente anaerobiche ed è codificato dai due isoenzimi GPD1 e GPD2 (Larsson et al. 1998).
INTRODUZIONE
Figura 8. Correlazione tra attività respiratoria, trasferimento di potenziale ossidoriduttivo tra citosol e mitocondrio e trasporto degli elettroni (Bakker et al. 2000)
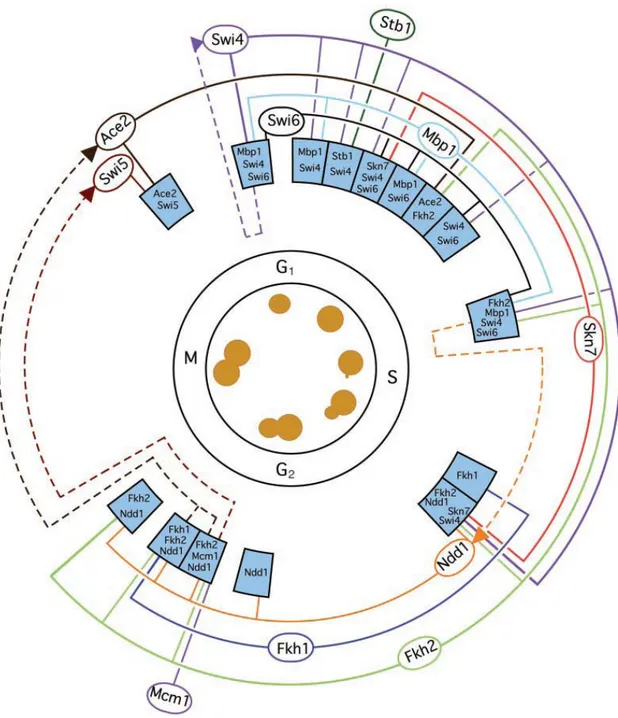
1.2 Regolatori dell’espressione genica
Affinché una cellula possa svolgere un corretto funzionamento in risposta ai cambiamenti ambientali, l’espressione genica deve essere regolata da specifiche proteine, regolatori trascrizionali, in grado di complessarsi tra loro per legarsi a determinate sequenze poste nella regione promotrice (P) di ogni gene bersaglio. Lee et al. (2002) hanno stimato che, su di uno stesso gene, possono trovarsi fino a 181 sequenze promotrici in grado di legare contemporaneamente fino a 38 proteine regolatrici. Il meccanismo di azione dei geni regolatori è un sistema a diversi livelli di complessità dove l’espressione fenotipica è strettamente connessa a networks di regolatori trascrizionali che, a loro volta, influenzano l’attività di altre proteine regolatrici
INTRODUZIONE
coinvolte nella modulazione di differenti vie biochimiche (Lee et al. 2002). Ad oggi sono stati identificati 6 tipologie di networks regolatori:
1. Autoregolatori (Ste2). Sono geni in grado di auto-indursi, le proteine da essi codifcati si legano alla regione promotrice dei geni stessi. Ciò comporta un aumento nella stabilità nel processo di traduzione del gene target accompagnato a una riduzione del “costo biosintetico cellulare” in risposta ai cambiamenti ambientali (Guelzim et al. 2002); 2. Regolatori multicomponenti (Swi4): sono geni che codificano pre
proteine che agiscono in circuiti come la glicolisi, coinvolgendo uno o più fattori. I loops feedback modulano l’espressione dei geni a monte di un particolare processo grazie all’attività di una proteina regolatrice sintetizzata alla fine della via biochimica in esame: questi sistemi sono molto stabili e oscillano tra due stadi alternativi “on/off” che attivano o disattivano il processo interessato. I loops feedforward sono sistemi che si avvalgono dell’attività di due regolatori trascrizionali interconnessi: il regolatore secondario che viene tradotto in risposta a particolari imput ambientali, e il regolatore primario che si lega alla regione di regolazione del gene target solo al raggiungimento di determinati livelli nell’accumulo della proteina regolatrice secondaria. Ciò genera un controllo temporale dei processi biochimici in esame (Shenn-Orr et al. 2002).
3. Regolatore singolo: sono geni che codificano per proteine in grado di modulare set di geni adibiti a una singola funzione biologica (Lee et al. 2002);
4. Regolatori multipli: sono geni multipli che che codificano per proteine che si legano tra loro costituendo dei complessi proteici in grado di modulare set di geni adibiti a una singola funzione biologica (Lee et al. 2002);
5. Regolatori a catena (Swi5): sono regolatori espressi in particolari fasi del ciclo cellulare. Legano i siti P dei geni target in maniera sequenziale generando un controllo temporale della traduzione che
INTRODUZIONE
avviene, in genere, durante lo stadio del ciclo cellulare successivo a quello del primo legame. Sono stati identificati 188 geni in grado di formare catene di 3-10 legami consecutivi ai siti P (Simon et al. 2001).
Figura 9. Il ciclo cellulare di lievito e i network genici che lo regolano (Lee et al. 2002).
INTRODUZIONE
S. cerevisiae presenta un gran numero di regolatori trascrizionali. In
questo lavoro verrà focalizzata l’attenzione sul complesso SWI/SNF che riveste un ruolo essenziale nella regolazione di moltissimi geni tra i quali le ADHs. SWI/SNF è un complesso multiproteico di 2 MDa scoperto nel 1994 da Peterson et al. durante un’indagine sul gene SUC2; agisce in substrati caratterizzati da fonti di carbonio semplici ed è costituito da 13 polipeptidi di cui 6 rappresentano il core attivo del complesso e 7 hanno funzione addizionale (Neigeborn and Carlson 1984): SNF2/SWI2, SNF1, SNF3, SNF5, SNF6, SNF12, SWI1/ADR6, SWI3, SWI4, SWI5, SWI6 e SWI11. Ogni polipeptide ha domini funzionali altamente conservati e il complesso è presente in molti organismi eucarioti come Drosophila e uomo (Workman and Kingston 1998).
La funzione principale del complesso è rimodellare la cromatina in modo da stimolare il legame delle proteine attivatrici e dei fattori di trascrizione TATA-box binding protein (TBP) al sito nucleosomale UASs (upstream activation site), facilitando così l’espressione genica in vivo (Imbalzano et al. 1994). Il nucleosoma rappresenta il livello primario dell’architettura della cromatina; in vitro inibisce l’inizio della trascrizione competendo con i fattori di attivazione per l’occupazione dei siti di legame del DNA (Workman and Buchman 1993).
In vivo SWI/SNF :
1. risente fortemente del tipo di terreno di coltura, rimodellando la cromatina solo in presenza di fonti carboniose semplici;
2. agisce come attivatore di particolari geni;
3. controlla i livelli di trascrizione dei geni per il mating type (MATα, HO e MCM1);
4. controlla la trascrizione dei geni SUC2 e GAL1;
5. controlla la trascrizione di geni richiesti per la biosintesi degli amminoacidi (Peterson and Herskowitz 1992);
6. controlla la trascrizione delle ADHs e gli elementi Ty (Happel et al. 1991),
INTRODUZIONE
7. controlla la trascrizione dei geni delle fosfatasi PHO5, PHO11, PHO12, PHO84 (Sudarsanam et al. 2006).
