1.3 La regolazione epigenetica
L’epigenetica è definita comunemente come “lo studio dei cambiamenti nella funzione genica che sono conservati attraverso la mitosi e la meiosi, non implicano un cambiamento nella sequenza genica e costituiscono quindi l’eredità epigenetica”. (Wu e Morris, 2001). L’eredità epigenetica può persistere nel tempo e fornisce un’alternativa alle mutazioni geniche come substrato per la selezione naturale. Essa può essere propagata da differenze nelle modificazioni post-traduzionali degli istoni, dalla deposizione nella cromatina di varianti istoniche o da differenze nella metilazione delle basi azotate che compongono la struttura del DNA (Zilberman e Henikoff, 2005).
La metilazione del DNA gioca un ruolo essenziale nella regolazione dello sviluppo delle piante poiché permette di “spegnere” geni precisi in specifiche regioni cromosomiche, anche piuttosto estese, attraverso la modificazione della struttura in eterocromatina che rende il DNA non accessibile dall’apparato trascrizionale. Le sequenze metilate sono in grado di legare specifiche proteine che a loro volta reclutano altri enzimi coinvolti nella modifica delle code istoniche capaci di indurre localmente una conformazione eterocromatinica. Ultimamente sono emerse molte prove che supportano il fatto che la demetilazione del genoma ha un effetto pleiotropico sulla morfologia delle piante, comprendeno trasformazioni omeotiche negli organi fiorali e un’alterata espressione del tempo della fioritura, come confermato sia dall’uso di mutanti che da geni antisenso. Come negli animali, la metilazione nelle piante è strettamente associata con la modificazione istonica e coinvolge proteine che legano il DNA e i rispettivi complessi di trascrizione della cromatina.
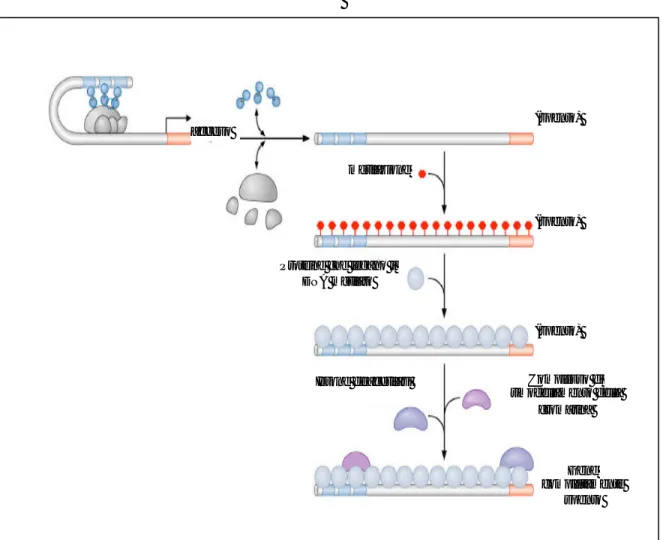
Fig. 1.3.1. Modello di silenziamento epigenetico.
Il gene dapprima trascrizionalmente attivo, viene “spento” grazie a vari meccanismi epigenetici: viene metilato, poi associato a proteine ed infine complessato in cromatina non accessibile diventando così completamente inattivo.
Gene completamente spento Complesso di rimodellamento della cromatina (spento) (spento) (spento) Istone deacetilasi acceso metilazione
Proteine che legano il DNA metilato
1.3.1 La metilazione del DNA nelle piante
La funzione genica può essere stabilmente modificata dalle componenti epigenetiche che possono sia silenziare che super-attivare stampi preferenziali di DNA. La metilazione del DNA è una delle modificazioni epigenetiche più comuni nelle piante superiori e negli animali, generalmente influenza la trascrizione direttamente o indirettamente: nel primo caso bloccando il legame di fattori di trascrizione, nel secondo caso attraverso proteine in grado di legare il DNA metilato e di attivare enzimi che rimodellano le code istoniche, diminuendo così la trascrizione (Finnegan et al., 2000). La metilazione del DNA nelle piante avviene grazie all'addizione covalente di un gruppo metile alla posizione 5 dell'anello pirimidinico delle citosine, ma anche nella posizione 6 dell'anello purinico delle adenine, questo secondo caso, più comune nei funghi che nelle piante, è esclusiva del genoma plastidico e non è stato sufficientemente indagato (Vanyushin, 2006).
