2
Le porteine ATP-binding-cassette (ABC) umane sono trasportatori transmembrana che appartengono ad una famiglia di 49 geni classificati in 7 sottofamiglie: ABCA, ABCB, ABCC, ABCD, ABCE, ABCF, ABCG1,2.
Le proteine di questa famiglia prendono parte a molti processi fisiologici:
1. a livello dell’epitelio delle mucose intestinali regolano il trasporto di composti nel lume3 e determinano una riduzione dell’assorbimento di farmaci somministrati per via orale4;
2. nel fegato proteggono gli epatociti da composti tossici riassorbiti con la bile5; 3. nei tubuli prossimali del rene partecipano all’escrezione e limitano il recupero
di molecole anfipatiche dalla pre-urina6,7;
4. controllano lo scambio tra madre e feto di molte sostanze attraverso la placenta.
Questi geni codificano per proteine di trasporto quali la Glicoproteina-P, la BCPR (breast cancer resistance protein), e le MRP (multidrug restistance-associated protein).
Il loro meccanismo d’azione prevede l’accoppiamento dell’idrolisi di una molecola di ATP al trasporto di substrati endogeni o xenobiotici.
Alcuni farmaci comunemente somministrati in terapia sono riconosciuti dalle proteine di questa famiglia come ligandi.
I geni ABC sono responsabili del fallimento di terapie chemioterapiche. Le proteine codificate contribuiscono alla MDR (Multidrug Resistance): sono in grado di legare ed estromettere dalle cellule tumorali molti farmaci, impedendo l’accumulo intracellulare dell’agente antineoplastico che non può così raggiungere una concentrazione sufficiente per inibire la crescita tumorale.
Inoltre le proteine ABC ed in particolare la P-gp sono coinvolte nella resistenza alle terapie del morbo di Parkinson e il morbo di Alzheimer.
3
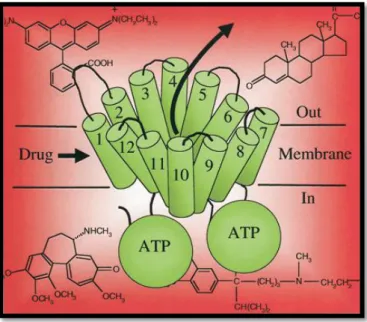
Figura 1: Gicoproteina-P
Struttura
La P-gp o Glicoproteina di permeabilità è una proteina di trasporto transmembrana che agisce come una pompa di efflusso ATP-dipendente codificata dal gene ABCB1, anche conosciuto come gene della resistenza multipla (MDR1), dotata di una specificità notevolmente ampia per numerosi composti8.
La P-gp è struturalmente costituita da 1280 amminoacidi organizzati in due unità ripetute, ciascuna di 610 residui, unite tra loro tramite una regione-linker di circa 60 aminoacidi9.
La minima unità funzionale consiste in 4 domini:
2 domini transmembrana (TMD), detti anche “membrane spanning domains” (MSD);
2 domini leganti il nucleotide (NBD).
La proteina ha una struttura pseudo-simmetrica10 costituita da due frammenti, ognuno dei quali include un dominio transmembrana (TMD) e un dominio di legame col nucleotide (NBD). I due frammenti interagiscono per formare un singolo trasportatore funzionale.
4
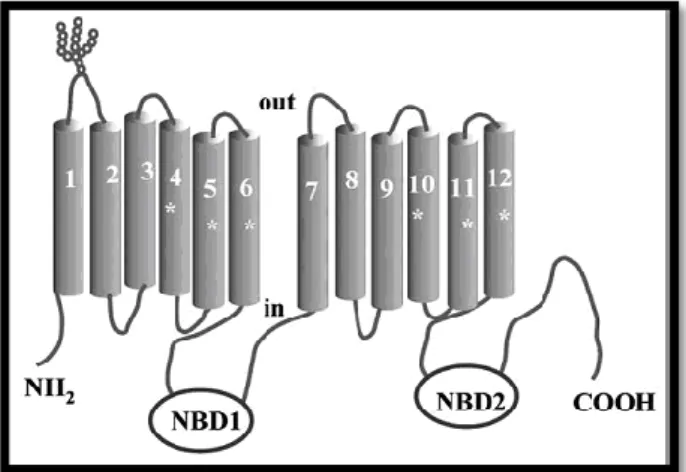
Figura 2: illustrazione schematica P-gp
I domini MSD sono composti da α-eliche multiple che attraversano la membrana ed interagiscono assieme per formare i siti di legame per il substrato ed il canale di trasporto attraverso il doppio strato lipidico.
Alcuni studi hanno dimostrato che i siti di riconoscimento della P-gp hanno una superficie estesa e flessibile e sono caratterizzati dalla presenza di amminoacidi in grado di interagire con i substrati.
Nei siti di riconoscimento sono presenti numerosi residui amminoacidici aromatici, i quali hanno la possibilità di formare legami elettrostatici, interazioni idrofobiche ed interazioni .
I domini NBD legano l’ATP; l’idrolisi del nucleotide ed il suo rilascio inducono cambiamenti conformazionali, spesso associati al trasporto del substrato attraverso la membrana17-19.
Come dimostrato da studi di binding, il legame della proteina con ATP non sembra essere necessario per il riconoscimento e l’interazione con il substrato. Tuttavia il processo di trasporto è obbligatoriamente accoppiato all’idrolisi del nucleotide, e sono necessari ambedue i siti di legame con ATP per garantire la funzionalità della proteina; infatti quando l’idrolisi è bloccata, la P-gp risulta inattiva.
Entrambi i siti per l’ATP hanno capacità idrolitica ma non agiscono simultaneamente.
5
Figura 3: struttura tridimensionale P-gp
Localizzazione
La P-gp è localizzata sulle membrane apicali del fegato, del rene, della placenta, sui villi degli enterociti intestinali20,21. Nell’intestino questa glicoproteina interagisce con il citocromo e mostra un’attività strategica nella regolazione del metabolismo e dell’assorbimento dei farmaci.
Inoltre risulta essere espressa in numerose barriere, come la barriera emato-encefalica, la barriera sangue-fluido cerebrospinale, e la barriera sangue-testicolo. Anche attraverso queste barriere modula l’assorbimento e l’escrezione di xenobiotici22.
Recentemente è stata individuata la P-gp anche a livello di compartimenti intracellulari; in particolare sembra agire a livello di vescicole citoplasmatiche, trasportando e concentrando all’interno di queste i farmaci-substrati, impedendo loro il raggiungimento del bersaglio intracellulare.
6 Barriera Ematoencefalica
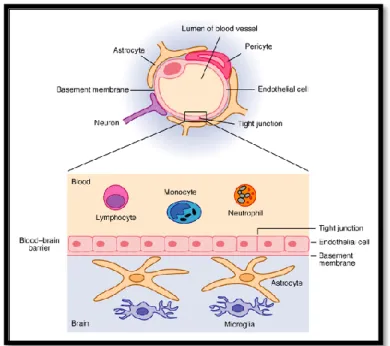
La barriera ematoencefalica è una struttura anatomica costituita dalle cellule endoteliali dei capillari del Sistema Nervoso Centrale con funzione di protezione dei tessuti cerebrali da elementi nocivi presenti nel sangue.
L’integrità della barriera viene controllata da astrociti, periciti e componenti della matrice extracellulare23.
Figura 4: barriera ematoencefalica
Nel’uomo adulto la vascolarizzazione capillare copre circa l’1% del volume cerebrale e l’area superficiale delle cellule dell’endotelio capillare è stimata essere di 10 metri quadrati.
La barriera ematoencefalica ha il ruolo di proteggere il cervello da xenobiotici tossici mantenendo separato il cervello dalla circolazione cerebrale.
Solamente composti xenobiotici e farmaci lipofili possono attraversare la barriera ed entrare nel cervello tramite diffusione semplice. Alcuni studi hanno dimostrato che esiste una relazione fra la lipofilicità di un farmaco e la sua permeabilità cerebrale24,25.
7
Nonostante la lipofilicità sia un parametro importante per determinare la penetrazione di un farmaco nel cervello, molti composti lipofili mostrano avere una penetrazione cerebrale ridotta.
Questa limitata permeabilità può essere attribuita anche ad altri fattori. Tra questi la capacità di formare legami ad idrogeno sembra influenzare l’accesso al SNC; infatti esiste una correlazione negativa fra la penetrazione di molecole lipofile ed il numero totale di legami ad idrogeno; i derivati che possono interagire con più legami ad idrogeno mostrano una permeabilità inferiore26.
Altro fattore che condiziona la permeabilità al SNC è rappresentato dalle dimensioni molecolari del farmaco27.
La possibile funzione di efflusso della P-gp non è stata messa in relazione al ridotto assorbimento cerebrale di questi composti lipofili negli animali fino al momento della scoperta della presenza della P-gp nei capillari del cervello28,29.
Grazie all’utilizzo di anticorpi monoclonali venne dimostrato che la P-gp è altamente espressa sulla superficie delle cellule endoteliali dei capillari cerebrali. Attualmente si ritiene che la ridotta penetrazione sia dovuta al trasporto mediato dalla P-gp.
Quando i farmaci che si comportano da substrati della glicoproteina diffondono dal sangue verso le cellule endoteliali vengono rapidamente riportate nel sangue. In questo modo la capacità di penetrazione ai tessuti cerebrali di una varietà di farmaci viene strettamente controllata, anche in condizioni fisiologiche.