SNF2
È una proteina di 194kDa appartenente al complesso SWI/SNF ed è direttamente coinvolta nel rimodellamento in vitro dei nucleosomi (Phelan et al. 1999). Contiene due domini importanti posti sulla coda N-terminale: un
bromo-domain un’ATP-asi/elicasi-simile. Mutazioni che coinvolgono uno o
entrambi questi domini creano il blocco dell’espressione di numerosi geni (Laurent et al. 1993).
SWI1 e SWI3
Sono proteine di 148kDa e 99kDa coinvolte:
• nella replicazione del DNA durante la fase S del ciclo cellulare; • nella formazione del setto cellulare;
• nella determinazione del mating-type in S. cerevisiae e S. plombe poiché influenzano l’espressione del gene MAT1 (Sommariva et al. 2005);
• negli eventi di ricombinazione genica.
SNF2, SNF5 e SNF6
Sono proteine di 194kDa, 102,5 kDa e 37,6kDa richieste per l’espressione di SUC2 e altri geni reprimibili dal glucosio.
SNF1, SNF3 e SNF4
Sono geni coinvolti in numerosi processi biochimici: • Sensing degli zuccheri ed “Effetto Crabtree”;
• Crescita invasiva in risposta alla disponibilità di azoto; • Biosintesi dei fosfolipidi;
INTRODUZIONE
• Regolazione delle proteine con funzione di mantenimento della struttura della parete cellulare;
• Attivazione dell’ADH2, Snf1 fosforila l’istone H3 attivando il promotore di ADH2, Adr1 (Young et al. 2002).
SNF12
Mutanti di lievito snf12∆ sono sensibili in modo letale alle alte temperature, generano perdita della funzione E1A e il blocco della trascrizione di INO1 e SUC2 e HO, geni regolati a loro volta da SWI2/SNF2. (Cairns et al.1996).
SWI5
È un fattore trans-acting coinvolto in numerosi processi:
• Definizione del mating type. Swi5 dei geni HO e di MCM1 che controllano l’espressione dei geni MATα come STE, SAG, e MFα;
• Sintesi degli amminoacidi. Swi5 riconosce i promotori di ARGs geni responsabili della sintesi dell’arginina (Cosma et al. 1999);
• Controllo dei trascritti genici durante le diverse fasi del ciclo cellulare in fase M; si lega nella regione 3’ dell’mRNA istonico dei geni del cluster M/G1 come i geni MATα e i geni SIC (EGT2, PCL9, TEC1, ASH1, SIC1, CTS1) garantendo così il passaggio tra le due diverse fasi del ciclo cellulare (Knapp et al. 1996).
SWI6
È un gene che codifica, come SWI5, per un fattore di trascrizione che regola una determinata fase del ciclo cellulare. Fa parte del complesso DSC/MBF (DNA syntesis control/Mlu1 cell cicle box binding factor complex) che svolge un ruolo chiave nell’espressione dei geni ORC (Origin recognition
INTRODUZIONE
SWI4 e SWI6
Sono geni che codificano per due fattori di trascrizione coinvolti nel processo di gemmazione. Affinché le cellule possano iniziare questo fenomeno devono raggiungere la grandezza adeguata che dipende dal tipo di mezzo di coltura utilizzato per la loro crescita (Asche et al. 2000). Swi4 e Swi6 monitorano i livelli di accumulo dell’mRNA dei geni per la gemmazione legandosi al dominio cataliltico AIRE della ciclina Cdks (Jeffrey et al. 1995).
SWI11
È un attivatore trascrizonale di 19 KDa che si lega nella regione N-terminale di SNF2 e precisamente alle 40 basi del bromo-domain BRG1 (Treich et al. 1995). È attivo in condizioni di stress ambientali. (Khavari et al 1993).
CCR4
Oltre al complesso SWI/SNF ai fini del nostro lavoro è necessario indagare l’attività di CCR4, una proteina specificatamente richiesta per l’espressione di molti geni non fermentativi compreso ADH2 (Denis 1984). Mutanti ccr4∆ conferiscono fenotipi sensibili alle temperature in condizioni di crescita non-fermentative, sopprimono mutazioni nei geni SUC2, GAL4, CRE1(SPT10) e CRE2 (SPT6) ed esprimono 15 volte in più il gene ADH2 rispetto ai ceppi wild type agendo direttamente su ADR1 (Denis 1984). Per esplicare la sua funzione CCR4 presenta degli effettori trascrizionali (SWI1/ADR6 e SWI3/TYE2), un’elicasi con attività ATPasica DNA-dipendente, simile a quanto osservato per SNF2, e un rimodellatore della cromatina analogo a SNF5 (Laurent and Carlson 1992). Differentemente da quanto osservato per SWI/SNF questo fattore di trascrizione si lega direttamente alle regioni promotrici del gene in esame senza ricorrere ad altre proteine intermediarie agendo in maniera più rapida e mirata in risposta agli stimoli ambientali (Neigeborn et al. 1986).
INTRODUZIONE 1.2.1 Alcol Deidrogenasi
In S. cerevisiae sono conosciuti 7 geni che codificano per Alcol deidrogenasi (ADHs) : ADHs (ADH1, ADH2, ADH3, ADH4, ADH7, SFA1 e ADH5) enzimi citosolici e mitocondriali coinvolti direttamente nelle reazioni di riossidazione del NADH. Inoltre sono note altre 13 proteine con attività alcol deidrogenasica putativa:
• ADDs: sette geni detti aril-alcol deidrogenasi putative di cui non è
ancora chiara la funzione (Delneri et al. 1999);
• BDH1 e BDH2 coinvolti nella formazione del 2,3-butandiolo (Gonzales
et al. 2000);
• SOR1, SOR2 e XHD1 codificanti un alcol deidrogenasi specifica per
prodotti derivanti da zuccheri (Richard et al. 1999);
• CDH1 e ADH6 codificanti enzimi NADH dipendenti (Larroy et al. 2000).
Reid e Fewson (1994) dividono le alcol deidrogenasi in 3 superfamiglie (Reid and Fewson 1994):
1. deidrogenasi/riduttasi a piccola catena;
2. deidrogenasi/riduttasi a media catena (MDR). Sono una super famiglia di enzimi con subunità di circa 350 residui, organizzate in dimeri o tetrametri con due domini attivi per ogni subunità: uno è detto dominio catalitico, l’altro è responsabile del legame del nucleotide (NAD+ o NADP+). Molti enzimi appartenenti a questa famiglia hanno lo zinco nel loro sito attivo: esso forma un motivo detto zinc-containing ADH. Esistono circa 21 potenziali enzimi MDR, 12 dei quali mostrano il motivo zinc-ADH: tra questi enzimi si trovano: Adh1, Adh2, Adh3, Adh5, Adh6, Adh7, Sfa1 e Sor1. Le MDR sono state identificate nei genomi di E. coli, S. cerevisiae, D. melanogaster e C. elegants in una famiglia di enzimi strutturalmente relazionati alle cinnamil-alcol deidrogenasi (Jornvall et al. 2001);
3. alcol deidrogenasi ferro-attivate.
