1 1.
La Glicoproteina
PLa Glicoproteina P (P-gP) è una proteina di membrana appartenente alla famiglia dei trasportatori ABC (ATP-Binding Cassette). Queste proteine transmembranali sono localizzate nelle cellule di diversi tessuti ed utilizzano l’energia di idrolisi dell’ATP per trasportare, contro gradiente di concentrazione, una grande varietà di substrati. Questa attività permette loro di regolare l’assorbimento, il metabolismo e la distribuzione di xenobiotici tra i diversi tipi di cellule e la matrice extracellulare; inoltre, limitando l’accumulo di farmaci all’interno della cellula, hanno un ruolo fondamentale nello sviluppo della resistenza farmacologica [1].
La P-gP è stata il primo trasportatore ABC ad essere scoperto a livello delle cellule endoteliali della barriera ematoencefalica (BEE) umana nel 1989 [2]. Successivamente è stata localizzata nelle cellule endoteliali dei capillari cerebrali di numerose specie tra cui topi, maiali, scimmie, ecc. L’elevata espressione di questa proteina a livello cerebrale indica che la P-gP svolge un ruolo fondamentale nella protezione del cervello da xenobiotici ed eventuali composti tossici lipofili, che possono attraversare la BEE mediante meccanismi di diffusione [3].
La P-gP è codificata da un gruppo di geni chiamati “Multidrug Resistance Genes”
(MDR o ABCB) [4]. Nelle cellule umane sono stati identificati 2 tipi di questi geni, MDR1 e MDR2. Questi codificano per 2 tipi di proteine con funzioni differenti: la prima conferisce
multiresistenza alle cellule in cui viene espressa, mentre la seconda è espressa principalmente a livello della membrana degli epatociti dove partecipa alla secrezione della fosfatidilcolina nella bile [4]. Entrambi i geni sono localizzati sul cromosoma 7 umano; MDR1 è costituito da 28 esoni, che codificano per i 1280 amminoacidi della P-gP [5].
Nei roditori la P-gP è codificata da 3 tipi di geni : Mdr1a, Mdr1b e Mdr2. I primi due hanno le stesse funzioni del gene MDR1 nell’uomo [4].
1.1.Struttura
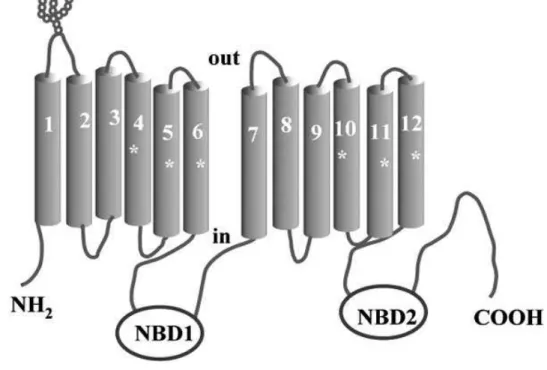
La P-gP è una proteina composta da 1280 amminoacidi, con un peso molecolare di circa 170 KDa [6], organizzata in 2 unità omologhe. Gli amminoacidi sono uniti a formare una lunga catena polipeptidica con un’estremità carbossi-terminale (-COOH) e un’estremità ammino-terminale (-NH2).
2 Ciascuna metà contiene un dominio transmembranale idrofobico (TMD) che attraversa 6 volte la membrana cellulare formando 3 loop intracellulari e 3 loop extracellulari. Ogni TMD è seguito da un dominio idrofilico, localizzato sul lato citoplasmatico, contenente il sito di legame per l’ATP (NBD). (Fig.1.1) L’idrolisi dell’ATP fornisce l’energia fondamentale per il trasporto di substrati attraverso la membrana cellulare [5].
La P-gP è N-glicosilata sul primo loop extracellulare; il grado di glicosilazione è differente da specie a specie. Sembra che gli zuccheri presenti non siano necessari per il trasporto dei substrati ma che rendano la struttura della proteina più stabile [7].
Figura 1.1. Struttura della P-gP
Sono state identificate 2 sequenze amminoacidiche caratteristiche conservate in tutti i membri della superfamiglia dei trasportatori ABC [8]. Queste si trovano all’interno di ogni dominio NBD e sono state identificate come Walker A e B. Tra queste regioni è presente una terza sequenza amminoacidica conservata che è implicata nel riconoscimento, legame ed idrolisi dell’ATP.