I genomi delle piante hanno un contenuto relativamente alto in 5-metilcitosina (5mC), tra il 5 e il 25% delle citosine totali, a seconda della specie, questo a differenza del genoma umano che ne contiene circa del 4%. (Rangwala e Richards, 2004). Analisi cromatografiche in liquido ad alta risoluzione (HPLC) hanno dimostrato che il contenuto di citosine metilate è correlato alla grandezza del genoma e alla sua complessità: un genoma relativamente semplice come quello di A.thaliana è composto da circa 120 megabasi (Mb) e possiede circa il 6% delle citosine metilate, mentre il genoma di mais (2500 Mb) ha mostrato che la metilazione interessa ben il 25% delle citosine (Xiao et al., 2006). La localizzazione cromosomale delle citosine metilate nelle piante è principalmente concentrata nelle regioni eterocromatiniche, cioè in quelle zone dove la cromatina è fortemente condensata. Nel genoma di Arabidopsis
thaliana, la metilazione delle citosine è abbondante in zone dove i geni sono disposti
in lunghe ripetizioni, come i geni dell’rRNA. La componente eterocromatica maggiormente ricca in citosine metilate è quella costitutiva, caratterizzata da vari livelli di organizzazione: dalla ripetizione in tandem di satelliti o corte ripetizioni (per esempio, 180 pb in Arabidopsis) fino a domini più complessi. La metilazione nel DNA genomico delle piante può interessare tre diverse tipologie di sequenze nucleotidiche, a differenza dei mammiferi dove è limitata a corte sequenze simmetriche di 50- 30 nt CG; si possono quindi avere: siti CG simmetrici, siti CnG o CnnG simmetrici, ma anche siti Chh asimmetrici (dove “n” è qualsiasi nucleotide e “h” è A, C, o T). (Rangwala e Richards, 2004). L'importanza relativa della simmetria nella regolazione dell'espressione è sconosciuta, ma permette una facile trasmissione durante i cicli di replicazione del DNA, sembrerebbe che questo tipo di metilazione giochi un ruolo cruciale nella regolazione dell'espressione genica (Finnegan et al. 2000). La modifica covalente di una citosina metilata simmetricamente viene ereditata da una delle due eliche figlie dopo la replicazione del DNA, fornendo una memoria epigenetica. Nelle eliche figlie, l'impronta della metilazione viene ristabilita con l'utilizzo come guida del pre-esistente gruppo metile associato all'elica complementare e mantenuto da metiltransferasi che utilizzano il DNA emimetilato come substrato preferenziale. In molti casi l'impronta della metilazione è stata persa nei genomi delle piante, questa perdita è spesso dovuta non da una diretta excisione delle citosine metilate, ma piuttosto da un fallito mantenimento dell’originario piano di metilazione dopo la replicazione del DNA, anche se vi sono casi più direttamente coinvolti con una regolazione mirata dell’espressione genica, in cui il processo di demetilazione attivo può intervenire su particolari sequenze geniche il cui stato di attivazione deve essere regolato molto attentamente (Bender et al., 2004). Come abbiamo detto, non tutte le metilazione sono simmetriche e la loro conservazione risulta chiaramente incompatibile con il modello di ereditabilità descritto sopra e
dovrebbe essere ristabilito de novo ad ogni divisione cellulare attraverso l'attività delle metiltransferasi. Alcuni autori ipotizzano che i livelli relativamente bassi di metilazione non simmetrica non abbiano un disegno preciso e siano un sottoprodotto del sistema di metilazione poiché tutte le citosine possono essere in qualche modo suscettibili alla metilazione de novo effettuata dalle metiltransferasi e possono essere efficientemente mantenuti solo i siti simmetrici. Il significato funzionale della metilazione non simmetrica non è ancora chiaro, ma nuovi studi suggeriscono il loro potenziale ruolo nel silenziamento genico. Dieguez et colleghi (1997) costruirono una versione del promotore 35S Cauliflower Mosaic Virus che era privo dei siti CnG and CnnG ma conservava le citosine metilate non simmetriche. L'attività di questa sequenza mutata risultava marcatamente ridotta nei protoplasti di tabacco se confrontata con il promotore originale anche se lo era meno rispetto al promotore con tutte le citosine metilate (Dieguez et al. 1997). Questo suggerisce che la metilazione non simmetrica può contribuire al silenziamento genico in assenza dei siti metilati simmetricamente. (Goodrich e Tweedie 2002).
1.3.1 a FATTORI DI CONTROLLO DELLA METILAZIONE DEL DNA
Abbiamo descritto i tipi di metilazione che possono interessare il genoma, ma come vengono ereditati questi meccanismi? Il procedimento più conosciuto implica l’azione di enzimi citosin-metiltransferasi (CMT) in grado di catalizzare direttamente il trasferimento di un gruppo metile all’anello della citosina. Le CMT batteriche ed eucariotiche contengono una serie di motivi proteici conservati che costituiscono: il sito di legame per gruppo metile, un sito di legame del DNA bersaglio ed un sito attivo di cisteina che catalizza l’attacco nucleofilo alla posizione 6 dell’anello della citosina facilitando il trasferimento del gruppo metile alla posizione 5. Oltre al dominio catalitico, le CMT eucariotiche possiedono un’estesa sequenza
ammino-terminale (N-ammino-terminale) che riconosce la giusta regione del genoma da metilare (Bender, 2004). Sono state identificate molte citosin-metiltransferasi omologhe tra loro, alcune hanno dimostrato di possedere capacità di metilazione in vitro altre sono state individuate tramite test genetici. Il numero di questi enzimi è estremamente variabile tra i vari organismi, alcuni, come Arabidopsis, possiedono 10 o più omologhi di citosin-metiltransferasi mentre altri, come C. elegans e S. Cerevisiae, non hanno nessun segnale di attività enzimatica, infatti, hanno rivelato di non possedere citosine metilate all’interno del loro genoma (Goll e Bestor, 2005). Possiamo raggruppare le citosin-metiltransferasi in quattro famiglie basandoci sulla loro omologia del dominio catalitico carbossi terminale (C-terminale), anche se gli enzimi dei funghi mostrano una grande divergenza. Nelle piante sono state strutturalmente distinte quattro classi di citosina-metiltransferasi: MET, CMT, DRM e DNMT2.