INTRODUZIONE
enzimatica delle ADHs che catalizzano reazioni di ossidoriduzione in grado di convertire un’aldeide in un alcol e viceversa con il conseguente consumo di NAD+/NADH. E’ stato osservato che questi enzimi sono coinvolti, oltre che nel processo fermentativo, nello step finale dell’Erlich pathway la via biochimica che sovraintende al catabolismo degli amminoacidi per la conversione di un aldeide in una lunga catena o un alcol complesso (Dikinson et al. 2002).
ADHs e Sfa1 sono presenti in organismi sia eucarioti sia procarioti e, in lievito, si presentano in forma tetramerica, mentre, in piante e vertebrati, si trovano sottoforma di dimeri (Branden et al. 1975) De Bolle et al. (1995) hanno caratterizzato la struttura tridimensionale delle ADHs in S. cerevisiae stabilendo che il core di queste proteine è costituito da domini idrofilici ripiegati a β-foglietto in contatto tra loro mediante una serie di ponti salini che incrementano la stabilità termica dell’intero complesso proteico (De Bolle et al. 1995).
Alcol deidrogenasi 1 (Adh1). È stata individuato da Racker nel 1955. E’ la
principale alcol deidrogenasi di lievito. È attiva nel citosol in forma di omeotetrameri o eterotetrameri, ciascuno associato a due atomi di zinco, uno con funzione strutturale e l’altro necessario al processo enzimatico; è prodotta notevolmente in lievito nelle cellule che crescono su glucosio o saccarosio e rappresenta circa l’1% delle proteine cellulari totali. Catalizza la conversione di acetaldeide in etanolo durante il metabolismo fermentativo con la rigenerazione del cofattore NAD+ necessario alla processo glicolitico.
In passato l’ADH1 è stato identificato come un gene costitutivo e Denis and Jung nel 1983, hanno stabilito che esso è sintetizzato costitutivamente ma i suoi livelli di mRNA sono regolati e la traduzione di Adh1 diminuisce 6-10 volte dopo il trasferimento dei ceppi di S. cerevisiae su fonti di carbonio non fermentabili, in terreni di coltura con glucosio al 10%, o durante la fase stazionaria di crescita (Denis at al. 1983). Infatti, in presenza di etanolo nel mezzo di coltura, il gene ADH1, è trascritto circa 10 volte meno rispetto a quanto osservato su glucosio al 2%; inoltre, sempre in presenza di etanolo, se
INTRODUZIONE
il promotore ADH1 è posto a regolare il gene per il citocromo C, il suo mRNA diminuisce confermando che ADH1 è regolato trascrizonalmente a seconda del mezzo di coltura usato. Denis et al. (1983) hanno confermato che il decremento della proteina Adh1 su etanolo è osservabile 3-4 ore dopo di il passaggio da glucosio ad una fonte di carbonio non fermentabile ma i trascritti già esistenti di questa proteina non sono degradati. Questo significa che anche in presenza di etanolo-glicerolo l’ADH1 ha comunque un’attività, anche se residuale, supportata dall’ammontare della sintesi di altri ADHs funzionali che non subiscono repressione trascrizionale in presenza di questa particolare fonte carboniosa (Denis et al. 1983). Infine, in presenza di glucosio 1-2%, si osserva una dimuzione del livello della proteina Adh1 che avviene circa 20 ore dopo l’inoculo ossia alla fine della fase logaritmica e all’inizio della fase stazionaria della curva di crescita, anche se il glucosio è esaurito dopo 16-18 ore. Da ciò si deduce che ADH1 è trascritto per la maggio parte nelle prime 14-15 ore dall’inizio del processo di crescita (Denis et al. 1983). Mutanti
Adh1∆ presentano una crescita ridotta su glucosio in condizioni aerobiche.
A livello strutturale è stato osservato che il gene ADH1 non contiene introni (Hall and Bennetzen 1982). Hall and Bennetzen hanno sequenziato questo gene e le sue sequenze fiancheggianti in 3’ e 5’ determinando che:
• In 5’ si trova una sequenza ricca in purine (circa il 67,5%), che costituiscono la regione del promotore e che si trovano localizzate in posizione -224/-239 e -373/-407. In vivo queste sequenze sono coinvolte nell’attacco dell’RNA polimerasi II e sono un segnale per mantenere alto il livello di trascrizione di questo gene (Gallwitz and Sures 1980).
• subito dopo il codone di inizio è presente una zona dove l’85% della sequenza nucleotidica è composta da A o T ed è altamente conservata nelle ADHs sia in lievito sia in altri organismi eucarioti (Gallwitz and Sures 1980). Questa particolare regione sembra essere coinvolta nel riconoscimento e nell’attivazione dell’RNA polimerasi ma non sembra
INTRODUZIONE
essere coinvolta nella regolazione dell’evento di trascrizione (Hall and Bennetzen 1982);
• ADH1 è controllato dai fattori di trascrizione Rap1 e Gcr1 a loro volta controllati da HPR1 in caso di mezzi contenti glucosio e da SWI1, SWI2 e SWI3 in presenza di etanolo-glicerolo o glicerolo-lattato (Peterson and Herskowiz 1992);
• la sequenza codificante i 347 amminoacidi di Adh1 è di 1041 nucleotidi in S.cerevisiae. Gli amminoacidi che hanno funzione strutturale sono: la treonina in posizione 58 con un ruolo essenziale nel mantenimento della struttura terziaria e della funzione proteica; l’isoleucina o la valina in posizione 151 sono residui altamente conservati in lievito e in altri eucarioti; l’isoleucina in 313, la valina in 338 e l’istidina in 20 conferiscono la specificità nell’attività proteica (Wills and Jornvall 1979). • sono presenti 2 codoni non-senso UGA 22 nucleotidi dopo la tripletta di fine della traduzione e, successivamente, altre 10 triplette non-senso addizionali sono reperibili nella zona che va da +1060 e +1139;
• in 3’ sono osservabili tre diverse sequenze continue di 8 nucleotidi TAAATAAA/G dette “sequenze TATAAA-like” dopo la fine del sito di terminazione in posizione +1070/+1074, +1114 /+1117 e +1133/+1136 rappresentano il sito di terminazione dell’RNA polimerasi III, e sono sequenze target per i geni MATa1 e la gliceraldeide-3-P deidrogenasi. (Eklund et al. 1976). Successivamente si trovano sequenze ripetute AATAAG costituenti la coda poly-(A) in modo analogo a quando osservato negli mRNA animali.
Il gene ADH1 è utilizzato per indagini scientifiche di diversa natura:
• Hanoi et al. (2004) hanno utilizzato il promotore di ADH1 come controllo dei geni esogeni batterici XYL1 e XYL2 che modificano la via di fermentazione dello xilosio con il consumo di una solo molecola di NADH. Le cellule che presentano questa modificazione mostrano un
INTRODUZIONE
aumento nella produzione degli enzimi legati al metabolismo del glicerolo come la glicerolo-3-fosfatasi, l’Adh2, Ald4 e Ald6;
• Taherzadeth et al. (1997) hanno accertato che Adh1 è un enzima di conversione del furfurale in acido furoico e/o acido furfurilico nella degradazione dei materiali lignocellulosici in condizioni aerobiche;
• La funzione del gene ADH1 è infine utilizzato per la produzione su scala industriale di glicerolo grazie all’aggiunta di bicarbonato (Neuberg and Reinfurth 1918).