Farmaci antiistaminici di prima generazione inducono spesso effetti secondari a livello centrale (es. sedazione), mentre i farmaci di seconda generazione non hanno questi effetti. Studi in vitro su cellule endoteliali cerebrali di topo hanno dimostrato che tutti i farmaci antiistaminici di seconda generazione sono substrati della P-gp, mentre quelli di prima generazione non mostrano affinità per questa proteina30,31. E’ stato allora ipotizzato che l’attività di trasporto della P-gp nella barriera ematoencefalica possa esser la spiegazione per la mancanza di effetti centrali dei moderni farmaci antiistaminici.
La glicoproteina P sembra anche esser coinvolta nella modulazione della risposta immunitaria. Essa media la secrezione di numerose citochine dai linfociti T e la migrazione delle cellule dendritiche dalla periferia verso i linfonodi in modo tale da dare inizio alla risposta immunitaria mediata dai linfociti T32.
8
Recenti studi indicano che tale proteina svolge un ruolo, indipendente dall’efflusso di famaco, nell’inibizione della apoptosi33-38. La modulazione dell’efflusso delle citochine, “signaling lipids” e il pH intracellulare sono stati suggeriti esser vie tramite le quali la P-gp potrebbe indurre la resistenza all’apoptosi cellulare35.
La up-regulation della P-gp negli astrociti e nei neuroni osservata nei casi di malattie neurodegenerative potrebbe esser un importante meccanismo per proteggere le cellule cerebrali.
Barriera Sangue-Fluido Cerebrospinale
La barriera sangue-fluido cerebrospinale sembra contribuire in maniera significativa al controllo del trasporto fra sangue e cervello; ha una superficie tre volte inferiore rispetto alla barriera ematoencefalica, e per questo viene considerata di minor importanza nella regolazione del trasporto di sostanze al cervello; tuttavia ha caratteristiche differenti a quelle della BEE e il loro rispettivo contributo nello scambio sangue-cervello non può esser giudicato limitatamente all’ampiezza della superficie.
Questa barriera sembra influenzare la penetrazione di xenobiotici nel cervello attraverso:
una maggiore organizzazione delle tight junction che si traduce in un significativo aumento della permeabilità;
la presenza di enzimi metabolizzanti, come isoenzimi del citocromo P450;
l’espressione di trasportatori quali P-gp e MDR1 (proteina codificata da geni appartenenti alla superfamiglia ABC).
9
Figura 5: attività della P-gp e della MDR1 nella barriera sangue-fluido
cerebrospinale
Le proteine P-gp e MDR1 a livello dell’epitelio coroidale mediano il trasporto in opposte direzioni: la P-gp è localizzata sulla membrana apicale dell’epitelio dei plessi coroidali e trasporta i substrati all’interno del liquido cerebrospinale, mentre MDR1 agisce sulla membrana basolaterale mediando l’efflusso verso il sangue. Quindi la P-gp a livello dei plessi coroidali induce un aumento della quantità di substrato nel liquido cerebrospinale, e nello stesso tempo è responsabile dell’efflusso dalle altre zone cerebrali.
Placenta
La placenta è l’organo adibito a mettere in stretta comunicazione il sangue materno e il sangue fetale, mantenendo separate le due circolazioni.
La sua attività risulta essenziale per il mantenimento della gravidanza e lo sviluppo del feto.
Il suo principale scopo è la regolazione dello scambio di nutrienti e gas fra madre e feto e la rimozione di prodotti di rifiuto fetali; allo stesso tempo i composti chimici a cui viene esposta la madre possono penetrare attraverso la placenta39,40; la placenta, infatti, è il primo organo del feto che viene a contatto con xenobiotici e farmaci. La capacità di una molecola di attraversare tale organo è in funzione della liposolubilità, peso molecolare, grado di ionizzazione e legame con proteine plasmatiche.
10
Molecole non ionizzate, lipofile, e con un peso di 600 Da possono attraversare la placenta per diffusione passiva, mentre gli xenobiotici con struttura sufficientemente sovrapponibile a quella dei composti endogeni, possono esser riconosciuti come substrati41,42.
Grazie a studi immunoistochimici è stata scoperta la presenza della P-gp nei microvilli del sinciziotrofoblasto della placenta umana nel primo trimestre di gravidanza43, ed ulteriori studi hanno dimostrato che la P-gp viene espressa durante l’intero periodo di gestazione44,45
11
Fattori di regolazione
L’espressione di molti trasportatori ABC è sotto lo stretto controllo di recettori nucleari orfani; questi costituiscono una famiglia di fattori di trascrizione che agiscono come eterodimeri in grado di legare il promotore ed indurre l’espressione genica.
Greick et al. scoprirono un complesso gruppo di regolazione di molti siti di legame per i recettori nucleari attivati da ligando, il PXR, recettore X del pregnano (NR112) nella regione 5’-upstream del gene MDR1 umano.
PXR è un membro di una superfamiglia di fattori di trascrizione attivati da ligando; può esser attivato da steroidi naturali, come progesterone e pregnenolone, glucocorticoidi sintetici e antiglucocorticoidi, ma anche da un’ampia gamma di xenobiotici inclusi composti alimentari, tossici e numerosi farmaci comunemente impiegati (per esempio steroidi, agenti antitumorali, inibitori delle proteasi dell HIV, glucocorticoidi, ed anticonvulsivanti).
Studi su fegato ed intestino hanno dimostrato che PXR agisce su geni coinvolti nel metabolismo di xenobiotici (fase I e II) e nel trasporto di efflusso. PXR viene, quindi, considerato un importante regolatore delle difese contro xenobiotici a livello cellulare e molecolare46.
12
I sistemi controllati dal PXR includono l’isoforma 2 del polipeptide trasportatore di anioni organici (SLCO1A4), la pompa di escrezione dei sali biliari (ABCB11), le isoforme 2 e 3 della multidrug resistance- associated protein, MRP2 e MRP3 (ABCC2 e ABCC3), ed infine la P-gp47-50.
Espressione di questi recettori nucleari è riscontrabile all’interno delle cellule dell’endotelio dei capillari cerebrali51
ed è stato notato che l’esposizione a PCN (pregnenolone 16alfa-carbonitrile) e dexametasone, entrambi ligandi per la PXR, incrementa i livelli di espressione della P-gp ed aumenta anche il trasporto mediato dalla glicoproteina di un derivato fluorescente della ciclosporina A nei capillari del cervello del topo51.
Questi studi hanno messo in evidenza le prove dell’espressione della PXR nel cervello e la sua capacità di regolare le pompe di efflusso di xenobiotici a livello della barriera emato-encefalica.
E’ stato quindi dimostrato che la riduzione dell’efficacia di farmaci per terapie sul sistema nervoso centrale che sono substrati della P-gp potrebbe esser dovuta all’attivazione della PXR nella barriera emato-encefalica.
13
P-pg e le malattie neurodegenerative
La P-GP esercita la propria funzione protettiva anche nella barriera ematoencefalica (BEE), e recenti studi hanno evidenziato l’esistenza di una correlazione fra l’attività e/o l’espressione della P-gp e malattie del sistema nervoso centrale (SNC) come il morbo di Azheimer (AD), il morbo di Parkinson(PD) e l’epilessia52.
E’ riportato che alcuni farmaci anti-epilettici come fenitoina e carbamazepina sono substrati per la PG-gp. Conseguentemente, l’iperespressione intrinseca o acquisita di questa proteina nella BEE può portare alla resistenza verso questi farmaci limitandone la penetrazione nel cervello e di conseguenza l’efficacia della terapia53. L’iper-espressione della P-gp può esser legata all’insorgenza dell’epilessia, alla terapia con farmaci o polimorfismo del gene ABCB1, che codifica per la proteina54. La carbamazepina è capace di indurre l’espressione della pompa di efflusso ed enzimi che metabolizzano il farmaco nell’intestino, riducendone così l’assorbimento55,56
.
Gottesman e collaboratori hanno riportato che il polimorfismo del gene C3435T nell’esone 27, che interessa il tempismo di ripiegamento della P-gp e l’inserimento nella membrana, porta ad una P-gp con alterati siti di interazione col farmaco57 e una conseguente riduzione della biodisponibilità orale di alcuni farmaci antiepilettici. L’iper-espressione della P-gp nei vasi della barriera ematoencefalica, che interessa l’accesso al cervello di farmaci antiepilettici, può essere considerato come una delle cause della resistenza ai farmaci.
Il Parkinson è una malattia caratterizzata dalla perdita delle cellule dopaminergiche nella “substantia nigra” e che manifesta aggregazioni proteiche conosciute come corpi di Lewy; questi sono composti principalmente da -sinucleina, una proteina nucleare solubile che in condizioni patologiche sviluppa oligomeri insolubili58. Sebbene non sia ancora noto se l’-sinucleina sia un substrato della P-gp, è stato ampiamente dimostrato che la P-gp è in grado di espellere alcuni pesticidi59 e tossine considerate responsabili dell’inizio della malattia di Parkinson60
. Infatti, MPP+, una neurotossina coinvolta nella sindrome, rappresenta un substrato della P-gp61.