1. CITOSIN-METILTRANSFERASI1 (MET1)
La Metiltransferasi1 (MET1) di Arabidopsis è una famiglia composta da quattro geni con la posizione dell'introne conservata, con un dominio catalitico C-terminale e un lungo dominio N-terminale collegato ad una sequenza ricca in glicina e lisina, che non è una semplice alternanza delle due (Goll e Bestor, 2005). La MET1 agisce come il principale enzima di mantenimento della metilazione specificatamente nei siti CG: quando il gene MET1 è posto in antisenso o viene mutato si ha una forte riduzione della metilazione CG che comporta una serie di difetti morfologici come: taglia ridotta, ritardo nella fioritura e cambiamenti omeotici fiorali. (Bender 2004).
2. CROMOMETILASI (CMT)
La classe delle cromometilasi (CMT) sembra essere una peculiarità del regno delle piante, caratterizzata da un cromodominio inserito nella sequenza del motivo catalitico C-terminale, è deputata alla metilazione delle sequenze simmetriche CnnG (Bender, 2004). I siti CnnG dopo la replicazione del DNA sono emimetilati simmetricamente e la loro metilazione può essere protratta semplicemente. Il genoma
di A. thaliana include tre cromometilasi omologhe: CMT1, CMT2 e CMT3 ritrovate anche in riso e broccoli. Le funzioni di CMT1 e CMT2 sono sconosciute, anche se retroelementi inseriti all’interno della CMT1 in alcuni ecotipi di A. thaliana, sembrano non avere effetti, al contrario di mutazioni nel locus CMT3 che portano ad una marcata riduzione della metilazione in CnnG e alcuni effetti minori sulla metilazione CnG e di sequenze asimmetriche. Nelle piante, è probabile che la metilazione CnnG sostituisca o rinforzi la metilazione CnG in sequenze senza un elevato contenuto di siti CnG. L'inattivazione di CMT3 porta anche ad un’attivazione significativa dei transposoni CACTA, in modo marcato nei doppi mutanti met1 cmt3 rispetto ai mutanti singoli anche se i gravi difetti della metilazione CnnG non si traducono in fenotipi anomali anche dopo generazioni successive di autofertilizzazione. La metilazione CnnG potrebbe essersi evoluta dalla replicazione dei trasposoni che avrebbe sviluppato promotori privi di siti CnG, diventando così immuni alla metilazione. La metilazione CnnG rinforza il sistema di metilazione CnG, e i transposoni riattivati dalla demetilazione di siti CnG e CnnG, producono trascritti di RNA che hanno come obiettivo metilare e silenziare gli elementi sorgente attraverso vie mediate da RNA e DRM. (Goll e Bestor, 2005)
3. CITOSIN-METILTRANSFERASI CON DOMINIO RIARRANGIATO (DRM)
Il genoma di A. thaliana codifica per diverse proteine DRM: la DRM1 e la DRM2, queste proteine decretano l’inizio della metilazione di sequenze Chh, CnG, e CnnG tramite un processo chiamato metilazione del DNA-RNA (RdDM). RdDM sembra affidata alla generazione di RNA come segnali di inizio per la metilazione di sequenze di DNA omologhe. DRM1 e DRM2 operano con la CMT3 per perpetuare la metilazione dei siti asimmetrici, ma non hanno effetti sul mantenimento della metilazione CnG. Sono stati identificati altri geni DRM: DMT10 e DMT106 rispettivamente in Arabidopsis e Zea mays, questi geni hanno perso due motivi critici
(PC e ENV). DMT10 mostra il 34% di identità con DRM2 nel dominio C-terminale. Il ruolo delle loro proteine non è chiaro, ma la loro funzione sembra essere analoga a quella delle Dnmt3 dei mammiferi in cui rappresentano un fattore regolatorio derivante da una citosin-metiltransferasi che se viene liberato può evolvere rapidamente come transposone se sottoposto a pressioni difensive (Goll e Bestor, 2005).
4. DNMT2
La classe delle Dnmt2 delle DNA metiltransferasi, non sembra giocare un ruolo nello stabilire o mantenere il pattern di metilazione del DNA nonostante sia una delle classi maggiormente conservate. Queste proteine non mostrano un’attività in vitro significativa, e la perdita di funzionalità di Dnmt2 dovuta a mutazione non mostra nessuna riduzione nell’ammontare della metilazione del DNA o riduzione della metilazione de novo (Cao et al., 2000). Dnmt2 è una proteina relativamente piccola mancante del grande dominio N-terminale, caratterizzante le famiglie Dmnt1 e Dmnt3 (Dong et al., 2001), con questo sembrano essere molto più vicine alle citosin-metiltransferasi batteriche. L’alta conservazione di sequenze e di strutture, la diffusione filogenetica e l’espressione ubiquitaria della Dnmt2 sembra rimandare a importanti funzioni, ma i dati genetici e biochimici indicano che non è una metiltransferasi convenzionale. (Goll e Bestor, 2005).