Alcol deidrogenasi 2 (Adh2). È stata isolata da Ebisuzaki e Barron nel 1957,
è attiva nel citosol in forma di tetramero, è repressa dal glucosio, interviene nella produzione di alcuni esteri e catalizza la reazione che converte l’etanolo in acetaldeide. Questa proteina è più termostabile di Adh1 con la quale ha un’omologia dell’89%, ma differentemente da essa, ha un’alta affinità per l’etanolo, è espressa in assenza di fonti di carbonio fermentabili e nelle cellule petite (Hall and Wills 1986). La presenza di etanolo incentiva processi biochimici quali la respirazione ossidativa e la gluconeogenesi; ADH2 è un gene glucosio-repressibile che ha forte affinità per l’etanolo che utilizza come fonte di carbonio e di energia per il funzionamento cellulare (Wills 1976). Denis et al. (1983) hanno condotto analisi sull’espressione di questo gene marcando la proteina con metionina [35S] e monitorandola per immunoprecipitazione. In presenza di etanolo ADH2 viene codificata circa 2 ore dopo l’inoculo, mentre ADH1 viene completamente represso. Il corrispondente gel di poliacrilammide mette in evidenza che Adh2 ha una mobilità elettroforetica maggiore rispetto a Adh1.
ADR2 è il gene che regola l’espressione di ADH2. Esso si presenta sottoforma di due alleli ADR2-S e ADR2-F che codificano due proteine (Adr2s e Adr2r) con diversa modalità di separazione elettroforetica rispettivamente una corsa lenta e una veloce. L’analisi di diploidi ADR2/adr2 ha evidenziato
INTRODUZIONE
che l’enzima è omologo per il 90% a Adh1 e la proteina si presenta sottoforma di tetramero (Williamson et al. 1983, Ciriacy 1975);
ADR1 codifica un fattore di trascrizione che controlla l’espressione di ADH2 senza interferire con l’attività di Adr2. La sua espressione è fortemente influenzata dal mezzo di coltura utilizzato; infatti la presenza di glucosio determina bassi livelli di espressione del gene ADR1 con conseguente riduzione dell’espressione di ADH2. A livello molecolare Adr1 controlla l’espressione di Adh2 legandosi a due regioni di controllo UAS1 e UAS2, che sono elementi cis-acting poste nel promotore di ADH2 (Shuster et al. 1986). Mutanti adr1∆ bloccano l’espressione di ADH2 mantenendo il gene strutturale intatto (Walther and Shuller 2001).
Alcol deidrogenasi 3 (Adh3). Isolata da Heck nel 1969 è attiva in presenza
di glucosio (Bakker et al. 2000). Presenta un’omologia amminoacidica intorno all’80% con Adh1 e Adh2. Adh3 è codificata da un gene nucleare (ADH3), ma la proteina è localizzata nella matrice mitocondriale, dove svolge, in condizioni anaerobiche, un ruolo chiave nel trasferimento del potenziale ossidoriduttivo del NADH mitocondriale dalla matrice al citosol. Nella cellula, l’attività di ADH3 si accompagna a quella della NADH deidrogenasi interna (NDI) che trasferisce gli elettroni dal NADH intramitocondriale all’ubichinone (Bakker et al. 2000). Mutanti adh3∆ sono ceppi petite rho- che mostrano una crescita ridotta in anaerobiosi, mentre in condizioni aerobiche, non presentano particolari alterazioni fenotipiche, in accordo con la funzione di mantenimento del potenziale redox del mitocondrio in assenza di attività respiratoria.
Mutanti Petite Scoperti da Boris Ephrussi negli anni ’40, sono particolari ceppi di lievito che non sono in grado di crescere su terreni che consentono solo la respirazione, ad esempio quelli contenente alcol-etilico, acetato o glicerolo. Su glucosio 2%, lattato 0,1% o acetaldeide, generano colonie di piccole dimensioni e da questa caratterisitica ne derivano il nome. Il fenotipo petite è dovuto a mutazioni del genoma mitocondriale che determinano deficienze
INTRODUZIONE
nella capacità respiratoria. Rispetto ai ceppi wild type, i mutanti petite, dopo un’ indagine al microscopio elettronico, presentano mitocondri poco definiti e corpi amorfi durante la fase stazionaria (Bowers et al. 1967) mentre, a livello biochimico-molecolare, sono citocromo-deficienti e mostrano accumulo di citrato alla fine del TCA perché risentono del fenomeno della repressione indotta da glucosio.
I ceppi petite vengono distinti in petite nucleari, e petite citoplasmatici a seconda della localizzazione della mutazione: nucleare o mitocondriale. I
petite citoplasmatici si dividono a loro volta in petite neutrali mit-, che
presentano mutazioni puntiformi o piccole delezioni, petite neutrali rho-, che hanno grosse delezioni nell’mRNA e non danno luogo alla sintesi proteica,
petite neutrali rho0 che hanno la perdita completa del genoma mitocondriale e
petite soppressivi che non presentano modifiche nelle sequenze geniche
mitocondriali ma le proteine risultano non funzionali.
In S. cerevisiae si possono ottenere mutanti petite in condizioni di stress come ad esempio di calore. A temperature prossima alla massima temperatura di crescita del lievito si osserva l’induzione di mutanti rho- durante le prime fasi di crescita logaritmica in quanto più il ceppo di lievito si avvicina alla fase stazionaria maggiore sarà la sua resistenza a stress di varia natura (Wiemken 1990).
Da un’indagine comparativa tra i ceppi rho+ (wild type) e rho- Esser et al (1982) hanno osservato che, in terreni di coltura che non risentono dell’effetto Pasteur, i ceppi rho+ hanno una produzione di etanolo intorno al 15% e solo il 10% dell’energia prodotta dal processo respirativo viene convertita in nuova biomassa. Diversamente i ceppi rho- producono il 14% di etanolo ma il 78% del substrato viene convertito in nuova biomassa. Da queste osservazioni è stato possibile attribuire ai mutanti petite un valore economico/commerciale; infatti Gillham nel 1987 realizzò i primi mutanti rho- geneticamente stabili per massimizzare la produzione la biomassa in S.
cerevisiae in bireattori areati (Gillham 1987). Riprendendo i ceppi dalla
INTRODUZIONE
con genotipo adc e adm che non hanno attiva l’ADH1 e l’ADH3 ma presentano attivi i geni ADR1 e ADR2 (Megnet and Lustford 1968). Questi ceppi sono in grado si sopravvivere su mezzo contente glucosio perché si osserva una residuale attività di ADH attraverso test ottici.