1.2.Ruolo e meccanismo d’azione
La P-gP è una glicoproteina di membrana la cui funzione principale è quella di trasportare diversi substrati dall’interno all’esterno della cellula riducendo così la concentrazione di determinate sostanze nello spazio intracellulare.
3 Il meccanismo di trasporto è dipendente dall’idrolisi dell’ATP che fornisce l’energia necessaria per il trasferimento del substrato fuori dalla cellula [5]. Se da una parte questa attività protegge i tessuti dalla potenziale tossicità di xenobiotici e farmaci citotossici, dall’altra, limita l’accumulo di farmaco nella cellula, con conseguente insorgenza di resistenza. Lo sviluppo della MDR (Multidrug Resistance) è un fattore molto importante che spesso limita l’efficacia di numerose terapie farmacologiche come ad esempio la chemioterapia antitumorale. In alcuni tipi di neoplasie è stata osservata una iperespressione della P-gP, che è risultata la principale responsabile della MDR [9]. Questo fenomeno è un tipo di resistenza acquisita non solo da cellule neoplastiche ma anche da microorganismi verso farmaci che hanno struttura e meccanismo d’azione differenti.
Sono stati proposti diversi meccanismi per spiegare il ciclo di trasporto della P-gP. Il più conosciuto è il modello “ATP switch”, proposto da Higgins e Linton [10] per descrivere il trasporto della vinblastina fuori dalla cellula ad opera della P-gP. In questo modello, il trasporto è descritto come un processo multistep in cui si hanno cambiamenti conformazionali dei TMDs e dei NBDs della P-gP e idrolisi di 2 molecole ATP. I NBDs assumono 2 conformazioni principali: quella a “dimero chiuso” che lega ATP e quella a “dimero aperto” facilitata dall’idrolisi dell’ATP con rilascio di Pi e ADP. Lo “switch” tra conformazione aperta/chiusa provoca un cambiamento conformazionale nei TMDs necessario per il trasporto del substrato fuori dalla cellula. Il ritorno dalla conformazione chiusa a quella aperta dopo l’idrolisi dell’ATP riporta la proteina allo stato iniziale, pronta per iniziare un nuovo ciclo di trasporto.
Il meccanismo “ATP switch” può essere descritto in 4 step [10]:
Step I. Il ciclo di trasporto ha inizio con il legame del substrato ai TMDs sul lato
citoplasmatico; i NBDs si trovano nella conformazione a “dimero aperto”. Questi subiscono un cambiamento conformazionale che facilita il legame dell’ATP e la formazione del “dimero chiuso”. La P-gP allo stato iniziale ha una ridotta affinità per l’ATP, mentre nei TMDs è presente un sito ad alta affinità per il substrato sul lato citoplasmatico. Il substrato si lega a questo sito e determina un cambiamento conformazionale che facilita l’interazione dell’ATP con i siti di legame presenti a livello dei NBDs con conseguente passaggio alla conformazione chiusa.
Step II. La formazione del dimero chiuso e il legame di 2 molecole di ATP induce un
4 Il passaggio attraverso la membrana richiede che il sito di legame ad alta affinità per il substrato, accessibile dal lato citoplasmatico, sia convertito in un sito a bassa affinità sul lato extracellulare, in modo che il substrato venga rilasciato all’esterno. Per questi cambiamenti è necessaria una fonte di energia. E’ stato dimostrato che il legame dell’ATP e la formazione del dimero chiuso, senza l’idrolisi, fornisce energia sufficiente per il trasporto del substrato [10].
Step III. 2 molecole di ATP sono idrolizzate in ADP+Pi; i NBDs passano ad uno stato
intermedio tra la conformazione aperta a quella chiusa. Sembra che il rilascio del substrato nello spazio extracellulare sia responsabile di questo cambiamento ma il meccanismo non è stato ancora chiarito [10].
Step IV. Nell’ultimo passaggio si ha il rilascio prima di Pi, poi di ADP. La proteina ritorna alla sua configurazione iniziale, pronta per compiere un nuovo ciclo di trasporto.
Il modello “ATP switch” proposto da Higgins e Linton per il trasporto della vinblastina è rappresentato nella figura seguente. (Fig.1.2)
L’attività ATPasica della P-gP è Mg++ dipendente [7].