Così da quando è stata dimostrata una correlazione in vivo fra l’invecchiamento e la riduzione della funzionalità della pompa, una disfunzione dell’attività della P-gp
14
nella barriera ematoencefalica è considerata una delle cause dell’incipit del morbo di Parkinson e del morbo di Alzheimer62.
Bartels e collaboratori dimostrarono che la riduzione della funzionalità della P-gp era dipendente dall’età, stabilendo così una stretta relazione fra invecchiamento e suscettibilità alla malattia di Parkinson62. Questo studio ha portato all’identificazione delle regioni cerebrali, materia bianca e regioni orbitofrontali, nelle quali la funzione della pompa è ridotta.
Il morbo di Alzheimer è un disordine neurodegenerativo caratterizzato da una progressiva perdita della funzione cognitiva; evolve in diverse forme di demenza che mostrano placche insolubili di -amiloide (A) e ammassi neurofibrillari52. La secrezione di A è l’evento iniziale nella patologia, mentre l’aggregazione di A è implicato nella neurodegenerazione successiva63.
Sembra che la P-gp svolga un ruolo cruciale nell’eliminazione di A dal cervello52. Un recente studio ha indicato che la proteina -amiloide potrebbero esser un substrato endogeno della P-gp64,65. Per muovere dal cervello al sangue, la proteina A deve attraversare sia la membrana citoplasmatica basolaterale sia la membrana apicale dell’endotelio capillare.
Nel cervello affetto da Alzheimer i livelli di deposizione di -amiloide sono inversamente proporzionali al livello di P-gp nella vascolarizzazione cerebrale66. Non è chiaro se questo rifletta un rapporto di causa-effetto, tuttavia dati preliminari su roditori indicano che i segnali di esposizione a -amiloide aumentano la degradazione della glicoproteina nei capillari cerebrali in vitro ed in vivo.
Questi dati sembrano suggerire un circolo vizioso di feed-back positivo nel quale l’aumento del livello di A nel cervello riduce l’espressione della P-gp, e di conseguenza promuove l’ulteriore incremento di A a livello cerebrale.
Tenendo presente questi effetti la proteina di trasporto P-gp potrebbe essere considerata un bersaglio per la prevenzione e la terapia di questo disordine del SNC65.
15
Meccanismo d’azione
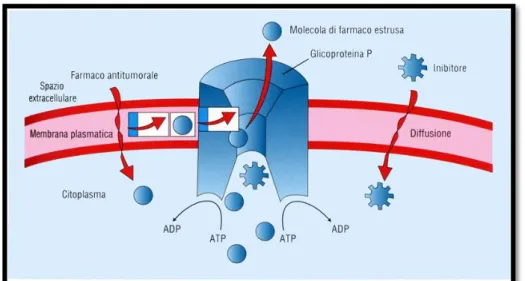
La P-gp è iper-espressa in diverse linee cellulari tumorali ed è responsabile dell’efflusso dei farmaci al di fuori della cellula52
. Utilizza l’energia dell’idrolisi dell’ATP per estrudere i composti grazie ad un complesso processo di trasporto67
. La fase iniziale del processo di trasporto coinvolge il sito di legame ad alta affinità per i farmaci e contemporaneamente il legame con ATP nei NBD.
Il legame con il farmaco e l’ATP sono accoppiati; l’accoppiamento dei legami al substrato ed al nucleotide causa cambiamenti conformazionali del sito di legame, e si verifica il rilascio del farmaco.
Per descrivere il meccanismo di trasporto sono stati suggeriti 3 modelli:
Modello a poro: i farmaci che legano la P-gp nel citosol vengono trasportati fuori attraverso una proteina canale68,69.
Modello flippasi: la P-gp conduce i farmaci dal compartimento interno citoplasmatico a quello esterno contro un gradiente di concentrazione. L’idrolisi dell’ATP fornisce l’energia per far muovere le molecole del foglietto citoplasmatico della membrana a quello esterno. Il substrato si disperde sul lato citoplasmatico della membrana e si lega alla glicoproteina; in seguito all’idrolisi dell’ATP la molecola viene “flippata” sullo strato esterno e diviene libera di rompere il legame dalla proteina.
Modello “hydrophobic vacuum cleaner”: le molecole riconosciute dalla P-gp nel doppio strato lipidico entrano nella proteina in un sito membranoso ed escono attraverso la cavità centrale, costituita dal ripiegamento di una o più subunità.
16
Figura 7: possibili meccanismi d’azione della P-gp
Substrati-Modulatori-Inibitori
Substrati e Modulatori
La P-gp interagisce con un ampia varietà di composti, nonostante vi siano numerose diversità strutturali.
I substrati della P-gp differiscono fra loro sia per struttura sia per funzione, ma presentano alcune caratteristiche comuni come un alto grado di idrofobicità, un basso peso molecolare, compreso fra 250 e 900 Da, una carica positiva a pH neutro e la capacità di attraversare la membrana citoplasmatica per diffusione passiva.
Fra i substrati si ritrovano farmaci appartenenti a numerose categorie terapeutiche, quali antineoplastici, antiaritmici, immunosoppressori, antiistaminici, antivirali e farmaci utilizzati nelle cure delle malattie di Parkinson ed Alzheimer.
Queste molecole, dopo essere penetrate nella cellula, vengono riportate nella regione extracellulare dalla proteina mediante trasporto attivo, e quindi contro gradiente di concentrazione70.
I modulatori, attraverso un’interazione allosterica negativa, riducono la capacità della proteina di formare il legame con i substrati.
E’ stato dimostrato, attraverso l’impiego di radioligandi, che queste molecole alterano il legame col substrato in maniera non competitiva (così viene ridotta la
17
massima densità recettoriale (Bmax) per il legame col substrato ma non la costante di equilibrio di dissociazione (Kd)), interagendo con la P-gp in un sito diverso.
Inibitori
Gli inibitori interferiscono con la fase di legame con il substrato o con il nucleotide, bloccando così il trasporto da parte della P-gp52.
L’attività degli inibitori ha perfezionato la comprensione del meccanismo di trasporto della P-gp.
Figura 8:interazione di un inibitore con la P-gp
L’ampio numero di composti che interagisce con la P-gp ha portato ad ipotizzare la presenza di vari siti di legame70,71. Sono stati individuati almeno 4 distinti siti d’interazione sulla proteina; tre siti sono stati riconosciuti essere quelli adibiti al trasporto in quanto interagiscono sia con i substrati e che con i modulatori. Al contrario, il quarto è un sito di regolazione sul quale alcune molecole come elacridar e nicardipina sembrano agire come modulatori.
La formazione di un legame in uno dei quattro siti provoca negli altri un cambiamento conformazionale70.
18
Studi SAR hanno rivelato gli aspetti strutturali strettamente correlati alla capacità inibitoria delle molecole ed hanno permesso la sintesi di diverse classi di inibitori. Il verapamile, un bloccante dei canali del calcio, rappresenta un farmaco appartenente alla I generazione di inibitori. Gli effetti secondari cardiaci ne hanno limitato l’impiego come inibitore della P-gp; tuttavia è stato considerato come modello per la progettazione di nuove molecole.
verapamile
La sostituzione del gruppo dimetossifenil-4-ciano-4-isopropilbutile del verapamile con una funzione azapentaciclica72 ha dimostrato il ruolo fondamentale svolto dalla sovrapposizione degli orbitali del residuo aromatico nel legame con la P-gp.
Queste scoperte hanno condotto alla realizzazione di numerose molecole dotate di anelli aromatici in grado di inibire efficacemente la P-gp senza indurre effetti cardiovascolari73.
La natura del linker spaziatore non si è rivelata critica ma deve essere garantita la presenza di un atomo di azoto protonato ad una distanza ottimale fra i due gruppi aromatici per riconoscere il sito di legame74. Una parziale riduzione della flessibilità ha migliorato l’assorbimento e la selettività75
.
Anche la trifluoperazina ha mostrato la capacità di contrastare la MDR; così sono stati sviluppati bioisosteri del nucleo fenotiazinico quali le fenossazine e gli acridoni76. Inentrambe le classi la presenza di un’ammina terziaria legata ad un nucleo triciclico attraverso una catena metilenica ha portato ad un incremento dell’inibizione della P-gp.
19
trifluoperazina fenossazina acridone
Studi SAR hanno permesso la progettazione e la sintesi di inibitori di nuova generazione come lonafarnib, zosuquidar, e il suo derivato laniquidar.
lonafarnib zosuquidar laniquidar
La molecola acridonecarbossammide presenta attività inibitoria nei confronti sia della P-gp sia della BCRP (appartenente alla superfamiglia ABC) ed è stata sfruttata per la realizzazione di studi SAR riguardo il riconoscimento delle due proteine77, in particolare l’importanza dell’ isochinolina e il frammento N-anilide legato al nucleo N-azaxantone.
La presenza di piperazina o piperidina legata al nucleo N-azaxantone migliora l’inibizione della P-gp grazie alle caratteristiche basiche, che inducono la protonazione a pH fisiologico, facilitando l’accumulo intracellulare di farmaco78. Il composto antranilammidico è in grado a di invertire la MDR grazie alla presenza sia di una funzione tetraidroisochinolina sia di una funzione antranilammidica su un anello fenilico.
20
Una maggiore idrofilia garantisce un mantenimento dell’attività inibitoria nei confronti dei due bersagli farmacologici74.