Alcol deidrogenasi 4 (Adh4). Codificata dal gene ADH4 è membro delle
iron-activated ADH family (Williamson et al. 1987). È indotta dall’etanolo e non è sensibile alla repressione esercitata dal glucosio (Saliola et al. 1990). Si esprime a bassi livelli, ha un’omologia del 76% con ADH1, è attiva sotto forma di dimeri e catalizza la conversione di acetaldeide in etanolo in condizioni di carenza di zinco. L’analisi della sequenza genica ha rivelato una stretta somiglianza con le ADHs batteriche attivate dal ferro. Adh4 differisce dalle altre alcol deidrogenasi in quanto non sembra direttamente coinvolta nel processo di fermentazione del glucosio in ceppi wild type; infatti mutanti
Adh4∆ non mostrano particolari diversità produttive rispetto ai fenotipi rho+;
ciò ha indotto a pensare che questo sia un gene ADH1-like e che agisce soltanto in particolari condizioni di stress (Sakurai et al. 2004). Questa ipotesi è stata confermata da studi su mutanti Adh1∆ che mostrano solo un ritardo temporale per il raggiungimento dei massimi livelli produttivi nella produzione finale di etanolo: questo fenomeno non è frutto di una reversione mutazionale ma piuttosto dall’incremento dell’espressione del gene ADH4; infatti la duplicazione di questo gene può sopperire a questa deficienza, ripristinando un fenotipo wild type (Sakurai et al. 2004). Studi analoghi sono stati condotti su Kluyveromyces marxianus da Lertwattanasakul et al. (2007): questo ceppo di lievito è utilizzato su scala industriale per la produzione di etanolo in quanto è in grado di crescere ad alte temperature su fonti di carbonio poco costose come il lattosio: serve per la produzione di bioingredienti, proteine cellulari, β-galattosidasi, poligalatturonasi e, in maniera analoga a S. cerevisiae, è utilizzato per la lievitazione del pane (Caballero et al. 1995).
INTRODUZIONE Alcol deidrogenasi 5 (Adh5). È una proteina codificata dal gene ADH5 che
presenta un’omologia di sequenza del 70-80% con Adh1, Adh2 e Adh3. Essa è stata individuata attraverso il sequenziamento del genoma del lievito e sembra essere coinvolta, come Adh1, nella conversione dell’acetaldeide in etanolo. La formaldeide deidrogenasi glutadione dipendente (Sfa), cidificata dal gene SFA1, agisce come Sfa e come Adh5. E’ coinvolta in una molteplicità di processi biochimici come il catabolismo degli amminoacidi fenilalanina e triptofano e la rottura di alcol a lunga catena (Wehner et al. 1993).
Alcol deidrogenasi 6 e 7 (Adh6 e Adh7). Si tratta di due enzimi operanti
sotto forma di dimeri e facenti parte della famiglia delle cinnamil-alcol-deidrogenasi. La loro attività risulta non essenziale al metabolismo cellulare, anche se concorrono al mantenimento dei livelli di NADPH cellulari. Entrambi infatti utilizzano NADP+ invece di NAD+ come cofattore. La delezione dei geni ADH6 e ADH7 non provoca la morte cellulare in S. cerevisiae. Si pensa che questi geni, in maniera simile a quanto osservato nelle ADHs cinnamil-alcol deidrogenasiche delle piante, siano coinvolti nella sintesi di alcoli a lunga catena, nella ligninolisi e nel controllo del NADP(H) nell’omeostasi cellulare. A livello commerciale questi enzimi vengono utilizzati nel miglioramento delle di qualità organolettiche delle bevande alcoliche.
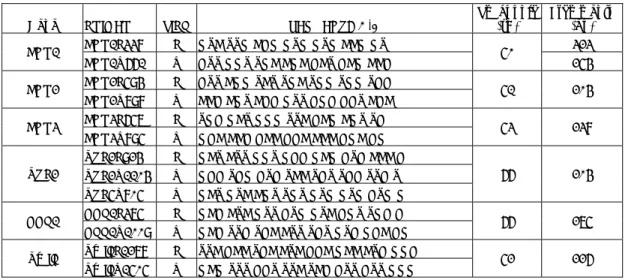
INTRODUZIONE GENE
Cofattore della proteina
Localizzazione nel genoma Nome Sistematico (accessione) ADH1 NADH ChrXV (da 160594 a 159548) YOL086C
ADH2 NAD+ ChrXIII (da 874336 a 873290) YMR303C
ADH3 NADH ChrXIII (da 434787 a 435914) YMR083W
ADH4 NADH ChrVII (da 14910 a 16307) YGL256W
ADH5 NADH ChrII (da 533756 a 534811) YBR145W
ADH6 NADPH ChrXIII (da 912141 a 911059) YMR318C
ADH7 NADPH ChrIII (da 309067 a 310152) YCR105W
SFA1 NADH ChrIV (da 159605 a 160765) YDL168W Tabella. 1 I geni ADHs
1.3 I Mutanti ADHs
L’ alcol allilico (AA) o alcol etilenico è un composto di formula HO–CH2–
CH=CH2, con massa molecolare pari a 58,08 g/mol e una densità di 0,854
g/ml, che si presenta come un liquido incolore e solubile in acqua, caratterizzato da un odore pungente.
I suoi effetti inibitori sono stati ampiamente studiati nei batteri
Escherichia coli e Lactobacillus casei (Rees et al. 1993). Nei lieviti Candida albicans, Candida utilis, Pichia pastoris e S. cerevisiae produce lesione alla
membrana citoplasmatica, ai vari organelli cellulari e all’organizzazione del citoscheletro (Lemar et al. 2005.).
Studi condotti su topo (Serafini-Cessi, 1972) hanno messo in evidenza che in vivo l’alcol allilico è metabolizzato attraverso due differenti vie ossidative: una che porta alla formazione di acrolenia e l’altra che genera la formazione di epossidi come il glicidolo e successivamente il glicerolo (Patel
INTRODUZIONE
et al 1980).
La trasformazione dell’alcol allilico in acroleina avviene in presenza di NAD+ ed è uno step biochimico mediato dagli enzimi alcol deidrogenasi (ADHs). L’acroleina, acrilaldeide o 2-propenale è il più piccolo membro della serie delle aldeidi etileniche o olefiniche, ed è un composto con massa molecolare di 56,06 g/mol. La sua formula chimica è la seguente:
La tossicità dell’acroleina dipende dalla sua struttura chimica: la presenza dell’atomo di ossigeno causa la polarizzazione della molecola e rende quest’ultima in grado di reagire con reagenti nucleofilici e altri enzimi che presentano gruppi sulfidrilici o tiolici come centri di reazione, ad esempio le ADHs (Izard 1967).
In C. albicans, è stato osservato che trattamenti con alcol allilico 10mM generano la destabilizzazione nella superficie della parete cellulare bloccando la formazione delle pseudo-ife (Lemar et al. 2005).
Per quanto riguarda S. cerevisiae l’acroleina modifica le cinetiche cellulari, il bilancio redox e la mobilità elettroforetica delle proteine, attiva i meccanismi di degradazione delle riserve intracellulari di glutadione, causa l’ ossidazione dei lipidi destabilizzando le membrane cellulari e riduce l'attività delle ADHs (Wills e Phelps 1978).
Cellule in grado di sopravvivere in terreni contenenti alcol allilico, sono quelle che non lo trasformano in acroleina. Diversi mutanti resistenti all’alcol allilico sono stati isoltai e caratterizzati; la maggior parte delle mutazioni riguarda i geni ADH. Le mutazioni determinano nella proteina la sostituzione
INTRODUZIONE
di uno o più residui amminoacidici nella sequenza proteica, la modificazione nelle caratteristiche elettroforetiche (Hall e Wills 1987), una diversa affinità per il substrato, una diversa affinità per il NAD+ , mutazioni che riguardano il promotore e i fattori di trascrizione che regolano l’espressione genica.