1.3.2 La struttura della cromatina
Per molti anni il ruolo della cromatina è stata meramente relegato a “congegno strutturale di impacchettamento del DNA”, oggi sappiamo che agisce come un potente meccanismo di regolazione post-traduzionale, generalmente influenzando l’accessibilità dei fattori di trascrizione ai cis-elements. L’impacchettamento della cromatina è determinato dalla modificazione covalente dei nucleosomi, dalla presenza di varianti istoniche e il legame con proteine non istoniche associate alla cromatina. (Higgs et al., 2007).
Fig. 1.3.3. Struttura e modificazioni della cromatina: dal massimo grado di condensa zione inattivo (cromosoma) alla cromatina distesa attiva.
Ibridazioni in situ a florescenza hanno mostrato che nei nuclei interfasici che i geni possono stare entro il territorio del cromosoma (A) oppure al fuori sui loops estesi della cromatina (B). (Modificato da: Higgs et al., 2007)
Cromatina inacessibile Cromatina acessibile
Segnali attivatori
Telomero Telomero Centromero
Territorio del cromosoma
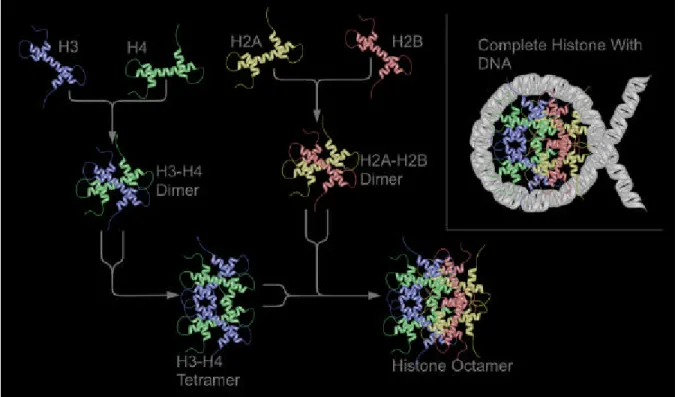
Le proteine istoniche sono cariche positivamente e formano particolari strutture dette nucleosomi, che rappresentano l'unità fondamentale della cromatina. Il nucleosoma possiede un core particolare circondato da circa 145-147 pb di elica sinistrogira di DNA che lo avvolge per 1,65 giri ed è separato e collegato alle particelle del core adiacenti da DNA linker di 20-80 pb. La particella core ha forma sferica e consiste di un ottamero contenete due molecole di ogni proteina istonica: H2A, H2B, H3, e H4. Le interazioni tra gli stessi core istonici e tra le proteine istoniche con il DNA sono mediate attraverso un dominio ripiegato detto “Histone Fold Motif” (HFM), organizzato in tre o quattro a -eliche, a1, a2, a3 e aC o aN , separate da corte regioni dette anse, che mediano la dimerizzazione istonica e le interazioni con il DNA, non sequenza-specifiche. Le interazioni tra gli istoni nel nucleosoma avvengono con orientamento testa-coda: a1 di un peptide e a3 di un altro, mentre la regione a2 è impegnata in estesi contatti idrofobici. Il dominio N-terminale del core istonico è altamente ricco di residui di lisina che formano una catena sporgente detta coda. Le code si ritrovano decisamente conservate tra le specie sia perché sono siti di modificazioni istoniche, sia perché sono necessarie per la formazione di strutture ad ordine superiore della cromatina: ad esempio la coda H4 protrude dal nucleosoma ed entra in contatto con il dimero H2A/H2B di nucleosomi adiacenti. Molte delle strutture dei nucleosomi sono state studiate in sistemi animali, la valutazione nel regno delle piante ha evidenziato alcune differenze a livello di conservazione: ad esempio gli istoni H3 e H4 sono ben conservati entro i due regni, mentre gli istoni H2 sono decisamente più conservati solo entro il regno delle piante, ma questo significato non è chiaro (Goodrich e Tweedie, 2002).
Fig. 1.3.4. Struttura del core nucleosoma.
I quattro singoli istoni si associano a formare strutture via via più complesse fino a formare l’ottamero proteico in grado di complessarsi con la struttura del DNA. (Tratta da: Luger, et al., 1997)
L’istone linker H1 è molto meno conservato e variabile rispetto al core istonico ed è associato all’aumento della compattezza della cromatina. Una singola molecola di H1 è legata all’esterno del core, vicino al DNA centrale del nucleosoma e interagisce con circa 20 pb del DNA-linker, in modo da creare due super-avvolgimenti completi dell’elica di DNA intorno alla particella dell’ordine di 10 nm detto a “collana di perle”. Questo avvolgimento di DNA intorno al nucleosoma, comunque spiega solo un settimo degli avvolgimenti della molecola di DNA lineare, fibre maggiori sono state osservate in preparazioni in vitro semplicemente aumentando la concentrazione di Mg2+ in grado di ripiegare la fibra da 10 nm a 30 nm di diamentro. Nei nuclei
interfasici, infatti, è più facile osservare configurazioni di fibre di diametro maggiore di 30 nm, in cui i nucleosomi sono compattati tra di loro in una superelica con circa sei nucleosomi per giro, con l’istone H1 come linker interno a stabilizzarne la
struttura. La compattazione superiore al livello di fibre 30 nm, presenta oltre 40-50 giri di impacchettamento, generalmente termina in strutture ad alto ordine non ben conosciute.