Acridonecarbossammide (elacridar) antranilammide
Per quanto riguarda lo spaziatore alchilico, sono stati inseriti anelli pirrolidinici, cicloesilici, ed arilpiperazinici; il migliore risultato è stato ottenuto con la funzione arilpiperazinica e un nucleo antranilammidico metossi-sostituito79,80.
Recenti studi condotti su composti di origine naturale quali la curcuma ed il curcumino hanno evidenziato la loro capacità di interagire con la P-gp, inducendo effetti completamente opposti.
La curcuma incrementa l’attività della proteina di trasporto inducendo up-regulation dell’espressione genica e aumento dei livelli di mRNA.
Il curcumino, al contrario, inibisce la P-gp riducendo i livelli di mRNA81.
curcumino
Anche i flavonoidi e le furanocumarine, oltre ad inibire il citocromo P3A4, agiscono come modulatori sulla P-gp82.
21
I flavonoidi sono composti polifenolici presenti nella nostra alimentazione; hanno numerose proprietà benefiche per la salute umana in quanto sono sostanze ad attività antiossidante, antiinfiammatoria, antivirale, antitumorali83.
I flavonoidi hanno mostrato la capacità di inibire alcune proteine leganti ATP. Molecole come quercetina e camferolo legano direttamente il dominio NBD della P-gp di topo84.
L’inserimento di un gruppo alchilico nella struttura dei flavonoidi aumenta l’idrofobicità e induce un aumento dell’inibizione.
I flavonoidi non sostituiti interagiscono in maniera bifunzionale con i due domini NBD della proteina, mentre analoghi sostituiti in posizione 6 o 8 con un prenile, legano anche il sito di legame con i substrati nel dominio TMD84.
Tabella 1
R1 R2 R3
quercetina -OH -OH -H
camferolo -H -OH -H
Nonostante la bassa tossicità, sono state riscontrate alcune limitazioni nell’uso in terapia di tali sostanze85. La loro attività sembra esser moderata, ed hanno un ampio spettro di attività biologiche secondarie, inibiscono altre proteine ATPasi e hanno azione anti-estrogena.
Per contrastare efficacemente le MDR sono necessarie dosi che indurrebbero la comparsa di effetti collaterali tossici.
22
È stata sfruttata la natura pseudodimerica e la molteplicità dei siti di legami della P-gp attraverso interazioni polivalenti, per migliorare la selettività e la potenza dei flavonoidi.
Queste interazioni polivalenti nei sistemi biologici sono caratterizzate dalla simultanea formazione di legami multipli con un’entità biologica86.
23
P-gp nella chemioterapia
La Multidrug Resistance (MDR) è uno dei problemi principali nella chemioterapia dei tumori. Il meccanismo di resistenza meglio caratterizzato è quello mediato dall’iper-espressione della P-gp, che trasporta al di fuori delle cellule una grande varietà di farmaci antitumorali, risultando in un ridotto accumulo intracellulare di farmaco.
Inibitori di prima generazione
In una pubblicazione di Tsuruo et al.87 per la prima volta si rivelò che il verapamile, un bloccante dei canali del calcio, potesse invertire la resistenza inibendo l’efflusso di farmaco mediato dalla pompa.
Tuttavia l’inversione della MDR da parte del verapamile era associata ad effetti collaterali cardiovascolari52.
L’agente immunosopressivo Ciclosporina A è stato scoperto essere un potente modulatore della MDR ma sfortunatamente interferisce con l’attività di metabolizzazione dell’enzima CYP 3A488.
Farmaci quali gli inibitori della calmodulina, le chinine antimalariche, ed anche il Tamoxifene (antitumorale) appartengono alla prima generazione di inibitori della P-gp89,90. Tuttavia la loro inaccettabile tossicità li ha preclusi dall’uso clinico.
Questi composti sono caratterizzati da una bassa potenza e specificità, dovuta alla loro attività primaria, così richiedono elevate dosi, che sfortunatamente hanno determinato la comparsa di diversi effetti collaterali91,92.
Questi composti mancano di selettività nei confronti di trasportatori di tipo ABC e di altri sistemi enzimatici (CYP450) con conseguente comparsa di importanti interazioni farmacocinetiche93.
24 ciclosporina A
Inibitori di seconda generazione
La seconda generazione di inibitori include analoghi del verapamile (dexverapamil e dexiniguldipine) che mostra una minore attività cardiovascolare, un derivato della ciclosporina (valspodar), ed il biricodar, derivato dal tacrolimus91.
Questi modulatori sono più potenti di quelli di prima generazione sebbene continuino a presentare interazioni farmacocinetiche critiche dovute all’inibizione del Citocromo P450 e riduzione dell’escrezione biliare con conseguente aumento della tossicità94,95.
25 Inibitori di terza generazione
La progettazione di inibitori della P-gp di terza generazione si è avvalsa di studi SAR70 ed ha condotto alla sintesi di composti dotati di maggiore efficacia e selettività e privi degli effetti secondari tipici delle precedenti generazioni di inibitori52.
Farmaci come acridonecarboxamide, antranilammide, ciclopropildibenzosuberano, l’inibitore della farnesil transferasi, e diarilimidazolo sono alcuni inibitori di terza generazione52.
acridonecarboxamide
antranilammide
26
inibitore della farnesil transferasi
INTRODUZIONE ALLA PARTE
SPERIMENTALE
28
La glicoproteina-P (P-gp) è una pompa ATP-asica che appartiene alla famiglia dei trasportatori di membrana ABC. La P-gp oltre ad essere a livello della cellula tumorale, risulta essere espressa a livello della barriera emato-encefalica (BEE), nell’epitelio intestinale e nel fegato dove sembra svolgere un ruolo importante nella regolazione della secrezione di molecole lipofile e nell’estrusione di xenobiotici dall’organismo.
Dato l’importante ruolo svolto dalla P-gp, la sua inibizione rappresenta un target interessante sia per prevenire la MDR, che si instaura durante il trattamento di patologie infettive o tumori, che per aumentare l’efficacia terapeutica di farmaci ad azione centrale.
Ad oggi sono stati caratterizzati numerosi agenti capaci di modulare la P-gp come ad esempio il verapamile (un bloccante dei canali del calcio), la ciclosporina A ed alcuni antagonisti della calmodulina.
verapamile ciclosporina A
Tutti questi composti producono però dei risultati contrastanti in vivo a causa della loro bassa affinità di legame alla P-gp che quindi richiede la somministrazione di dosi elevate con conseguente comparsa di effetti tossici. È inoltre importante sottolineare il fatto che questi agenti chemiosensibilizzanti sono substrati oltre che della P-gp anche per altri trasportatori e sistemi enzimatici e possono quindi presentare imprevedibili interazioni farmacocinetiche se somministrati in associazione a chemioterapici.
29
Il verapamile, ad esempio, a causa della bassa selettività per la P-gp e della sua attività intrinseca calcio-antagonista, è risultato esser cardiotossico alle dosi efficaci come P-gp inibitore, mentre la ciclosporina A è risultata nefrotossica.
Per superare questi limiti è stata successivamente sviluppata una seconda generazione di modulatori della P-gp che comprende farmaci come il dexverapamile ((+)-verapamile) ed il biricodar.
biricodar
I farmaci appartenenti a questa famiglia di modulatori sono più potenti e meno tossici dei loro predecessori, ma influenzano in modo significativo il metabolismo e l’eliminazione dei farmaci chemioterapici provocando quindi una marcata tossicità che costringe ad una drastica riduzione del dosaggio del chemioterapico. Inoltre, molti modulatori appartenenti alla seconda generazione agiscono come substrati per altri trasportatori, soprattutto per quelli appartenenti alla famiglia delle proteine ABC; la loro inibizione potrebbe pertanto diminuire la capacità delle cellule sane e dei tessuti di proteggersi dagli agenti citotossici.
Successivamente, in seguito a studi di SAR è stata sviluppata una terza generazione di inibitori della P-gp. Alcuni di questi composti sono attualmente in fase clinica e comprendono derivati antranilammidici come il tariquidar e l’elacridar. Quest’ultima generazione di composti presenta un’elevata potenza ed affinità per il trasportatore P-gp ma anche per la proteina BCRP (proteina resistente al cancro al mammella).
30
tariquidar elacridar
Recentemente, presso il laboratorio in cui è stata svolta questa tesi di laurea sono stati sintetizzati alcuni derivati ariletilfenilici di tipo A che in test preliminari condotti su linee cellulari tumorali che sovraesprimono la P-gp (cellule Caco-2, carcinoma umano del colon, e cellule MCF7/Adr, carcinoma umano della mammella resistente all’adriamicina) hanno mostrato una buona attività inibitoria su questo trasportatore. In particolare, per quanto riguarda i derivati di tipo A è stato osservato che l’attività inibitoria sulla P-gp è fortemente influenzata da due fattori principali:
1. la presenza di gruppi metossilici sui sistemi aromatici A e C; 2. il tipo di eteroatomo presente nella molecola (X).