I mutanti, che crescono sia in mezzi di coltura contenenti glucosio, glicerolo o etanolo, presentano un incremento nela quantità di NADH e NAD+ nel citoplasma (Wills 1976) e a partire dall’alcol allilico attivano la formazione di altri alcoli con la conseguente diminuzione della produzione di acroleina (Wills and Phelps 1978).
Mutanti resistenti all’alcol allilico possono essere utlizzati come modello per investigare i meccanismi metabolici di vari microorgansimi e per condurre analisi genetiche di popolazione al fine di identificare fenomeni di microevoluzione molecolare in maniera analoga a quanto osservato per il sistema della β-galattosidasi di E.coli (Hall 1984). Sono stati isolati: mutanti per le ADHs utilizzando un mezzo di coltura contenente 1 mM di alcol allilico (AA) e YPD con 1% di glucosio che su gel di poliacrilamide presentano solo la banda Adh2 (mutanti adc) (Wills 1976); mutanti che non producono Adh2 (mutanti adr). Sono ottenuti in YPG 4% di glicerolo e 5mM di alcol allilico. Essi presentano esclusivamente attività per Adh1 e Adh3 e un decremento nella produzione di NAD+; mutanti non producono Adh3 (mutanti adm) ottenuti utilizzando terreno di coltura YPG con glicerolo al 4% e 50mM di alcol allilico. Studi recenti hanno messo in evidenza che anche nei mitocondri vi sono siti di riconoscimento per l’alcol allilico (Lemar et 2005). Mutanti petite di lievito sono in grado di sopravvivere in terreni contenenti fino a 60mM di alcol allilico; essi presentano un cilo vitale più lungo (Mazzoni et al. 2006).
Wills (1976) ha osservato che nei ceppi mutanti cambia la risposta all’ambiente spostando l’equilibrio tra alcol allilico e acroleina nella direzione dell’alcol allilico, cambiando così il bilancio NADH/NAD+ (Wills 1976).
Tra i numerosi casi di studio su mutanti resistenti ad alcol allilico si ricordano i lavori di:
INTRODUZIONE
1. Hall e Wills (1986). Utilizzando il ceppo di S. cerevisiae SC 288C hanno selezionato su alcol allilico i mutanti:
• LTR (low temperature resistant mutants) che presentano una temperature ottimale di crescita di 19°C.;
• HTR (high temperature resistant mutants) che presentano una temperatura ottimale di crescita di 37°C.
Questi mutanti sono diversi dagli altri casi di ceppi sensibili alla temperatura perché non presentano una disfunzione enzimatica o una denaturazione dell’enzima ma si ha un vero e proprio cambiamento chimico come risposta al cambiamento ambientale. La resistenza all’alcol allilico è strettamente correlata con la quantità di NADH e NAD+ presente nel citoplasma cellulare e l’attività di Adh1 si modifica in base alle modificazioni nel substrato di crescita; nei ceppi mutanti l’aggiunta di etanolo riduce gli effetti deleteri dell’alcol allilico reprimendo la crescita di colonie mutanti per ADH1 mentre l’acetaldeide incrementa la suscettibilità del lievito all’alcol allilico in quanto stimola la produzione di etanolo e NAD+ ad opera di ADH1 (Hall and Wills 1986);
2. Megnet nel 1967 ha condotto studi su Schizosacchaormyces plombe resistente ad AA. Egli ha osservato che l’alcol allilico non inibisce la crescita delle cellule wild type ma le inattiva in tempi diversi a seconda della loro sensibilità a questo particolare substrato. Differentamente da quanto osservato in S. cerevisiae, in S. plombe non si osservano differenze nei profili proteici e nell’attività di ADHs tra wild type e mutanti (Megnet 1967). Ciò che risulta modifcato sono le Km di ADHs per etanolo e il NAD+ con il conseguente accumulo di acetaldeide,
glicerolo e NADH nel mezzo di coltura. A differenza dei WT i ceppi mutanti però non riescono a utilizzare il glicerolo come fonte di carbonio a causa della bassa contrazione di NAD+ cellulare.
3. Mazzoni et al. (2005) hanno individuato in Kluyveromyces lactis cresciuto in presenza di AA ed etanolo una mutazione, detta aar900,
INTRODUZIONE
che rende inattivo ADH4 stimolando l’espressione di ADH3. Le colonie mutanti mostrano un incremento della resistenza ai cationi monovalenti quindi questa mutazione coinvolge anche geni diversi oltre alle ADHs (Mazzoni et al. 2005);
4. Dmytruk et al. hanno osservato che i ceppi di Hanesula polymorpha su metanolo che sono anche resistenti all’alcol allilico (Dmytruk et al. 2007). Questi ceppi presentano un decremento dell’attività dell’alcol ossidasica (AOX). Questi mutanti sono utilizzati per gli studi di applicabilità di enzimi modificati nello sviluppo di biosensori.
INTRODUZIONE 1.4 Morfologia del mutante
1.4.1 Isolamento di mutanti resistenti all’alcol allilico dal ceppo diploide 1014 di Saccharomyces cerevisiae
Le colonie resistenti ad alcol allilico sono state selezionate in terreno YPD 2% ed alcol allilico 4mM. Dopo 4 giorni di incubazione a 28° C i cinque mutanti isolati sono stati ricontrollati per la resistenza all’alcol allilico e conservati a 4°C. La frequenza comparsa della mutazione è circa 5x10-7.
Fig10: Morfologia del ceppo wildtype e dei ceppi mutanti su piastre di YPD (la freccia indica le colonie di piccola dimensione) (Nucci, 2006).
A livello morfologico (Fig.10), le colonie wildtype hanno dimensione grande e superficie liscia mentre le colonie mutanti presentano un evidente dimorfisimo e una superficie rugosa. Da colonie di grande dimensione si originano colonie sia di grande dimensione sia di piccola dimensione mentre da colonie di piccola dimensione si originano sempre colonie piccole. La comparsa di colonie di piccola dimensione ha una frequenza che varia tra il 10% e il 20% .
INTRODUZIONE
Fig11 Morfologia delle colonie mutanti scelte: AA60, AA95 e AA95petite (Nucci, 2006).
La colonie di piccola dimensione non sono mutanti rho- ma hanno un
comportamento petite-simile. Esse mostrano crescita su terrento YPG, un basso livello di attività mitocondriale e consumi di ossigeno fortemente ridotti sia in terreno YPD sia in YPG.
I ceppi resitenti sono stati caratterizzati attraverso l’analisi delle tetradi del ceppo ibrido ottenuto dall’incrocio tra un ceppo resistente e un ceppo
wildtype (Hy = AA95 X 1014). La frequenza della mutazione ha un rapporto di
segregazione 2:2.
1.4.2 Isolamento di revertanti sensibili all’alcol allilico dal ceppo mutante AA95
Le colonie revertanti sono state selezionate in terreno YPD 2%. Dopo 3 giorni di incubazione a 37° C le colonie isolate sono controllate per la sensibilità all’alcol allilico e conservate a 4°C. La frequenza comparsa della mutazione è circa 1 x10-3 .