Fig. 1.3.5. Fibra di DNA di 30 nm.
I nucleosomi sono compattati tra loro in una superelica con circa sei nucleosomi per giro. Avvoglimenti superiori, osservabili nei nuclei interfasici, portano a strutture complesse in cui le singole componenti sono difficilmente rintracciabili. (Tratta da: Robinson et al., 2006).
Studi recenti sulle piante hanno identificato che la cromatina è anche organizzata a formare loops che escono dall’impalcatura cromosomale verso il nucleo interfasico e questi loops possono corrispondere a domini di strutture cromatinici. Nel nucleo interfasico possimo inoltre distingure due forme di cromatina a seconda del grado di condensazione: l’eucromatina e l’eterocromatina, quest’ultima è suddivisibile ulteriormente in facoltativa e costitutiva. L’eterocromatina costitutiva è riconoscibile come regioni altamente condensate associate ai telomeri e centromeri ed è caratterizzata da regioni a replicazione tardiva, un basso contenuto di geni, un alto contenuto di sequenze ripetute e bassa attività trascrizionale; mentre l’eterocromatina facoltativa è reversibile e il suo stato dipende dallo stadio di
sviluppo o dal tipo cellulare. L’eucromatina, invece risulta meno compatta e viene condensata solo durante la mitosi, è ricca di geni e spesso è in stato di trascrizione attiva. Il recente completamento del sequenziamento del genoma di Arabidopsis ha permesso studi dettagliati sulla regione eterocromatica del cromosoma 4 e ha confermato queste teorie (Fransz et al., 2000). Oltre agli istoni vi sono importanti proteine componenti di cromatina come la DNA topoisomerasi II, che costituisce la maggior parte dell’impalcatura del cromosoma metafasico, le coesine e le condensine che sono coinvolte nella condensazione dei cromosomi durante la divisione cellulare e in alcuni casi nella regolazione genica durante l’interfase. Coesine e condensine sono state recentemente caratterizzate in Arabidopsis e sono state identificate anche come importanti nell’integrità del cromosoma durante la mitosi e la meiosi. Altri importanti componenti cromatinici includono i fattori di rimodellamento discussi successivamente. Infine l’RNA è sempre più riconosciuto come un importante componente cromatinico, particolarmente a riguardo della regolazione genica. Per esempio, l’inattivazione del cromosoma X dei mammiferi e la formazione del pericentromero eterocromatinico del topo richiedono tutte molecole di RNA. Nelle piante l’RNA si è rivelato importante nella metilazione de novo del DNA, ma non sono stati descritti componenti strutturali cromatinici a RNA (Wassenegger, 2000).
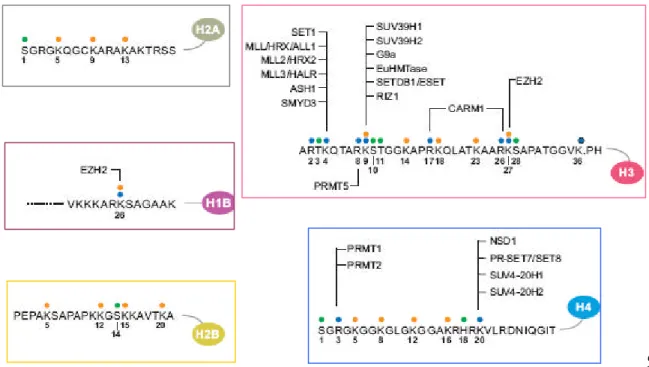
1.3.3 La modificazione delle code istoniche
Le code N-terminali degli istoni sono altamente conservate negli eucarioti e contengono in maggior misura residui in Lys, Arg, e Ser che sono bersagli di modificazioni post-traduzionali come acetilazione, metilazione, fosforilazione e ubiquitinazione. Queste modifiche possono originare altre modificazioni, influenzare direttamente la cromatina o alterare la composizione in proteine associate alla cromatina in un particolare locus (Springer et al. 2003). Le modificazioni delle code sono cruciali non solo per la loro natura ma anche a seconda della posizione in cui avvengono: ad esempio la metilazione alla lisina9 dell’istone H3 (H3-Lys9) è associata al silenziamento del promotore sul cromosoma X inattivo dei mammiferi, mentre la metilazione della lisina4 ne attiva il gene (Boggs et al., 2002). Infine troviamo casi in cui possono essere coinvolte più modificazioni successive anche se i meccanismi più importanti nello sviluppo delle piante coinvolgono principalmente l’acetilazione, deacetilazione e la metilazione delle code istoniche.
Fig. 1.3.6. Modificazioni covalenti post -traduzionali della cromatina.
Gli istoni possono essere modificati nei residui indicati da metilazione (pallino blu), acetilazione (arancione) o fosforilazione (verde). Gli enzimi responsabili son indicati sotto o sopra i residui modificati conosciuti. (Tratto da: Higgs et al., 2007, modificato).