5 Non tutti i trasportatori ABC compiono lo stesso ciclo di trasporto, i.e. in alcuni casi è richiesta l’idrolisi di una sola molecola di ATP. Il passaggio tra la conformazione aperta a quella chiusa sembra però fondamentale per tutti i meccanismi di trasporto [10].
1.3.Substrati della P-gP
La P-gP può trasportare numerosi substrati molto diversi tra loro per struttura e funzione ma con alcune caratteristiche comuni quali:
• un alto grado di idrofobicità,
• un basso PM (tra 250 e 900 Da),
• la tendenza ad essere carichi positivamente a pH neutro,
• la presenza di un gruppo amminico terziario,
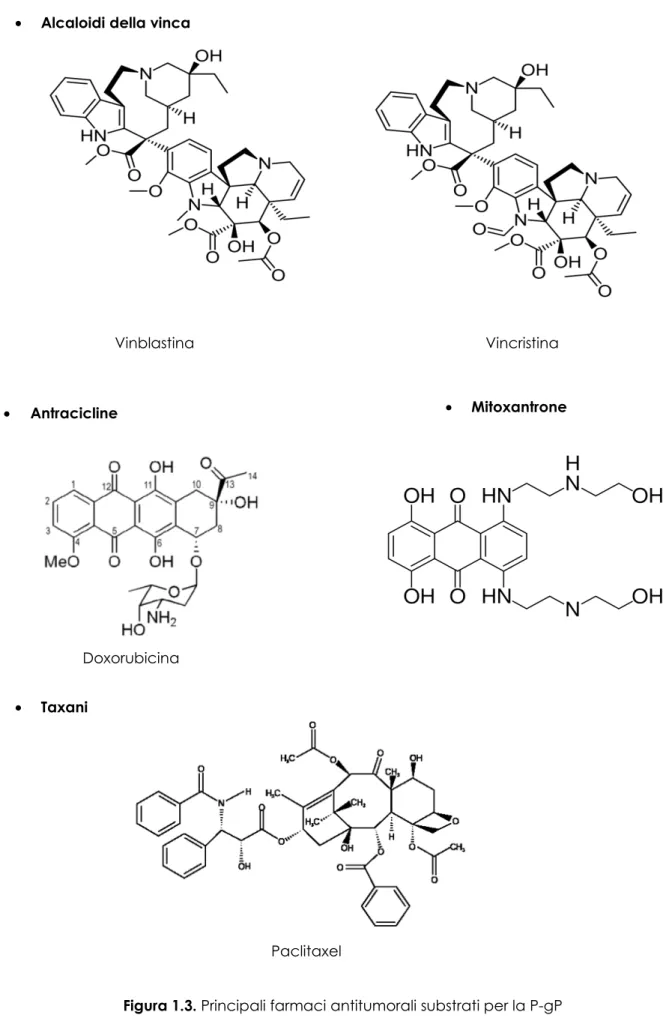
• la capacità di attraversare la membrana cellulare per diffusione semplice [7]. La P-gP può trasportare substrati esogeni o endogeni come ad esempio citochine, steroidi, bilirubina, ecc. Molti dei substrati della P-gP sono anche substrati dell’ isoforma 3A4 del citocromo P450, uno dei principali enzimi implicati nei processi metabolici [11]. I farmaci antitumorali sono stati i primi ad essere identificati come substrati della P-gP; tra questi troviamo gli alcaloidi della vinca (vincristina, vinblastina), le antracicline (doxorubicina, daunorubicina), il mitoxantrone e i taxani. (Fig.1.3) Un ruolo della P-gP nel limitare l’ingresso cerebrale di farmaci quali la daunorubicina, la vinblastina e il paclitaxel è stato dimostrato con esperimenti condotti su topi che esprimono i geni mdr1a e mdr1b e su topi knock-out [3]. Nelle specie mancanti dei 2 geni che codificano per la P-gP è stata osservata una concentrazione cerebrale di farmaco superiore rispetto a quella rilevata nelle altre specie.