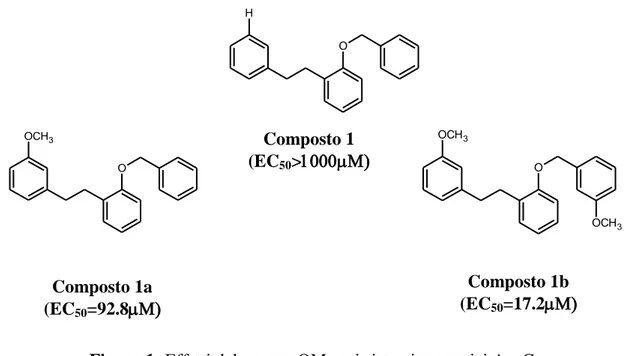
In particolare è stato osservato che l’inserimento di un OMe in posizione 3 del sistema aromatico A (1a) sembra essere critico per l’attività modulatoria sulla P-gp. Dai dati mostrati in figura 1 si osserva come l’analogo non sostituito 1 risulti essere praticamente inattivo (EC50>1000); l’ulteriore aggiunta di un gruppo OMe sull’anello aromatico distale C (1b) porta ad un ulteriore incremento dell’attività dimostrando che tale tipo di raggruppamento è fondamentale per garantire una buona attività inibitoria. X= O; NH; S R= H; OMe R1= R2= H R1= R2= OMe R1= H; R2= OMe R X B A C R1 R2
A
31 H O OCH3 O Composto 1 (EC50> OCH3 O OCH3 Composto 1a (EC50=92.8 Composto 1b (EC50=17.2
Figura 1: Effetti del gruppo OMe sui sistemi aromatici A e C
In uno studio precedente su questa serie di composti era stato inoltre osservato che la sostituzione dell’ossigeno presente nello scheletro arilossietilfenilico con un atomo di azoto da origine alla serie di tipo 2. In particolare il composto 2b risulta 36 volte più attivo dell’analogo ossigenato 1b. Questo risultato indica che anche tale modifica strutturale incrementa l’attività inibitoria nei confronti di P-gp.
OCH3 N H OCH3 N H OCH3 R1 R2 Composto 2b (EC 50= 0.48 Serie 2
L’indagine chimico farmaceutica è proseguita andando a valutare l’inserimento di un ulteriore gruppo metossilico sull’anello C sia di derivati arilossietilfenilici (di tipo
32
complessivamente positivi sull’attività portando ad un ulteriore incremento dell’attività inibitoria. OCH3 X B A C R2 R1 Tabella 1
Composto X R1 R2 Inibizione del trasporto della [3H]-vinblastina EC50(µM) Attivazione ATPasi 1a O H H 92.8 N 1b O 3-OCH3 H 17.2 N 1c O 2-OCH3 H 12 N 1d O 4-OCH3 H 5.2 N 1e O 2-OCH3 3-OCH3 0.25 S 1f O 3-OCH3 4-OCH3 0.65 N 1g O 3-OCH3 5-OCH3 2.5 N 2b NH 3-OCH3 H 0.48 N 2c NH 2-OCH3 H N.D. N.D. 2d NH 4-OCH3 H N.D. N.D. 2e NH 2-OCH3 3-OCH3 0.085 N 2f NH 3-OCH3 4-OCH3 0.18 N 2g NH 3-OCH3 5-OCH3 0.9 N elacridar 2.0 N ciclosporinaA 80 N verapamile 20 S N.D.= Not Determinated
33 OCH3 OCH3 O OCH3 OCH3 6 OCH3 N OCH3 OCH3 C H3 4 OCH3 H3CO N H OCH3 OCH3 5 OCH3 H3CO H3CO N H OCH3 OCH3 7 OCH3 N C H3 OCH3 3
Con l’intento di comprendere meglio quali siano i requisiti strutturali responsabili dell’attività inibitoria di questi derivati, in questa tesi di laurea mi sono occupato della sintesi dei composti 3-7, in cui sono state apportate le seguenti modifiche strutturali:
la metilazione dell’atomo di azoto amminico (composti 3,4) ;
l’inserimento di gruppi metossilici sull’anello A per ottenere il derivato dimetossifeniletilanilinico 5, il derivato dimetossifeniletilfenossilico 6 ed il composto trimetossifeniletilanilinico 7.
34
Lo schema 1 mostra la via di sintesi dei composti 3,4.
Il composto 8a è viene fatto reagire in ambiente acido con HCHO al 37% e NaBH3CN per ottenere il composto 9; il quale reagisce, successivamente, con gli opportuni cloruri di benzile 10a,b in presenza di carbonato di potassio e ioduro di potassio per dare i composti finali 3 e 4, rispettivamente.
SCHEMA 1 OCH3 NH2
+
HCHO OCH3 N CH3 H 8a 9 I OCH3 N CH3 H 9 OCH3 Cl 10a II II OCH3 OCH3 Cl OCH3 N C H3 OCH3 3 OCH3 N OCH3 OCH3 C H3 4 10b Reagenti e condizioni:I: AcOH, NaBH3CN, THF, t.a., 4 h.; II: K2CO3, KI, CH3CN, t.a., 12 h. La sintesi dei prodotti 8a,c,d viene descritta nello schema 2.
35
Per reazione dei cloruri di benzile 10a,c,d con trifenilfosfina a riflusso si ottengono i sali di fosfonio 11a,c,d.
La successiva reazione con la 2-nitrobenzaldeide commerciale porta all’ottenimento della miscela cis/trans dei nitrostilbeni 12a,c,d. Questi sono stati sottoposti a riduzione tramite idrogenazione in presenza di Pd/C per ottenere i prodotti 8a,c,d.
SCHEMA 2 R1 R2 Cl H3CO PPh3 R1 R2 PPh3 + Cl -H3CO
+
O N+ O -O NO2 OCH3 R1 R2 OCH3 R1 NH2 R2+
10a,c,d I 11a,c,d II cis/trans 12a,c,d III 8a,c,d 10a: R1=R2=H 10c: R1=OCH3; R2=H 10d: R1=R2=OCH3 Reagenti e condizioni: I: CH3CN, riflusso, 12 h. II: DBU, CH3CN, riflusso, 12 h.
36
Lo schema 3 mostra la reazione di amminazione riduttiva che ha permesso l’ottenimento dei composti finali 5 e 7.
La 3,4-dimetossibenzaldeide commerciale viene sottoposta a reazione di amminazione riduttiva con la 3,4-dimetossifeniletilanilina 8c e la 3,4,5-trimetossifeniletilanilina 8d in presenza di NaBH4 per fornire i prodotti finali 5 e 7.
SCHEMA 3 H3CO R OCH3 NH2
+
O OCH3 OCH3 N H OCH3 H3CO R OCH3 OCH3 I 8c,d I 8c: R=H 8d: R=OCH3 5,7 Reagenti e condizioni:37
Il composto finale 6 è stato ottenuto seguendo la procedura descritta nello schema 4. L’aggiunta di PPh3 ad una soluzione di 2,3-dimetossibenzilcloruro 10b in CH3CN fornisce il sale di fosfonio 13, il quale prende parte alla reazione di Wittig con la salicilaldeide commerciale; la miscela cis/trans del composto 14 viene ridotta tramite idrogenazione, utilizzando Pd/C come catalizzatore.
La soluzione del fenolo 15 in DMSO viene addizionata dell’appropriato cloruro di benzile 10c in presenza di KOH con lo scopo di ottenere il prodotto 6.
SCHEMA 4 OCH3 OCH3 Cl PPh3 OCH3 OCH3 PPh3 + Cl
-+
OH OCH3 OCH3 OCH3 OH OCH3 OCH3 OCH3 Cl OCH3 O OCH3 OCH3 OCH3 O OH+
I II 10b 13 II cis/trans 14 III 15+
IV 10c 6 Reagenti e condizioni:I: CH3CN, reflusso, 12 h.; II: DBU, CH3CN, reflusso, 12 h.; III: H2, Pd/C, EtOH assoluto, 12 h.; IV: KOH, DMSO, t.a., 12 h.
38
Nello schema 5 viene indicata la sintesi dei cloruro benzilici 10b-d necessari per la formazione dei prodotti finali 3-7.
I composti commerciali 16b-d vengono sottoposti a reazione di riduzione tramite l’agente riducente NaBH4, ottenendo gli alcoli 17b-d. Questi vengono fatti reagire con SOCl2 in CHCl3 per ottenere i composti 10b-d che sono stati utilizzati per le reazioni successive senza ulteriore purificazione.
SCHEMA 5 R2 R3 OCH3 O R1 NaBH4 R2 R3 OCH3 OH R1 SOCl2 R2 R3 OCH3 Cl R1
+
+
I II 17b-d 10b-d 16b-d II 16b: R1=R2=H; R3=OCH3 16c: R1=R3=H; R2=OCH3 16d: R1=R2=OCH3; R3=H Reagenti e condizioni: I: NaBH4, MeOH, H2O, t.a., 2 h.
40
MATERIALI E METODI
La struttura dei vari composti è stata controllata tramite spettrometria di massa ed 1
H-NMR. Degli spettri 1H-NMR abbiamo riportato i particolari più rilevanti. I dati spettrali di tutti i composti sono in accordo con le strutture assegnate.
Gli spettri di risonanza magnetica nucleare sono stati ricavati utilizzando uno spettro Varian Gemini 200 MHz. Le soluzioni sono al 5% circa in CDCl3 e CD3OD.
I chemical shift sono stati espressi in ppm (scala di δ).
Nel nostro laboratorio di Chimica Analitica sono state eseguite le analisi elementari : la differenza, tra i valori teorici e quelli ottenuti, è compresa in un intervallo di ±0.4%.
Le evaporazioni sono state eseguite tramite un evaporatore rotante. Le disidratazioni delle fasi organiche sono state portate a termine utilizzando Na2SO4.