1.3.3 a ACETILAZIONE E DEACETILAZIONE DEGLI ISTONI
Vi sono diverse ipotesi sugli effetti biologici dell’acetilazione delle code istoniche: 1. Ogni reazione di acetilazione neutralizza una carica positiva e potrebbe
potenzialmente indebolire l’interazione dell’ottamero del core istonico con la carica negativa del DNA, questo porterebbe ad una destabilizzazione del nucleosoma, permettendo ai regolatori trascrizionali di accedere al DNA. 2. L’acetilazione potrebbe interferire con l’impacchettamento di alto ordine
della cromatina riducendone l’accessibilità per le proteine regolatorie.
3. L’acetilazione potrebbe agire come uno specifico segnale in grado di alterare specifiche interazione istone-proteina.
Negli eucarioti le ricerche si sono focalizzate sull’acetilazione degli istoni H4 e H3, questo perché l’istone H4 è il bersaglio predominante dell’acetilazione in animali, funghi e protisti, seguito da H3 e infine da H2B e H2A, nelle piante invece l’istone maggiormente acetilato è H3 (Graessle et al., 2001). In generale gli istoni associati a DNA trascrizionalmente attivo sono iper-acetilati, l’analisi dello stato di acetilazione degli istoni associati al gene della plastocianina di pisello (PetE) è un esempio di questa correlazione: l’iperacetilazione delle forme H3 e H4 sono limitate al tessuto che esprime il gene e sono specificatamente localizzate sul promotore e nelle regioni
enhancer. Anche se analisi del livello di acetilazione degli istoni in fagiolo e in orzo
hanno mostrato che i livelli dell’istone H4 variano durante il ciclo cellulare, mentre quelli dell’H3 rimangono costanti suggerendo che l’acetilazione H4 è correlata con la replicazione piuttosto che con la trascrizione. Le code di tutti e quattro gli istoni del
core vengono solitamente acetilati nei residui di lisina, e ogni istone può avere dei siti
multipli di acetilazione anche se vi è un ordine preferenziale. Nei mammiferi l’istone H4 può essere acetilato in 4 siti (Lys5, Lys8, Lys12, and Lys16) e l’acetilazione della
Lys16 è preferenziale mentre in Medicago s., Arabidopsis, tabacco e carota sono stati ritrovate cinque isoforme acetilate (da mono a penta-acetilati nella Lys 5, 8, 12, 16 e 20), questo stabilisce un’ovvia differenza tra gli altri sistemi eucariotici dove l’istone H4 si trova modificato solo a livello di quattro isoforme, anche se non è stata identificato l’enzima capace di modificare H4Lys20. (Graessle et al., 2001).
I gruppi acetilici sono aggiunti dall’enzima Istone-Acetiltransferasi (HAT), la cui attività è stata ritrovata in un gran numero di proteine diverse; ne sono state classificate due famiglie sulla base della presenza e posizione del dominio conservato: la famiglia GCN5/PCAF contiene un bromodominio C-terminale mentre la famiglia MYST possiede un cromodominio N-terminale. I vari enzimi HAT sono coattivatori globali della trascrizione che contribuiscono alla regolazione di molti geni anche se non è ancora del tutto chiaro come riconoscano la regione specifica della cromatina con cui si interfacciano, sembrerebbe, però, che questi enzimi non funzionino singolarmente ma pittosto siano complessati in grandi subunità come TFIID e SAGA (Spt-Ada-Gcn5-acetiltransferasi), e in molti casi, i bersagli probabili sono contenuti su distinti componenti proteici che legano il DNA. Vi sono altri domini delle HAT, che possono giocare un ruolo importante nel mantenimento dell’acetilazione. L’acetilazione degli istoni è reversibile grazie all’enzima Istone Deacetilasi (HDAC), nelle plantule di mais sono stati individuati tre tipi di HAT e quattro distinti tipi di HDAC (Graessle et al. 2001). Lo stato di acetilazione nelle cellula riflette l’oscillazione dell’attività dei due enzimi HAT e HDAC. L’emivita del gruppo acetato varia da pochi minuti a poche ore a seconda del tipo di cellula e della localizzazione sul cromosoma. La cromatina molto condensata come in mitosi è molto più refrattaria all’acetilazione. Nel lievito Saccharomyces cerevisiae, l’intervento, il fattore di rimodellamento della cromatina Swi/Snf ATP-dipendente occorre prioritariamente all’acetilazione degli istoni, presumibilmente aiuta nell’accesso l’HAT (Berger, 2002). Ciò concorda con l’accessibilità alla nucleasi che precede
l’iper acetilazione durante l’attivazione del gene della plastocianina in pisello. Questo probabilmente perché possono essere attivati reversibilmente altri meccanismi di rimodellamento, anche se è il solo esempio conosciuto. Come le HAT, le HDAC sono state trovate in grandi complessi proteici, che risultano essere corepressori piuttosto che coattivatori. I complessi repressori sono numerosi e molti tra di essi condividono subunità di proteine con caratteristiche simili alle HADC e alle loro proteine bersaglio. Le piante contengono un’insolita classe di HDAC, HD2, in aggiunta agli omologhi delle famiglie Rpd3 e HDA1 trovate negli altri eucarioti (Wu et al. 2000). HDAC hanno differenti specificità, ad esempio HD1 di pisello deacetila preferenzialmente l’istone H4 nella Lys16 più che nella Lys5, mentre HD2 preferisce le H4-Lys5/8 alla H4-Lys16 (Clemente et al. 2001). HAT e HDAC giocano un ruolo chiave nel meccanismo di controllo nella regolazione della trascrizione, e alcuni studi preliminari ipotizzano la loro importanza nell’orchestrare lo sviluppo delle piante.