6 • Alcaloidi della vinca
• Antracicline
• Taxani
Doxorubicina
• Mitoxantrone
Paclitaxel
Figura 1.3. Principali farmaci antitumorali substrati per la P-gP
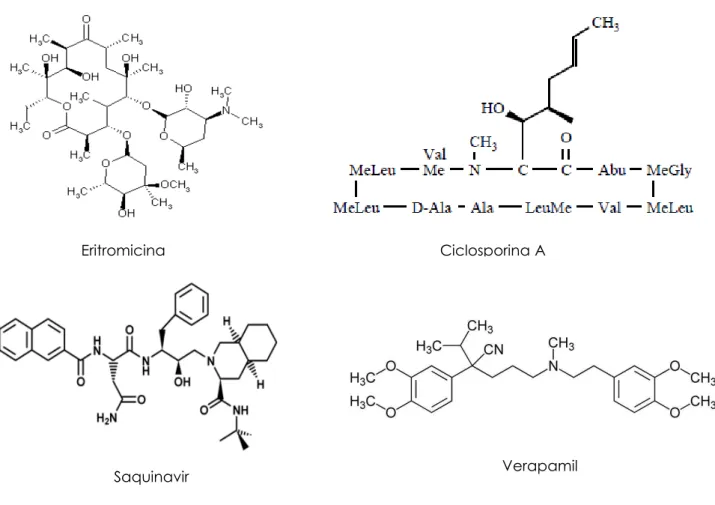
7 Altri substrati per la P-gP sono gli immunosoppressori (ciclosporina A), farmaci antivirali come gli inibitori delle proteasi dell’HIV (saquinavir), antibiotici (i.e. eritromicina, tetracicline, fluorochinoloni), calcio antagonisti (verapamil), antidepressivi (i.e. amitriptilina, citalopram), antistaminici, antiepilettici (fenitoina, carbamazepina),
glucorticoidi (prednisolone, desametasone), farmaci oppioidi (morfina, fentanil,
metadone), ipolipemizzanti (atorvastatina). La formula di struttura di alcuni di questi composti è riportata nella figura seguente. (Fig.1.4)
Figura 1.4. Altri substrati della P-gP
Nella classe degli antistaminici è stato osservato che la prima generazione di farmaci, come ad esempio la difenidramina, è caratterizzata dalla comparsa di sedazione a livello del SNC, mentre la seconda generazione, tra cui la terfenadina, non mostra questo effetto.
Eritromicina Ciclosporina A
8 Studi in vitro su cellule endoteliali cerebrali di topo hanno dimostrato che i farmaci appartenenti alla seconda generazione di antistaminici sono substrati per la P-gP e che questo ne limita l’accumulo a livello del SNC e di conseguenza ne riduce anche gli effetti sedativi; gli antistaminici di prima generazione non sono invece substrati per la P-gP e rimangono per lungo tempo a livello cerebrale [12].
1.4.Distribuzione tissutale
La P-gP è localizzata in diversi tessuti dove svolge un ruolo chiave nella eliminazione degli xenobiotici e nella protezione dagli effetti citotossici di queste sostanze. In particolare si trova localizzata in organi escretori o con funzione di barriera (i.e. fegato, rene, BEE, intestino, ecc).
1.4.1. Barriera emato-encefalica (BEE)
La BEE è una barriera fisica e metabolica che separa il cervello dalla circolazione sistemica; è composta da uno strato di cellule endoteliali dei capillari cerebrali. La ridotta penetrazione di sostanze a livello della BEE è dovuta alla presenza di “tight junctions” tra cellule endoteliali adiacenti [13]. Queste giunzioni impediscono la libera diffusione dei soluti idrofili, che normalmente passano la membrana per via paracellulare. Possono arrivare al cervello piccole molecole apolari tramite un meccanismo di diffusione facilitata, catalizzata da numerose proteine trasportatrici [13].
I trasportatori ABC, come la P-gP, hanno un ruolo fondamentale nell’eliminazione dal SNC di farmaci lipofili e di numerose sostanze neurotossiche che passano la BEE mediante meccanismi di diffusione. Se da un lato questa attività fornisce protezione al cervello da numerose sostanze, dall’altra riduce l’efficacia di terapie utilizzate contro disturbi neurodegenerativi come il morbo di Parkinson e di Alzheimer. Farmaci utilizzati per queste patologie, come ad esempio la levodopa usata nel trattamento del Parkinson, sono substrati della P-gP; il loro accumulo a livello centrale è ridotto e di conseguenza anche la loro efficacia terapeutica (vedi capitoli successivi).
9 Il cervello è uno dei siti di replicazione del virus HIV ed è un tessuto bersaglio di farmaci antiretrovirali. I farmaci anti-HIV hanno però limitati effetti a livello centrale in quanto sono substrati per la P-gP che li trasporta dal cervello alla circolazione sistemica.