INTRODUZIONE
Fig12: Morfologia dei ceppi revertanti selezionati su YPD a 37°C . Le frecce indicano le colonie di maggiore dimensione. Il ceppo revertante RV 5-1
(Nucci, 2006).
Morfologicamente i ceppi revertanti hanno colonie dimorfiche con una superficie liscia riconducibile a quella dei ceppi wildtype.
1.5 Obiettivo del lavoro
Il lavoro sperimentale di questa tesi ha avuto l’obiettivo di chiarire il ruolo della mutazione insita nella regione codificante il gene ADH1 su ceppi di Saccharomyces cerevisiea selezionati su alcol allilico. L’indagine, sul comportamento fenotipico dei ceppi e sull’espressione dei geni delle ADHs, è stata condotta con strumenti molecolari ed ha rigurdato l’analisi dell’espressione dei geni e l’attività della proteina. Inoltre è stato indagato il complesso di regolazione del gene ADH2 (SWI/SNF) e l’espressione del gene CCE1 codificante un’enzima di restrizione inplicato nel riconoscimento dei segnali di duplicazione genica. I ceppi indagati sono stati il ceppo
INTRODUZIONE
MATERIALI E METODI
2. MATERIALI E METODI
2.1 MATERIALI
Acqua
• Per i terreni è stata utlizzata H2O deionizzata microfiltrata con il
dionizzatore ELGA mod. 5124;
• Per i tamponi ulizzati nell’estrazione e nella corse delle proteine è stata utilizzata H2O Milli Q microfiltrata con il dionizzatore ELGA mod. 5124
dotato di filtro Millipore da 0,22 μm;
• Per le analisi condotte con RNA e cDNA è stata utilizzata H2O DEPC
ottenuta nel seguente modo:
1. 1L di acqua, è stato filtrato con Millipore da 0,22 μm e sterilizzato in autoclave.
2. Una volta raffreddata, all’acqua è stato aggiunto 1 mL di DEPC (dietilpirocarbonato).
3. Il contenitore è stato posto sotto cappa aspirante, e lasciato in agitazione a temperatura ambiente overnight.
4. L’acqua DEPC è stata nuovamente sterilizzata in autoclave per rimuovere ogni traccia di DEPC.
5. L’acqua DEPC è stata quindi aliquotata in tubi eppendorf sterili da 2 mL e conservata a –20°C.
Terreno di coltura solido YPD + agar:
• D-glucosio 2%
• estratto di lievito 10 g/l
MATERIALI E METODI
• bacto-agar 18 g/l
• acqua 1 l
Per i terreni solidi YPD utilizzati per la crescita di ceppi resitenti ad alcol allilico è stata aggiunta una concentrazione di alcol allilico pari a 4 mM.
Terreno di coltura YPD liquido:
1. YPD 2% • D-glucosio 2% • estratto di lievito 10 g/l • peptone 20 g/l • acqua 1 l 2. YPD 0,1%: • glucosio 0,1% • estratto di lievito 10 g/l • peptone 20 g/l • acqua 1 l
i terreni sono stati sterilizzati in autoclave per 30’ a 0.7 atm.
Tampone TE + Triton
• Tris-HCl pH 8 10 mM
• EDTA pH 8 1mM
• Triton X 4 μl/ml
La soluzione viene fatta fresca per ogni nuova estrazione di proteine. Tutti i soluti sono diluiti in H2O Milli Q.
Gel di acrilamide nativo al 5%
• Acrilammide 40%
• Bis-acrilammide 2%
• Tris-HCL pH 8,8 1,5 M
MATERIALI E METODI
• TEMED 12 μL
La soluzione è stata portata a un volume finale di 12 ml con H2O Milli Q, è
stata colata nell’apposito stampo e lasciata solidificare. I gel ottenuti sono stati conservati in frigorifero a 4°C per una notte.
Tampone di corsa per gel di acrilamide
• Tris-HCl 3g
• Glicina 14,4 g
I reagenti sono stati sciolti in acqua e portati ad un volume finale di 1 L con H2O Milli Q.
Sample Buffer per le proteine
• Tris-HCl (1M, pH 6,8) 0,5 mL
• glicerolo 2,5 mL
• β-mercaptoetanolo (14M) 0,1%
• blu di bromofenolo (0,2%) 200 μL
La soluzione è stata portata ad un volume finale di 10 ml, quindi agitata al Vortex per 5 min e frazionata in aliquote da 1 ml conservate a 4° C.
Soluzione per la colorazione dei gel di poliacrilammide
• PMS (40 mg/mL) 10 μL
• Tris-HCl 1M pH 8,8 500 μl
• NAD+ (20 mg/mL in Tris-HCl 0,1M pH 8,0) 100 μL
• NBT (50mg/mL) 20 μL
• etanolo 96% 100 μL
La soluzione è stata portata ad un volume finale di 5 ml con H2O Milli Q e
agitata energicamente al vortex subito prima dell'impiego (l'NBT tende a precipitare rapidamente in soluzione acquosa).
MATERIALI E METODI
Buffer S
• Sorbitolo 0,9 M
• EDTA pH 7,5 0,1 M
• NaP pH 7,5 0,05 M
Zimoliasi 100T L’enzima liofilizzato è stato sciolto in acqua fino ad una
concentrazione di 2 mg/mL, e conservato a –20°C in tubi eppendorf da 1,5 ml.
dNTP sono stati acquistati dalla ditta Amersham Biosciences (NTP Set 100 mM Solutions) contenente dATP, dCTP, dGTP, dTTP, 4 x 25 μmol, e disciolti
per preparare la seuente stock solution.
• H2O DEPC 360 μL
• dATP 10 μL
• dTTP 10 μL
• dGTP 10 μL
• dCTP 10 μL
La soluzione è stata conservata a –20°C in tubi sterili da 0,5 mL.
Taq DNA Polymerase della ditta Amersham Biosciences.
L’enzima, acquistato in tubi criogenici da 250U, è stato frazionato in aliquote da 10μL ciascuna e conservato a –20°C insieme al 10x Reaction Buffer e alla soluzione di MgCl2 25mM inclusi nella confezione.
Primer
I primer da utilizzare nelle amplificazioni PCR sono stati acquistati dalle ditte Invitrogen e Primm. Ciascun primer è stato diluito fino alla concentrazione di 0,1 ng/μL aggiungendo il volume di acqua DEPC indicato dalla ditta fornitrice. Lo stock così ottenuto è stato conservato a –20°C. Per ogni primer è stata preparata una ulteriore diluizione 1:10 dello stock fino ad una concentrazione di 0,01 ng/μL per l’uso in PCR e a sua volta conservata a –20° C.
MATERIALI E METODI
Costruzione dei primer per PCR
Tutti i primer specifici utilizzati sono stati costruiti sulle sequenze geniche di
S. cerevisiae disponibili sul sito web http://www.yeastgenome.org/, rispettando
i seguenti criteri:
1. Lunghezza compresa tra 20 e 25 nucleotidi (nt).
2. Massimo numero possibile di CG nelle prime 4 basi al 3’. 3. Rapporto CG / AT vicino al 50%.
4. Basso livello di “hairpin” e omeodimeri.
5. Similarità in temperatura di “melting” (Tm) tra i primer di ogni coppia. 6. Basso livello di eterodimeri tra primer di ogni coppia.
