3.
M
ATERIALI
E
M
ETODI
3.1 Animali
In tutti i protocolli sperimentali sono state utilizzate planarie (Platelminti, Tricladi) asessuate, appartenenti al clone GI di Dugesia japonica (Orii et al., 1993). Gli animali sono stati allevati in acqua di ruscello autoclavata alla temperatura di 18°C, nutriti con fegato di pollo e tenuti a digiuno per una o due settimane prima del loro utilizzo negli esperimenti.
3.2 Isolamento del clone
DjPHB2 mediante 3’-5’RACE
La sequenza completa del clone DjPHB2 è stata ottenuta mediante l’impiego della tecnica della 3’-5’ RACE, che permette l’amplificazione della sequenza genica di interesse, tra una regione già caratterizzata di un cDNA e le
estremità 3’ o 5’, mediante l’utilizzo del SMART RACE cDNA amplification kit (Clontech). I primers sequenza-specifici, progettati sulla base della sequenza EST presente in banca dati, sono i seguenti:
⋅ Per la 5’-RACE, il primer antisenso (AS1):
5’-GCTTCCTTCGGCATTGACTATT-3’
⋅ Per la 3’-RACE, il primer senso (S1):
⋅ 5’-GTTTTCTAAAACAATACGGCGAAGCA-3’
I prodotti ottenuti da questa metodica sono stati ulteriormente amplificati con una Nested PCR, utilizzando primers sequenza specifici. In particolare, per la Nested PCR sull’amplificato della 5’-RACE, è stato utilizzato il primer antisenso (AS2):
5’-TTGGGCTCGCTGAGCTTCTTGTT -3’
⋅ Per la Nested PCR sull’amplificato 3’-RACE, è stato utilizzato il primer senso (S2):
5’-GCATCAGAGAAAATCGAAGAAC -3’
3.3 Clonaggio dei prodotti di amplificazione in plasmide e
screening
bianco/blu delle colonie
I prodotti di amplificazione ottenuti dalla Nested PCR sono stati purificati da gel utilizzando il kit DNA purification Wizard (Promega) e clonati nel plasmide pGem-T Easy (Promega). Il plasmide viene fornito già linearizzato e possiede una deossitimidina sporgente ad entrambe le estremità 3'. Questo
È possibile ottenere una reazione di ligation fra plasmide e frammenti di DNA, utilizzando la T4 DNA ligasi (Promega), un enzima che forma ponti fosfodiesterici in corrispondenza dei punti di interruzione. La quantità di prodotto di PCR necessaria per la reazione di ligation è in funzione della concentrazione, della lunghezza del vettore plasmidico e della lunghezza del DNA esogeno. Dopo aver effettuato una breve centrifuga del vettore pGEM-T Easy per raccogliere il contenuto sul fondo del tubo, è stata messa a punto la seguente reazione di ligation (volume finale 10 µl):
Buffer 2x Rapid Ligation, T4 DNA Ligasi 5 µl
Vettore pGEM-T Easy (50 ng) 1 µl
Prodotto di PCR X µl
T4 DNA Ligasi (3 Weiss units / µl) 1 µl
H2O deionizzata a volume
La reazione è stata lasciata a 4°C per tutta la notte.
In seguito, i prodotti di ligation sono inseriti in cellule di Escherichia coli (ceppo DH5α) competenti: queste cellule, una volta messe in coltura, in presenza di ampicillina, potranno crescere soltanto se correttamente trasformate con il plasmide pGem-T Easy, che possiede un sito di resistenza verso questo antibiotico.
In particolare, il protocollo utilizzato è il seguente: devono essere preparate due piastre LB / ampicillina / IPTG / X-Gal per ogni reazione di ligation, più due piastre per determinare l’efficienza di trasformazione. Tutte le piastre devono essere equilibrate a temperatura ambiente prima di effettuare la piastratura. I tubi contenenti le reazioni di ligation devono essere centrifugati per far precipitare il contenuto sul fondo. 2 µl di ogni reazione devono essere
messi in tubi sterili da 1,5 ml, posti in ghiaccio. Un ulteriore tubo da 1,5 ml deve essere posto in ghiaccio, in cui deve essere messi 0,1 ng di plasmide non tagliato per determinare l’efficienza di trasformazione delle cellule competenti. Dopo aver scongelato ed agitato delicatamente i tubi contenenti le cellule competenti di E. coli, 50 µl di cellule sono trasferiti in ciascun tubo preparato precedentemente, 100 µl nel tubo per determinare l’efficienza di trasformazione. Dopo averli agitati delicatamente, i tubi vengono lasciati in ghiaccio per 20 minuti. A questo punto le cellule sono sottoposte ad uno shock termico, ponendo i tubi per 45 secondi a 42°C in bagnetto, quindi riportati in ghiaccio per 2 minuti. Lavorando a temperatura ambiente, vengono aggiunti 950 µl di SOC medium ai tubi contenenti le cellule trasformate con i prodotti della reazione di ligation, 900 µl in quelli contenenti le cellule trasformate con plasmide non tagliato. I tubi vengono messi in incubazione a 37°C per 1 ora e 30 minuti, in agitazione. Ogni coltura trasformata può essere, quindi, piastrata in duplice copia sulle piastre LB / ampicillina / IPTG / X-Gal, lasciate in incubazione overnight a 37°C per permettere alle cellule di proliferare e formare colonie.