Le TLC analitiche sono state eseguite per mezzo di lastre MERCK di gel di silice (G60) con indicatore di fluorescenza 20 x 20.2 mm. Le macchie sono state rilevate grazie ad una lampada UV (256nm).
Per le cromatografie su colonna è stato utilizzato gel di silice 70-230 mesch. Per la filtrazione su celite è stata utilizzata celite® 521.
41
SCHEMA 1
SINTESI DEL COMPOSTO
2-[2-(3-METOSSIFENIL)ETIL]-N-METIL-ANILINA (9)
OCH3 N CH3 H 9Ad una soluzione del derivato anilinico 8a (168 mg; 0.74 mmol) in THF (20 mL), è stata aggiunta goccia a goccia una soluzione di formaldeide commerciale al 37% (143 mg; 1.67 mmol), AcOH (333 mg; 5.55 mmol) e NaBH3CN (102.2 mg; 1.63 mmol); la miscela è stata lasciata in agitazione a temperatura ambiente per 4 h. Trascorso tale periodo il solvente è stato evaporato a pressione ridotta; il residuo è stato ripreso con AcOEt, lavato con una soluzione di NaOH 1N ed una soluzione satura di NaCl. La fase organica è stata essiccata ed evaporata a pressione ridotta fornendo il derivato 9 che è stato utilizzato nella reazione successiva senza ulteriori purificazioni.
RESA: 95%
1
H NMR (CDCl3): δ 2.68 (s, 3H, CH3); 2.86- 3.10 (m, 4H, CH2CH2); 3.80 (s, 3H, OCH3); 6.73-6.90 (m, 3H, Ar); 6.98-7.12 (m, 2H, Ar); 7.14-7.24 (m, 3H, Ar) ppm.
42
SINTESI GENERALE DEI COMPOSTI 3,4
Ad una soluzione dell’opportuno metossibenzilcloruro 10a,b (0.42 mmol) in CH3CN (10 mL) è stato aggiunto K2CO3 (357 mg) e una punta di spatola di KI. La miscela così ottenuta è stata lasciata sotto agitazione per 5 minuti e poi addizionata del composto 9 (150 mg; 0.62 mmol). La soluzione risultante è stata lasciata sotto agitazione, a t.a. per 12 h.
Trascorso tale periodo, il solvente viene evaporato ed il residuo così ottenuto è stato ripreso con AcOEt, filtrato su setto ed il filtrato è stato evaporato a p.r.
2-[2-(3-metossifenil)etil]-N-(3-metossibenzil)-N-metilanilina (3)
OCH3 N C H3 OCH3 3Il grezzo ottenuto è stato purificato tramite cromatografia flash, utilizzando come eluente n-Esano/Et2O (7:3).
RESA: 11%
1
H NMR (CDCl3): δ 2.58 (s, 3H,CH3N); 2.90-3.14 (m, 4H, CH2CH2); 3.73 (s, 3H,
OCH3); 3.76 ( s, 3H, OCH3); 3.99 (s, 2H,CH2N); 6.71-6.84 (m, 4H, Ar); 6.95-6.97 (m, 2H, Ar); 7.02-7.27 (m, 6H, Ar) ppm.
13
C NMR(CDCl3): δ 179.90, 159.86, 159.47, 152.55, 144.09, 140.90, 137.44,
130.03, 129.32, 126.90,124.10, 121.42, 121.00, 120.93, 114.23, 113.85, 112.77, 111.44, 62.12, 55.26, 42.22, 37.41,33.08 ppm.
43 ANALISI ELEMENTARE: C24H27NO2 C H N Calc. % 79.77 7.48 3.88 Trov. % 79.89 7.48 2.76
2-[2-(3-metossifenil)etil]-N-(2,3-dimetossibenzil)-N-metilanilina (4)
OCH3 N OCH3 OCH3 C H3 4Il grezzo ottenuto è stato purificato tramite cromatografia flash, utilizzando come eluente n-Esano/CHCl3 (1:9).
RESA: 13%
1
H NMR (CDCl3): δ 2.68 (s, 3H, NCH3); 2.90-3.01 (m, 4H, CH2CH2); 3.80 (s, 6H, OCH3); 3.86 (s, 3H, OCH3); 4.00 (s, 2H, CH2N); 6.72-6.89 (m, 4H, Ar); 6.92-7.09 (m, 4H, Ar); 7.14-7.22 (m, 3H, Ar) ppm.
44
SCHEMA 2
SINTESI GENERALE DEI COMPOSTI 11a,c,d
Ad una soluzione degli opportuni cloruri di benzile 10a,c,d (5.36 mmol) in CH3CN (62 mL) viene aggiunta la PPh3 (1,41 g; 5.36 mmol;). La miscela è stata lasciata a riflusso per 12 ore; trascorso tale periodo il solvente è stato evaporato ed il residuo purificato tramite cristallizzazione da CHCl3/Et2O.
Il composto è stato utilizzato nella reazione successiva senza ulteriori purificazioni.
Cloruro del 3-metossibenziltrifenilfosforano (11a)
OCH3 PPh3+ Cl -11a RESA: 98% 1 H NMR (CDCl3): δ 3.53 (s, 3H, OCH3); 5.44 (d, 2H, J = 16 Hz, CH2); 6.60-6.77
45
Cloruro del 3,4-dimetossibenziltrifenilfosforano (11c)
OCH3 OCH3 PPh3 + Cl -11c RESA: 71% 1 H NMR (CDCl3): δ 3.47 (s, 3H, OCH3); 3.75 (s, 3H, OCH3) 5.31 (d, 2H, J = 13.9 Hz, CH2), 6.58-6.71 (m, 3H, Ar), 7.54-7.74 (m, 15H, Ar) ppm.
Cloruro del 3,4,5-Trimetossibenziltrifenilfosforano (11d)
OCH3 OCH3 PPh3+ Cl -H3CO 11d RESA: 95% 1 H NMR (CDCl3): δ 3.49 (s, 6H, OCH3); 3.75 (s, 3H, OCH3); 5.48 (d, 2H, J = 14.1 Hz, CH2); 6.42 (d, 2H, J = 2.7 Hz, Ar); 7.57-7.81 (m, 15H, Ar) ppm.
46
SINTESI GENERALE DEI COMPOSTI 12a,b,c
Ad una soluzione dei sali 11a,c,d (3.82 mmol) in CH3CN e (4.5 mL) sono stati aggiunti DBU (0.43 mL) ed il composto commerciale 2-nitrobenzaldeide (577.2 mg ; 3.82 mmol). La miscela è stata lasciata a reflusso per 12 ore.
Trascorso tale periodo il solvente è stato evaporato ed il residuo ripreso con AcOEt e lavato con H2O, HCl e NaCl. La soluzione organica è stata essiccata con Na2SO4 e concentrata per evaporazione a p.r., ottenendo come grezzo la miscela degli isomeri cis/trans.
2-[(E/Z)-2-(3-metossifenil)vinil]anilina (12a)
N+O -O OCH3 cis/trans 12aIl composto è stato utilizzato nella reazione successiva senza ulteriori purificazioni.
RESA: 79%
1
H NMR (CDCl3) :δ 3.60 e 3.85 (s, 3H, OCH3, mix E/Z); 6.57- 7.15 (m, 3H, Ar, CH=CH); 7.26-7.78 (m, 6H, Ar); 7.94-8.10 (m, 1H, Ar) ppm.
47
2-[(E/Z)-2-(3,4-Dimetossifenil)vinil]anilina (12c)
N+O -O OCH3 H3CO cis/trans 12cIl composto è stato utilizzato nella reazione successiva senza ulteriori purificazioni.
RESA: 91%
1
H NMR (CDCl3): δ 3.53 e 3.82 (s, 3H, 3-OCH3, mix E/Z); 3.90 e 3.94 (s, 3H,
4-OCH3, mix E/Z); 6.64-6.70 (m, 2H, CH=CH); 7.05-7.11 (m, 2H, CH=CH); 7.30-7.58 (m, 9H, Ar); 7.61-7.77 (m, 5H, Ar) ppm.
2-[(E-Z)-2-(3,4,5-Trimetossifenil)vinil]anilina (12d)
N+O -O OCH3 H3CO H3CO cis/trans 12dIl composto è stato utilizzato nella reazione successiva senza ulteriori purificazioni.
RESA: 87%
1
H NMR (CDCl3): δ 3.78 (s, 3H, OCH3); 3.91 (s, 6H, OCH3); 6.76 (s, 2H, Ar); 7.01 (d, 1H, J = 15.9 Hz, CH=CH); 7.36-7.44 (m, 1H, Ar); 7.49 (d, 1H, J = 15.9, CH=CH); 7.56-7.64 (m, 1H, Ar); 7.57 (d, 1H, J = 7.7 Hz, Ar); 7.96 (d, 1H, J = 8.1 Hz, Ar) ppm.
48
SINTESI GENERALE DEI COMPOSTI 8a,c,d
Una soluzione della miscela cis/trans del composto 12a,c,d (3.51 mmol) in etanolo assoluto (15 ml) è stata sottoposta ad idrogenazione, utilizzando come catalizzatore Pd/C (50 mg) per 12h a t.a. Trascorse tale periodo, la miscela viene filtrata su setto con celite e l’etanolo viene evaporato a pressione ridotta.