Per determinare la giusta localizzazione dei trasportatori a livello delle cellule endoteliali cerebrali, sono state utilizzate numerose tecniche biochimiche come l’ibridazione in situ, l’immunoistochimica e l’immunofluorescenza [14].
La P-gP è localizzata a livello della membrana luminale (apicale) delle cellule endoteliali dei capillari cerebrali e trasporta i suoi substrati direttamente nella circolazione sistemica. (Fig.1.5) In questo modo l’accumulo di farmaci e altri substrati nel SNC è limitato [3].
Alcuni esperimenti condotti su topi privati del gene mdr1a hanno dimostrato che l’assenza di espressione di P-gP è correlata ad un aumento della concentrazione cerebrale di xenobiotici, che risulta essere da 10 a 100 volte superiore rispetto ai topi che esprimono il gene, con comparsa anche di effetti tossici [15].
La P-gP rappresenta quindi un potenziale bersaglio terapeutico per modulare la concentrazione di diversi farmaci nel cervello.
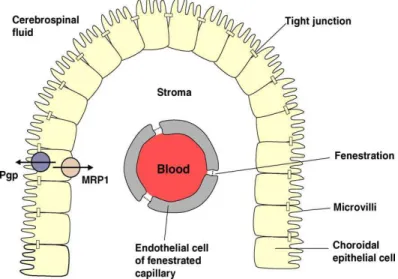
10 1.4.2. Barriera emato-liquorale (BEL)
La barriera emato-liquorale (BEL) separa il sangue dal liquido cerebro-spinale; è situata a livello dei plessi corioidei dei ventricoli cerebrali ed è pertanto costituita dalla parete dei capillari corioidei e dalle cellule epiteliali. I plessi corioidei sono formati da villi, capillari e un monostrato di cellule cilindriche che costituiscono l’epitelio che riveste l’interno delle pareti ventricolari. I capillari sono fenestrati, mentre le cellule dell’epitelio sono unite da giunzioni serrate che limitano l’ingresso di sostanze per via paracellulare mantenendo l’omeostasi chimico- fisica del liquido cerebro-spinale.
Diversi studi hanno dimostrato una localizzazione della P-gP a livello dell’epitelio corioideo, in particolare nella membrana apicale delle cellule epiteliali dove trasporta le sostanze in direzione del liquido cerebro-spinale [16].
A livello della membrana basale sono stati identificati altri tipi di trasportatori ABC tra cui le MRPs (Multidrug Resistance Proteins) che mediano il trasporto di sostanze dal liquido cerebro-spinale al sangue. (Fig.1.6) Si pensa che la P-gP, insieme agli altri trasportatori, sia in grado di coordinare la secrezione e il riassorbimento dei farmaci e di altri substrati nel SNC. La P-gP a livello della BEL sembra catalizzare un trasporto in direzione opposta rispetto a quello della BEE. Sono necessari ancora numerosi studi per comprendere meglio il ruolo della P-gP a livello della BEL [16].
11 1.4.3. Placenta
La placenta è l’organo che mette in comunicazione la circolazione materna con quella fetale; regola tutti gli scambi tra madre e feto. La sua funzione principale è rappresentata dal trasporto di ossigeno proveniente dal sangue materno a quello fetale e dall’eliminazione di CO2.
La placenta consente il passaggio di nutrienti al feto e l’eliminazione di sostanze tossiche che possono rappresentare un pericolo per il suo normale sviluppo. Alcune sostanze passano con facilità la placenta come ad esempio gli zuccheri, l’acqua, gli amminoacidi e vitamine, altri non sono in grado di oltrepassarla (i.e. ormoni). I farmaci in genere passano la placenta e possono risultare tossici per il feto, per questo è sconsigliata l’assunzione di particolari farmaci durante la gravidanza. Attraverso la placenta arrivano al feto anche gli anticorpi che il suo organismo non è in grado di produrre, in particolare le immunoglobuline G (IgG) della madre, che lo proteggono dal rischio di infezioni durante la vita intrauterina. La placenta secerne anche numerosi ormoni steroidei che influenzano il metabolismo materno e fetale.
Anche a questo livello si trovano espressi i trasportatori ABC, tra cui la P-gP, dove svolgono un ruolo di protezione nei confronti del feto da eventuali xenobiotici tossici assunti dalla madre o da terapie farmacologiche necessarie alla madre durante la gravidanza [17].