I criteri dal 3. al 6. sono stati rispettati avvalendosi di uno specifico programma informatico per l’analisi dei primer specifici, e disponibile sul web al sito http://www.idtdna.com/analyzer/applications/oligoanalyzer/default.aspx.
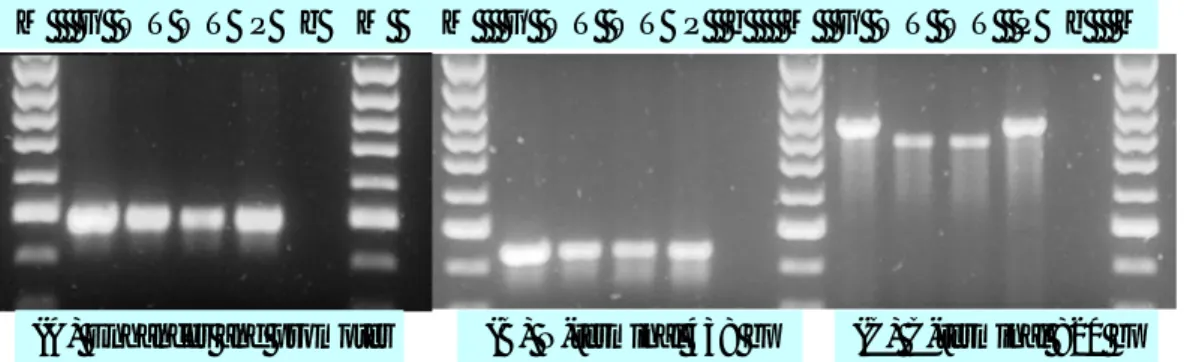
Gene PRIMER TIPO SEQUENZA 5'-3' Tm coppia (°C) Frammento (bp)
ADH1 ADH1F338 F GTA TTG ACG GTG GTG AAG GT 50 323 ADH1R661 R CTT GGT TGA AGA CAT CAG AAC 254
ADH2 ADH2F594 F CTT AGG TAT TGA TGG TGG TCC 51 204
ADH2R798 R AAC AGT ACC GTT CGC CCT ACA
ADH3 ADH3F657 F TGC GAT GGG TTA CAG AGT TC 53 238
ADH3R895 R GCA AAC CAA CCA AAA CGA CG
SNF2
SNF2F924 F GAT ATT GGT GCC GAG CTC AAA C
66 204 SNF2R1104 R GCC TCG CTC TAA TCT TCC TTC T
SNF5R805 R GAT GTA AGT TGT GTG GTG CTG G
CCE1 CCE1F375 F GAC ATA GTT CTG GTA CGT TGC C 66 275 CEE1R1009 R GAC TTC TCA ATT TCT GTC GCA CG
SWI1 SWI1F1278 F TTA CAA TCA ATA CCA GAA ATC GGC 52 226 SWI1R1505 R GAG TTT CCT TTA TAC CTT CTT GGG
Tabella 2. Sequenza, temperature di melting e dimensioni del frammento apmplificato
Loading Buffer: tampone di diluizione dei campioni per la visualizzazione degli
acidi nucleici su gel di agarosio.
MATERIALI E METODI
• Xilene cianolo 0,25%
• Glicerolo 50%
• EDTA 100mM
La soluzione è stata quindi agitata al vortex per 5 min, frazionata in aliquote da 1 mL e conservata a 4°C.
Marker (100bp e λHind)della ditta New England BioLabs
• marker 50 μg/μL 10μL
• tampone TBE 10μL
• H2O DEPC 40μL
La soluzione così ottenuta, e conservata a 4° C, è stata utilizzata nelle corse elettroforetiche caricandone direttamente 2 μl nel pozzetto desiderato.
Tampone TBE 5X
• Tris-HCl 54g
• EDTA pH 8 3,72g
• Acido borico 27,5g
I sali sono stati diluiti in acqua e portati ad un volume finale di 1litro. Il pH è stato aggiustato a 8,0. La soluzione è stata quindi filtrata per rimuovere eventuali corpi di aggregazione con un filtro Millipore da 0,22 μm.
Tampone TBE 0,5X
Il tampone TBE 0,5X utilizzato nelle separazioni elettroforetiche su gel di agarosio, è stato preparato di volta in volta per diluizione 1:10 del tampone TBE 5x.
MATERIALI E METODI 2.2 METODI
Preparazione delle colture
Il pre-inoculo è stato effettuato in Falcon sterili da 50 mL contenenti 5 mL di terreno; la colonia viene prelevata dalla piatre di Petri con uno stecchino da batteriologia sterile e disciolta nel terreno di coltura.
L’inoculo è ottenuto prelevando 0,5 mL di pre-inoculo e ponendoli in 25 mL di terreno in beuta sterile da 250 mL. Le colture così ottenute sono state poste in agitazione continua a 30°C overnight.
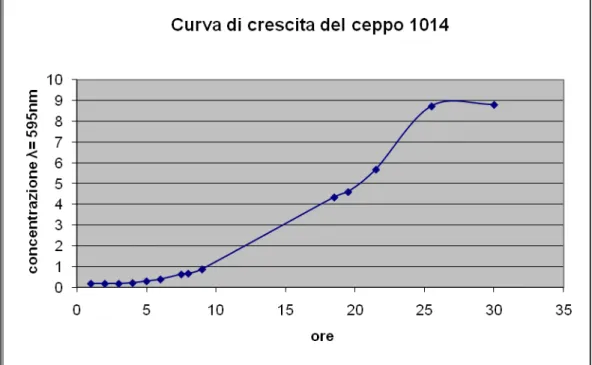
Determinazione delle curve di crescita
Lo sviluppo dei ceppi in coltura liquida è stato studiato monitorando l’aumento della torbidità del brodo colturale allo spettrofotometro: 1mL di terreno ci coltura liquido viene prelevato, a diversi stadi temporali di crescita, e caricato in una cuvette in plastica trasparente per misurazioni eseguibili nello spettro del visibile. Le letture vengono effettuate per comparazione ad una seconda cuvette caricata con il solo brodo culturale. L’assorbanza dei campioni è stata rilevata per lunghezze d’onda di 595 nm. I dati così ottenuti sono stati impiegati per tracciare curve di crescita specifiche per i diversi ceppi di lievito in esame ed i dati sono stati elaborati con l’ausilio di un foglio di calcolo Excell.
Estrazione delle proteine in condizioni non denaturanti
• 6 ml di coltura liquida di S. cerevisiae sono stati trasferiti in una provetta sterile da 1,5 mL e centrifugati per 2 minuti a 10.000 rpm;
• Il pellet così ottenuto viene pesato e sospeso in 150 μL di tampone di estrazione (TE + Triton). La soluzione così ottenuta viene trasferita in un’eppendorf da 2 mL di volume e posta in ghiaccio;
• Sono state aggiunte sfere in plastica acid washed del diametro di 0,5 mm in un volume approssimativamente ½ rispetto a quello della sospensione cellulare;