2-[2-(3-Metossifenil)etil]anilina (8a)
OCH3
NH2
8a
Il grezzo è stato purificato tramite cromatografia su colonna, utilizzando come eluente CHCl3.
RESA: 42%
1H NMR (CDCl3): δ: 2.74- 2.97 (m, 4H, CH2CH2); 3.78 (s, 3H, OCH3); 6.66-6.83
49
2-[2-(3,4-Dimetossifenil)etil]anilina (8c)
OCH3 H3CO NH2 8cIl grezzo è stato purificato tramite cromatografia su colonna, utilizzando come eluente CHCl3.
RESA: 20%
1
H NMR (CDCl3): δ 2.73-2.94 (m, 4H, CH2CH2); 3.81 (s, 3H, OCH3); 3.87 (s, 3H,
OCH3); 6.63-6.83 (m, 5H, Ar); 7.02-7.09 (m, 2H, Ar) ppm.
2-[2-(3,4,5-Trimetossifenil)etil]anilina (8d)
OCH3 H3CO NH2 H3CO 8dIl grezzo è stato purificato tramite formazione del cloridrato.
RESA: 23%
1
H NMR (CD3OD) :δ 2.87-3.07 (m, 4H, CH2CH2); 3.73 (s, 3H, OCH3); 3.81 (s, 6H, OCH3); 6.56 (s, 2H, Ar); 7.37-7.41 (m, 4H, Ar) ppm.
50
SCHEMA 3
SINTESI GENERALE DEI COMPOSTI FINALI 5,7
Ad una soluzione del composto 8c,d (0.24 mmol) in EtOH assoluto (3 mL) è stata aggiunta la 3,4-dimetossibenzaldeide commerciale (39.6 mg; 0.24 mmol) e la miscela è stata lasciata reagire per 24 h a riflusso.
Trascorso tale periodo, la miscela di reazione è stata addizionata goccia a goccia di una soluzione di NaBH4 (9.1 mg; 0.24 mmol) in H2O (0.5 ml) e lasciata a t.a. per 12 h.
Il solvente è stato evaporato ed il composto organico viene estratto con CH2Cl2, essiccato e concentrato per evaporazione a p.r.
2-[2-(3,4-dimetossifenil)etil]-N-[(3,4-dimetossi)benzil]-anilina
(5)
N H OCH3 OCH3 H3CO OCH3 5Il grezzo ottenuto è stato purificato tramite formazione del cloridrato.
RESA: 46%
1
H NMR (CDCl3) base libera: δ 2.72–2.92 (m, 4H, CH2CH2), 3.75 (s, 3H, OCH3),
3.85 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 4.21 (s, 2H, NCH2), 6.59-6.81 (m, 5H, Ar), 6.81-6.91 (m, 3H, Ar), 7.05-7.17 ( m, 2H, Ar) ppm.
13
C NMR (CDCl3): δ 159.81, 149.46, 149.14, 148.59, 147.68, 145.95, 134.64,
132.29, 129.27, 127.45, 125.87, 120.35, 119.89, 117.67, 112.35, 111.83, 111.41, 110.92, 56.20, 56.13, 55.98, 48.57, 35.12, 33.73 ppm.
51 ANALISI ELEMENTARE: C25H29NO4 C H N Calc. % 73.70 7.12 3.44 Trov. % 73.4 7.50 3.14
2-[2-(3,4,5-trimetossifenil)etil]-N-[(3,4-Dimetossi)benzil]-anilina (7)
N H OCH3 OCH3 H3CO OCH3 H3CO 7Il grezzo ottenuto è stato purificato tramite formazione del cloridrato.
RESA: 10%
1
H NMR (CDCl3) base libera: δ 2.72- 3.00 (m, 4H, CH2CH2); 3.71 (s, 3H, OCH3); 3.73 (s, 3H, OCH3); 3.79 (s, 3H, OCH3); 3.82 (s, 6H, OCH3); 4.31 (s, 2H, CH2NH); 6.52 (s, 2H, Ar); 6.56-6.70 (m, 2H, Ar); 7.01 (s, 1H, Ar); 7.18-7.31 (m, 3H, Ar) 7.40-7.44 (m, 1H, Ar) ppm.
13
C NMR (CDCl3): δ 153.29, 150.10, 149.05, 136.81, 136.50, 136.08, 132.93,
130.84, 129.05, 127.45, 125.07, 124.14, 122.13, 114.63, 110.97, 106.31, 60.94, 56.51, 56.18, 55.95, 37.19, 32.22 ppm.
52 ANALISI ELEMENTARE:
C26H31NO5 C H N
Calc. % 71. 37 7.14 3.20
53
SCHEMA 4
SINTESI DEL CLORURO DEL
2,3-DIMETOSSIBENZILTRIFENYLFOSFORANO (13)
OCH3 OCH3 PPh3 + Cl -13Ad una soluzione del cloruro di benzile 10b (1,604 g; 8.56 mmol) in CH3CN (100 mL), è stata aggiunta la PPh3 (2,321 g; 8.85 mmol). La miscela è stata lasciata a riflusso per 12 h e trascorso tale periodo è stata concentrata per evaporazione del solvente.
Il grezzo è stato purificato tramite precipitazione a caldo con CHCl3/Et2O.
RESA:85%
1
H NMR (CDCl3): 3.40 (s, 3H, OCH3); 3.63 (s, 3H, OCH3) 4.97 (d, 2H, J = 14.3 Hz,
54
SINTESI DEL COMPOSTO
2-[(E/Z)-2-(2,3-DIMETOSSIFENIL)VINIL]FENOLO (14)
OH OCH3
OCH3
cis/trans 14
Ad una soluzione del composto 13 (1.014 g; 2.26 mmol) in CH3CN (2.6 mL) è stata aggiunta l’opportuna salicilaldeide commerciale (276 mg; 2.26 mmol; 0.24 ml) in presenza di DBU (0.25 ml). La miscela è lasciata a riflusso per 12 h.
Trascorso tale periodo il solvente è stato evaporato ed il residuo è stato ripreso con AcOEt e lavato con H2O, HCl e NaCl. La soluzione organica viene essiccata con solfato di sodio ed evaporata.
Il composto è stato utilizzato nella reazione successiva senza ulteriori purificazioni.
RESA: 59%
1
H NMR (CDCl3): δ 3.86 (s, 3H, OCH3); 3.88 (s, 3H, OCH3); 6.78-6.86 (m, 1H,
CH=CH); 6.91-7.18 (m, 4H, Ar, CH=CH); 7.29 (dd, 1H, J = 8.0, 1.5 Hz, Ar); 7.43-7.44 (m, 2H, Ar); 7.59 (dd, 1H, J = 7.7, 1.6 Hz, Ar) ppm.
55
SINTESI DEL COMPOSTO
2-[2-(2,3-DIMETOSSIFENIL)ETIL]FENOLO (15)
OCH3
OH OCH3
15
Una soluzione della miscela cis/trans del composto 14 (683.1 mg; 2.67 mmol) in EtOH assoluto (12.03 ml) è stata sottoposta ad idrogenazione, utilizzando come catalizzatore Pd/C (45.76 mg). La miscela è stata lasciata reagire a t.a. per 12 h.; trascorso tale periodo la sospensione è stata filtrata su setto con celite e la soluzione è stata concentrata per evaporazione a p.r..
Il prodotto viene purificato tramite colonna cromatografica, utilizzando CHCl3 come eluente.
RESA: 13%
1
H NMR (CDCl3): δ 2.76-2.85 (m, 4H, CH2CH2); 3.89 (s, 3H, OCH3); 3.92 (s, 3H,
56
SINTESI DEL COMPOSTO FINALE
2-[2-(2,3-
DIMETOSSIFENIL)ETIL]-[(3,4-DIMETOSSIBENZIL)OSSI]-BENZENE (6)
OCH3 O OCH3 OCH3 OCH3 6Ad una soluzione del fenolo 15 (89.6mg; 0.35 mmol) in DMSO (0.7mL) è stata aggiunta una soluzione di KOH (43.2 mg) in DMSO (0.8 mL). La soluzione viene lasciata reagire per 15 minuti e successivamente è stato aggiunto il composto 10c (62.3 mg; 0.34 mmol). La miscela è stata lasciata in agitazione a t.a. per 12 h.
Trascorso tale periodo, il solvente è stato evaporato ed il residuo è stato ripreso con CH2Cl2 e lavato con H2O; la fase organica è stata essiccata e concentrata per evaporazione a p.r.
Il grezzo ottenuto è stato purificato tramite cromatografia su colonna, utilizzando come eluente n-Esano/AcOEt (7:3).
RESA: 31%
1
H NMR (CDCl3): δ 2.91-2.96 (m, 4H, CH2CH2); 3.71 (s, 3H, OCH3); 3.84 (s, 3H,
OCH3); 3.85 (s, 3H, OCH3); 3.89 (s, 3H, OCH3); 5.03 (s, 2H, CH2); 6.71-6.78 (m, 2H, Ar); 6.85-7.02 (m, 6H, Ar); 7.14-7.22 (m, 2H, Ar) ppm.
13
C NMR (CDCl3): δ 156.69, 152.76, 149.10, 148.70, 147.21, 136.26, 130.98,
130.13, 130.04, 127.18, 123.74, 122.01, 120.75, 119.89, 111.77, 111.06, 110.77, 110.17, 70.04, 60.68, 56.06, 55.93, 55.76, 32.15, 30.44 ppm.