La P-gP a livello della placenta si trova espressa nella membrana apicale delle cellule del sinciziotrofoblasto, rivolta verso il compartimento materno. Sembra che la proteina rimanga espressa per tutta la durata della gravidanza [17].
1.4.4. Tratto gastro-intestinale
A livello dell’intestino la P-gP è localizzata nella membrana dell’orletto a spazzola degli enterociti dove limita l’assorbimento di farmaci e di xenobiotici trasferendoli dalla cellula al lume intestinale e quindi favorendo poi la loro eliminazione tramite le feci. In questo modo la P-gP limita notevolmente l’assorbimento e quindi l’efficacia terapeutica di farmaci assunti per via orale.
12 A livello del fegato la P-gP è localizzata nella membrana canalicolare degli epatociti dove catalizza il trasferimento di sostanze nella bile.
Sia gli epatociti che gli enterociti esprimono simultaneamente la maggior parte dell’enzima metabolizzante i farmaci, il CYP3A4, e la P-gP. Questo porta ad una “alleanza” tra metabolismo e estrusione di sostanze dalle cellule, determinando una più alta disponibilità del farmaco ad essere metabolizzato [18].
E’ stato osservato che dopo somministrazione di digossina, substrato per la P-gP, in topi che esprimono questa proteina, circa il 16 % della dose veniva secreta nel lume intestinale. Non è stata osservata nessuna escrezione diretta invece in topi che mancano del gene che codifica per la P-gP [19].
1.4.5. Rene
A livello renale la P-gP si trova espressa nella membrana luminale dei tubuli renali prossimali dove media la secrezione di farmaci e xenobiotici nel lume tubulare e quindi favorisce la loro eliminazione tramite le urine.
La P-gP si trova localizzata in misura minore anche a livello dei testicoli, in particolare nella membrana apicale dei capillari che formano una barriera tra sangue e testicoli , dove facilita l’eliminazione di sostanze nel sangue, e anche nei linfociti CD4+ e CD56+ [20].
Come rivela la sua localizzazione anatomica, le conseguenze dell’azione della P-gP sono principalmente 3 :
• limitazione dell’assorbimento di farmaco a livello intestinale;
• eliminazione attiva di farmaco tramite le feci e/o le urine;
• limitazione della distribuzione di farmaco ai tessuti (i.e. SNC).
1.5.Altri trasportatori ABC
La superfamiglia dei trasportatori ABC comprende 49 tipi di proteine che sono state suddivise in 7 sottofamiglie. Tra queste le più importanti, oltre alla P-gP, sono le Multidrug Resistance- associated Proteins (MRPs) e la Breast Cancer Resistance Protein (BCRP).
13 1.5.1. Multidrug Resistance-associated Proteins (MRPs)
La famiglia delle Multidrug Resistance-associated Proteins (MRPs) comprende 9 proteine che come la P-gP hanno la funzione di trasportare numerose sostanze fuori dalle cellule utilizzando come fonte di energia l’idrolisi dell’ATP. La prima proteina di questo gruppo ed essere stata scoperta è la MRP1, identificata in alcune cellule tumorali [15].
La MRP1 è una proteina di 190 KDa [21], codificata dal gene Mrp1 (ABCC1) e costituita da 1531 amminoacidi distribuiti in 3 domini e 17 eliche transmembranali. (Fig.1.7)
Figura 1.7. Struttura MRP1
La sua struttura è molto simile alla P-gP: anche qui troviamo 2 siti di legame per l’ATP (NBD) situati sul lato citoplasmatico e anche questa proteina è N-glicosilata. Le eliche 10, 11, 16, 17 sembrano implicate nel legame con il substrato [21].
Le MRPs si trovano localizzate in diversi tessuti normali come ad esempio la BEE, la BEL, fegato, rene, testicoli e placenta [22]. MRP1, MRP3 e MRP5 si trovano localizzate a livello della membrana basolaterale; MRP2 e MRP4 nella membrana apicale delle cellule di diversi tessuti. L’espressione delle MRPs a livello dei capillari delle cellule endoteliali cerebrali che formano la BEE è stata individuata in diverse specie, inclusa quella umana [22].