AtHD1 codifica per il gene omologo in Arabidopsis della RPD3 di lievito e della
umana HD1, riduzione dei livelli di mRNA di AtHD1, attraverso un approccio antisenso, si risolvono nell’accumolo di H4 tetra-acetilato e sviluppo di effetti pleiotroici in piante transgeniche (Tian e Chen 2001). In studi simili, l’inibitore antisenso di AtRPD3A è stato trovato ritardare la fioritura nei transgeni di
Arabidopsis, suggerendo che l’acetilazione degli istoni può giocare un ruolo nel
controllo genico nel passaggio di fase del meristema. Carenze dell’enzima pianta-specifico HD2, AtHD2a, causate sperimentalmente con transcritti antisenso portano all’aborto dello sviluppo dei semi (Wu et al. 2000). Arabidopsis HDA6, un enzima simile-Rpd3, è stato identificato in uno screening basato sulla deregolazione della risposta auxinica nei transgeni, sebbene HDA6 sembri contribuire al silenziamento transgenico, non ci sono prove per il controllo di geni esogeni. Con 19 HDAC e 14 HAT omologhi, c’è molto da analizzare per ulteriori controlli dello sviluppo di questo meccanismo. (Goodrich e Tweedie, 2002).
1.3.3 b METILAZIONE DEGLI ISTONI
La metilazione interessa gli istoni nei residui di lisina (K): K4, K9, K27, K36 e K79 nell’istone H3, K20 e K59 nel dominio globulare nell’istone H4 e K26 dell’istone H1B. Sono, inoltre, state caratterizzate diverse proteine responsabili della metilazione dei residui specifici e tutte, tranne una, contengono un dominio SET (Serina-Treonina), e fanno quindi parte della famiglia delle metiltransferasi con dominio SET. L’eccezione è rappresentata dalla famiglia DOT1, i cui membri metilano K79 nella regione globulare dell’istone H3 e che non sono correlate strutturalmente con le proteine dominio-SET. La fuzione delle proteine SET è di trasferire gruppi metili dalla S-adenosil-L-metionina (AdoMet) al gruppo amminico di un residuo di lisina sull’istone o su altre proteine, lasciando la lisina metilata e il cofattore S-adenosil-L-homocisteina (AdoHcy). La metilazione in specifiche lisine di code istoniche è un segnale per il reclutamento di particolari complessi che sono diretti all’organizzazione della cromatina (Dillon et al., 2005). Nelle piante ci sono molte domini proteici SET (39 in Arabidopsis), e presumibilmente altri membri di questa famiglia saranno resposabili della metilazione in altri siti come H3-Lys4, H3-Lys27, e H4-Lys20. Comunque, l’attività dell’istone metiltransferasi (HMTasi) dipende non solo dai domini SET ma anche dalla presenza di regioni ricche in cisteina fiancheggianti (PRE-SET e POST-SET), implicando che non tutti i domini proteici SET hanno necessariamente un attività HMTasi. KRYPTONITE (KRY) è stato recentemente identificato come una funzionale HMTasi in Arabidopsis che metila H3-Lys9 (Jackson et al. 2002). KRY è stato trovato in uno screeninig per i soppressori del silenziamento genico dovuto alla metilazione del DNA, suggerendo un collegamento tra metilazione degli istoni e del DNA, inoltre questa connessione è stata anche ritrovata nel fungo Neurospora crassa, dove una mutazione nel dominio SET contenente HMTasi DIM-5 mostra una perdita della metilazione del DNA genomico (Tamaru e Selker, 2001). A supporto di questo, i mutanti kry perdono la metilazione
nelle sequenze CnG similmente ai mutanti che non hanno DNA metiltransferasi CMT3, questo implica che, in alcune estensioni, la metilazione del DNA conta sulla metilazione degli istoni. In Arabidopsis l’omologo di HP1, LIKE
HETEROCHROMATIN PROTEIN 1 (LHP1) è il potenziale collegamento tra i processi
perché può interagire sia con le CMT3 e gli istoni metilati. Mentre entrambi i mutanti
kry e cmt3 sono morfologicamente normali, mutazioni in LHP1 cambiano tutta
l’architettura della pianta, lo sviluppo fogliare e il tempo di fioritura (Goodrich e Tweedie, 2002).