57 ANALISI ELEMENTARE:
C25H28O5 C H
Calc. % 73,52 6,86
58
SCHEMA 5
SINTESI DEI COMPOSTI 17b-d
Ad una soluzione delle benzaldeidi commerciali 16b-d (12.04 mmol) in MeOH, portata a -10°C con un bagno di ghiaccio e MeOH, è stata aggiunta goccia a goccia una soluzione di NaBH4 (455.2 mg; 12.04 mmol) in H2O. La miscela è stata lasciata in agitazione per 2 ore a temperatura ambiente; trascorso tale periodo il solvente è stato evaporato e il residuo è stato estratto con CH2Cl2. La fase organica è stata essiccata e concentrata per evaporazione a p.r. ed il prodotto è stato utilizzato per la reazione successiva senza ulteriore purificazione.
(2,3-dimetossifenil)metanolo (17b)
OCH3 OCH3 OH 17b RESA: 88% 1 H NMR (CDCl3): δ 3.87 (s, 3H, OCH3); 3.89 (s, 3H, OCH3); 4.70 (d, J=5.9 Hz, 2H, CH2); 6.86-6.94 (m 2H, Ar); 7.02-7.09 (m, 1H, Ar) ppm.59
(3,4-dimetossifenil)metanolo (17c)
OCH3 OCH3 OH 17c RESA: 92% 1 H NMR (CDCl3): δ 3.88 (s, 3H, OCH3); 3.89 (s, 3H, OCH3); 4.62 (d, J=4.9 Hz, 2H, CH2); 6.82-6.88 (m, 2H, Ar); 6.94 (s, 1H, Ar) ppm.
(3,4,5-trimetossifenil)metanolo (17d)
OCH3 OCH3 OH H3CO 17d RESA: 76% 1 H NMR (CDCl3): δ 3.85 (s, 3H, OCH3); 3.86 (s, 6H, OCH3); 4.63 (s, 2H, CH2); 6.60 (s, 2H, Ar) ppm.60
SINTESI GENERALE DEI COMPOSTI 10b-d
Ad una soluzione del composto 17b-d (10.28 mmol) in CHCl3 a 0°C si aggiunge goccia a goccia SOCl2 (1.223 g; 10.28 mmol; 0.74 ml). La miscela è stata lasciata sotto agitazione per 12 h a t.a., e trascorso tale periodo, è stata lavata con H2O. La fase organica è stata essiccata e concentrata per evaporazione a p.r. ed il prodotto è stato utilizzato per la reazione successiva senza ulteriore purificazione.
2,3-Dimetossibenzilcloruro (10b)
OCH3 OCH3 Cl 10b RESA: 85% 1 H NMR (CDCl3): δ 3.87 (s, 3H, OCH3); 3.96 (s, 3H, OCH3); 4.57 (s, 2H, CH2); 6.85-7.07 (m, 3H, Ar) ppm.61
3,4-Dimetossibenzilcloruro (10c)
OCH3 OCH3 Cl 10c RESA: 70% 1 H NMR (CDCl3): δ 3.88 (s, 3H, OCH3); 3.90 (s, 3H, OCH3); 4.57 (s, 2H, CH2); 6.79-6.88 (m, 1H, Ar); 6.92-6.98 (m, 2H, Ar) ppm.
3,4,5-Trimetossibenzilcloruro (10d)
OCH3 OCH3 Cl H3CO 10d RESA: 84% 1 H NMR (CDCl3): δ 3.88 (s, 3H, OCH3); 3.90 (s, 3H, OCH3); 4.57 (s, 2H, CH2); 6.80-6.84 (m, 1H, Ar); 6.91-6.96 (m, 2H, Ar) ppm.63
1-Ross, D. D.; Doyle, L. A. Mining our ABCs: pharmacogenomic approach for evaluating transporter function in cancer drug resistance. Cancer Cell 2004, 6, 105– 107.
2-Pharm, A.-N.; Penchala, S.; Graf, R. A.; Wang, J.; Huang, Y. Pharmacogenomic Characterization of ABC Transporters Involved in Multidrug Resistance. In Multidrug Resistance: Biological and Pharmaceutical Advance in the Antitumour Treatment; Colabufo, N. A., Ed.; Research Signpost: Kerala, India, 2008; pp
19-62.
3-Stephens, R. H.; O Neill, C. A.; Warhurst, A.; Carlson, G. L.; Rowland, M. and Warhurs, G.; J. Pharmacol. Exp. Ther.; 2001, 296, 584-591.
4-Sparreboom, A.; Van Asperen, J.; Mayer, U.; Schinkel, A. H.; Smit, J. W.; Me, D. K.; Borst, P.; Nooijen, W. J.; Bejinen, J. H. and van Tellingen, O.; Proc. Natl. Acad. Sci. USA, 1997, 94, 2031- 2035.
5-Muller, M. and Jansen, P. L.; J. Hepatol.; 1998, 28, 344-354.
6-Schinkel, A. H.; Cancer Biol.; 1997, 8, 161-170.
7-Krishna, R. and Mayer, L. D.; Eur. J. Pharm. Sci.; 2000, 11, 265- 283.
8-Germann, U. A. P-glycoproteinsa mediator of multidrug resi stance in tumour cells. Eur. J. Cancer 1996, 32A (6), 927-944.
9-Chen, C. J.; Chin, J. E.; Ueda, K.; Clark, D. P.; Pastan, I.; Gottesman, M. M.; Roninson, I. B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986, 47 (3), 381-389.
10-Higgins, C. F. (1992). ABC transporters: from microorganisms to man. Annu Rev Cell Biol 8, 67−113.
64
11-Gottesman, M. M.; Fojo, T. and Bates, S. E.; Nat. Rev. Cancer.; 2002, 2, 48-58.
12-Campbell, J. D.; Koike, K.; Moreau, C.; Sansom, M. S. P.; Deeley, R. G. and Cole, S. P. C.; J. Biol. Chem.; 2004, 279, 463-468.
13-Klopman, G.; Shi, L. M. and Ramu, A.; Mol. Pharmacol.; 1997, 52, 323-334.
14-Loo, T. W. and Clarke, D. M.; J. Biol. Chem.; 2001, 276, 36877- 36880.
15-Yu, E. W.; McDermott, G.; Zgurskaya, H. I.; Nikaido, H. and Koshland, D. E.; Science, 2003, 300, 976-980.
16-Murray, D. S.; Schumacher, M. A. and Brennan, R. G.; J. iol. Chem.; 2004, 279, 14365- 14371.
17-Senior, A. E., Al-Shawi, M. K., & Urbatsch, I. L. (1995). The catalytic cycle of P-glycoprotein. FEBS Lett 377, 285−289.
18-Higgins, C. F., & Linton, K. J. (2004). The ATP switch model for ABC transporters. Nat Struct Mol Biol 11, 918−926.
19-Callaghan, R., Ford, R. C., & Kerr, I. D. (2006). The translocation mechanism of P-glycoprotein. FEBS Lett 580, 1056−1063.
20-Chan, L. M.; Lowes, S.; Hirst, B. H. The ABCs of drug transport in intestine and liver: efflux proteins limiting drug absorption and bioavailability. Eur. J. Pharm. Sci. 2004, 21, 25–51.
21-Colabufo, N. A.; Berardi, F.; Contino, M.; Niso, M.; Perrone, R. ABC pumps and their role in active drug transport. Curr. Top. Med. Chem. 2009, 9, 119–129.
22-Ohtsuki, S.; Terasaki, T. Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm. Res. 2007, 24, 1745–
65
1758.
23-Brightman, M. W.; Basic Science Aspects, New York Plenum, 1998, 1, 53–83.
24-Pardridge, W. M.; Endocr. Rev.; 1981, 2,103–123.
25-Rapport, S. I.; Raven Press, 1976, 17–42.
26-Chikhale, E. G., Ng, K.-Y., Burton, P. S., Borchardt, R. T.; Pharm. Res.; 1994 11, 412–419.
27-Levin, V. A.; J. Med. Chem.; 1980, 23, 682–684.
28-Thiebaut, F., Tsuruo, T., Hamada, H., Gottesman, M. M., Pastan, I., Willingham, M. C.; J. Histochem. Cytochem.; 1989, 37, 159–164.
29-Cordon-Cardo, C., O’Brien, J. P., Casals, D., Rittman-Grauer, L., Biedler, J. L., Melamed, M. R., Bertino, J. R.; Proc. Natl. Acad. Sci. USA, 1989, 86, 695–698.
30-Tamai, I., Kido, Y., Yamashita, J., Sai, Y., Tsuji, A.,; J. Drug Target, 2000b, 8, 383–393.
31-Chishty, M., Reichel, A., Siva, J., Abbott, N.J., Begley, D.J.; J. Drug. Target, 2001, 9, 223–228.
32-Pendse, S., Sayegh, M.H., Frank, M.H.,; Curr. Drug Targets, 2003, 4, 469–476.
33-Johnstone, R.W., Ruefli, A.A., Smyth, M.J.;Trends Biochem. Sci.; 2000a, 25, 1– 6.
34-Johnstone, R.W., Ruefli, A.A., Tainton, K.M., Smyth, M.J.; Leuk. Lymphoma, 2000b, 38, 1–11.