14 MRP1 è stata identificata in diversi tipi di cellule tumorali come ad esempio in alcuni tipi di leucemie e carcinomi esofagei, dove il suo livello di espressione è legato allo stato di avanzamento del tumore [15]. Le cellule che iperesprimono questa proteina sono resistenti ad una grande varietà di farmaci antitumorali tra cui l’etoposide, la doxorubicina, l’epirubicina, ecc; inoltre, la MRP1 lega sostanze coniugate con il glutatione (GSH) e con l’acido glucuronico: la vincristina e la daunomicina sono trasportate legate a GSH, MRP1 non è in grado di trasportare queste sostanze immodificate. Un altro substrato per la MRP1 è il leucotriene C4 (LTC4), un importante mediatore dell’infiammazione [14]. Questo è sintetizzato nei mastociti, nei basofili, nei microsomi epatici e nelle cellule endoteliali. La MRP1 è responsabile del rilascio di LTC4 dai mastociti in risposta alle immunoglobuline E (Ig-E) durante i processi infiammatori.
Anche i farmaci inibitori delle proteasi anti-HIV, come il ritonavir e il saquinavir sono substrati di tale trasportatore.
La MRP2 e la MRP3 hanno substrati simili alla MRP1 e anche queste proteine sono state localizzate in diverse cellule tumorali. La MRP4 trasporta principalmente nucleotidi ciclici, analoghi delle nucleobasi come ad esempio la 6-mercaptopurina e la 6-tioguanina usate come antitumorali, analoghi nucleotidici (PMEA) e nucleosidici (ganciclovir). Substrati fisiologici di questa proteina sono le prostaglandine, ormoni steroidei e l’acido urico. Anche le MRP5, 8 e 9 trasportano nucleotidi ciclici e farmaci antitumorali come la 5-fluorouracile e il metotrexato. Le MRP6 e 7 legano principalmente i taxani, il cis platino e le antracicline [15].
La funzione principale di queste proteine è quella di limitare l’accumulo di farmaci e xenobiotici nei tessuti per proteggerli da eventuali effetti tossici. Come la PgP anche le MRPs sono implicate nel meccanismo della MDR.
15 1.5.2. Breast Cancer Resistance Protein (BCRP)
La Breast Cancer Resistance Protein (BCRP) è il membro più recente della famiglia dei trasportatori ABC, costituita da 655 amminoacidi e con un peso molecolare di 72 KDa [15]. Conosciuta anche come ABCP (Placenta-specific ABC transporter), in quanto è stata identificata per la prima volta a livello della placenta umana [15], questa proteina è codificata dal gene
BCRP (ABCG2) localizzato sul cromosoma 4 umano.
All’estremità NH2-terminale è presente, sul lato citoplasmatico, un solo sito di legame e di idrolisi dell’ATP, mentre all’estremità COOH-terminale è localizzato un dominio transmembranale, formato da 6 α-eliche, implicato nel legame con il substrato [23]. (Fig.1.8)
Figura 1.8. Struttura BCRP
In genere tutti i trasportatori ABC contengono 2 siti di legame per l’ATP e 2 domini transmembranali per questo la BCRP può essere considerata un “emi-trasportatore”[24].
La BCRP è stata identificata per la prima volta a livello della placenta e in alcuni tipi di tumori alla mammella le cui cellule risultano resistenti a diversi tipi di terapie farmacologiche [15]. Studi di immunoistochimica, hanno messo in evidenza l’espressione di questa proteina in diversi tessuti sani: nella membrana canalicolare degli epatociti, nella membrana apicale delle cellule intestinali e delle cellule endoteliali dei capillari cerebrali che costituiscono la BEE e, infine, nelle cellule del sinciziotrofoblasto della placenta [24].
16 La BCRP trasporta i suoi substrati fuori dalla cellula idrolizzando ATP: ciò influenza notevolmente l’assorbimento, la distribuzione e l’eliminazione di diversi farmaci e xenobiotici, ma protegge anche i diversi tessuti da potenziali effetti citotossici di queste sostanze.
La BCRP è stata localizzata anche in numerose cellule tumorali, in particolare nei tumori al colon, in alcuni tipi di leucemie, nei carcinomi epatocellulari e nel melanoma [24]. La sua capacità di estrudere farmaci dalla cellula è alla base del fenomeno della MDR, una delle cause del fallimento di diverse terapie antitumorali.
I substrati per la BCRP sono sia sostanze endogene (i.e. ormoni steroidei, acido folico, protoporfirina IX), sia alcuni farmaci antitumorali, come le antracicline, gli inibitori della topoisomerasi I e gli antifolici [24].