1.3.4 Varianti istoniche
I complementi istonici di ogni specie non sono confinati nei cinque maggiori istoni (H1, H2A, H2B, H3, e H4) ma includono varianti aggiuntive, codificate da geni distinti, che possono essere sostituiti con gli istoni standard in particolari circostanze. L’alterazione del set di istoni associati al DNA ha un ruolo potenziale nel rimodellamento della cromatina. Sequenze differenti tra le varianti sono spesso sottili e non è chiaro il loro coinvolgimeto sulla reale eterogenicità, ma vi sono casi in cui si ha una buona specializzazione. Tutti gli eucarioti possiedono varianti del core istonico H3, localizzato preferenzialmente nell’eterocromatina del centromero, questa proteina CenH3 identificata nei mammiferi come CENP-A e in Drosophila Cid (Centromeres identifier), è presente anche nelle piante sembra giocare un ruolo nella formazione del cinetocore. Sono molte le variante istoniche documentate nelle piante: il database ChromDB (The plant chromatin database, http://www.chromdb.org) predice un totale di 40 geni istonici nel genoma di Arabidopsis (5 per H1, 13 per H2A, 11 per H2B, 13 per H3, 8 per H4) e diversi studi indicano che alcuni potrebbero contribuire a strutture cromatiniche specializzate. Uno studio combinato di immunofluorescenza e immunoprecipitazione rivela che la variante dell’istone linker di Arabidopsis H1-3 ha un distinto pattern di localizzazione cromosomale relativo all’istone H1-1 e H1-2 e sembra essere escluso dalle sequenze ripetute (Ascenzi e Gantt, 1999). Alcuni istoni delle piante mostrano sostanziali deviazioni dallo standard, ad esempio la proteina H2A(1) del grano ha una caratteristica estensione C-terminale di 19 amminoacidi, e H2B(2) ha un’insolita ripetizione nell’estensione N-terminale di 23 amminoacidi. Altri istoni insoliti sono espressi in Lilium longiflorum, specificatamente nelle cellule gametiche maschili (Ueda et al., 2000), queste proteine, che mostrano una
percentuale di identità del solo 40–50% con il core istonico nelle altre piante, possono contribuire alla condensazione specializzata o dell’espressione genica nei gameti maschili. Specifiche funzioni per le varianti istoniche delle piante rimangono per lo da stabilire per le prove contrastanti che ne derivano: ne è un esempio la variante H1-3, che sembrerebbe indotta da condizioni di siccità, ma di transcritti posti in antisenso non supportano questa teoria. (Goodrich e Tweedie, 2002)
1.3.5 Meccanismi epigenetici complessi che regolano lo
sviluppo della pianta
I meccanismi epigenetici descritti nei paragrafi precedenti interagiscono tra loro a creare una fitta rete di segnali decisivi nella regolazione dello sviluppo della pianta. Alcuni di questi possono intervenire durante la fioritura causando, ad esempio, presupposti che la ritardino come l’ipometilazione nel locus genico FLOWERING LOCUS A (FWA), o a paradossali cambiamenti omeotici fiorali dovuta ad una ipermetilazione svolta dall’enzima MET1 che causa il silenziamento del gene SUPERMAN in Arabidopsis. Altri meccanismi possono regolare l’espressione genica in risposta a diversi stimoli endogeni come stress, attacco di patogeni, temperatura e luce; ne sono chiari esempi gli studi su gli enzimi che portano alla modificazione delle code istoni come l’istone acetiltransferasi (HAT) HAC1, implicato nella regolazione trascrizionale del gene HSP17 in risposta allo shock termico, e altre HAT come HAF2 e GCN5 che hanno, rispettivamente, ruoli negativi e positivi in risposta alla luce e alla fotomorfogenesi, ma anche l’enzima istone deacetilasi (HDAC) HDA19 coinvolto nella regolazione dell’espressione di geni correlati alla patogenesi e per la resistenza a patogeni fungini (Pfluger e Wagner, 2007).
Uno dei meccanismi epigenetici meglio conosciuti controlla la vernalizzazione, cioè l’esposizione alle basse temperature, fondamentale per alcune specie di piante delle zone temperate, necessaria per il cambiamento di fase dal meristema vegetativo a quello riproduttivo. Diversi studi su Arabidopsis hanno portato all’individuazione di un gene chiave, il FLOWERING LOCUS C (FLC), represso dalle basse temperature tramite l’interazione di numerosi complessi genici capaci sia di creare modificazioni
delle code istoniche, che di inserire specifiche varianti istoniche H2A.Z nelle zone cromatiniche del locus genico (Dennis e Peacock, 2007). Altri studi genetici hanno mostrato l’importanza della metilazione nello stabilire e mantenere l’imprinting di alcuni geni. L’esempio più studiato riguarda i geni MEDEA (MEA), importanti per la germinazione dei semi. In questi geni è presente un’espressione differenziale tra i due alleli parentali, direttamente correlata allo stato di metilazione del DNA, epigeneticamente ereditata attraverso le divisioni nucleari durante i primi stadi di sviluppo dell’endosperma.
Negli stadi precoci dello sviluppo dell’endosperma gli alleli materni risultano ipometilati e si trovano poco espressi, mentre quelli paterni sono ipermetilati e già trascrizionalmente inattivi, in stadi successivi di sviluppo, a livello della cellula centrale, si ha l’azione di un attivatore specifico (DME), in grado di rimuovere la metilazione del gene derivante dall’allele materno, mentre quello paterno rimane metilato e trascrizionalmente silenziato (Huh et al., 2007).