Le colonie sono state sottoposte a screening bianco-blu: le colonie con plasmidi aventi l’inserto presentano colore bianco, quelle con plasmidi che si sono chiusi senza legare la sequenza amplificata dalla Nested PCR hanno colore blu. La differenza di colorazione delle colonie avviene grazie alla presenza nel plasmide del gene reporter della β-Galattossidasi (β-Gal), all’interno del quale è presente il polilinker. Se è stato clonato un inserto, il
precipitato di colore blu che caratterizza le colonie con cellule possedenti un plasmide privo di inserto. Le cellule che possiedono un plasmide con l’inserto e prive, quindi, di un enzima funzionale, non sono in grado di metabolizza l’X-Gal e la colonia rimane bianca. Nel terreno di coltura è anche presente Isopropil β-D-1-tiogalattopiranoside (IPTG), analogo del lattosio, che funge da attivatore della reazione catalizzata dalla β-Gal.
3.4 Estrazione di DNA plasmidico su piccola scala mediante lisi alcalina
(mini-prep)
Tale metodo permette di estrarre 2-20 µg di DNA plasmidico da cellule trasformate per successiva digestione con enzimi di restrizione.
A partire da una colonia di Escherichia coli recante il plasmide di interesse prelevata da piastra o da uno stock di batteri in glicerolo al 10%, viene effettuato, in maniera sterile, un inoculo in un tubo batteriologico da 10-15 ml contenente 3 ml di brodo di coltura LB (5 g yeast extract, 10 g NaCl, 10 g bactotriptone pH = 7/litro) con ampicillina (100 µg/ml). Il tubo è incubato per 12-16 ore a 37°C in agitazione affinchè la coltura batterica raggiunga la fase di crescita stazionaria. La coltura viene quindi centrifugata per ottenere un pellet di cellule batteriche che viene risospeso in 200 µl di soluzione 1 (10 mM Tris-HCl pH 7.5, 10 mM EDTA) contenente anche 400 µg/ml RNAsi I allo scopo di eliminare l’RNA. A questa sospensione si aggiungono 200 µl di soluzione 2 (0.2 M NaOH, 1% SDS). La reazione di lisi alcalina non deve procedere per una durata superiore a 5’, trascorsi i quali si aggiungono 200 µl di soluzione 3 (3M KAcetato, 11.5% acido acetico), necessaria a far precipitare le membrane e le pareti delle cellule lisate, insieme al DNA cromosomico. Dopo una
centrifugazione viene recuperata la fase acquosa, contenente il DNA plasmidico e le proteine batteriche. Si aggiungono 0.7 volumi di isopropanolo alla fase acquosa e si lascia 10 minuti a temperatura ambiente. In questo modo il DNA plasmidico precipita e sarà recuperato mediante centrifugazione. Seguono il lavaggio del pellet in EtOH-70% e la sua risospensione in H2O.
3.5 Estrazione di DNA plasmidico su media scala (midi-prep)
Per poter disporre di maggiori quantità di DNA si effettua un’estrazione del DNA plasmidico per mezzo di colonnine cromatografiche a scambio anionico, incluse nel kit NUCLEOBOND © AX-100 (Eppendorf). Con questa tecnica è possibile estrarre da 80 a 140 µg di DNA altamente purificato a partire da 100 ml di sospensione batterica.
3.6 Digestione di DNA con enzimi di restrizione
I cloni ricombinanti sono stati digeriti utilizzando l’enzima di restrizione EcoRI (Promega). Due siti di restrizione per questo enzima, localizzati lateralmente alla regione in cui viene inserito il DNA esogeno, sono presenti nel sito di clonaggio multiplo del vettore pGem-T Easy vector (Promega).
3.7 Estrazione di RNA totale e produzione di cDNA
RNA totale è stato estratto da planarie intatte mediante l’utilizzo del kit NucleoSpin RNAII (Macherey-Nagel). 1µg di RNA totale è stato retrotrascritto mediante l’utilizzo del kit Superscript First Strand Synthesis System for RT-PCR (Invitrogen).
3.8 Produzione
dsDjPHB2
Per il silenziamento genico, sono state adoperate piccole molecole di RNA a doppio filamento (dsDjPHB2) trascritte dal clone DjPHB2. Queste molecole una volta iniettate nell’animale, vengono processate all’interno delle cellule diventando molecole a singolo filamento e, appaiandosi specificamente con l’mRNA endogeno complementare, ne favoriscono la degradazione da parte di enzimi specifici impedendo che possa esprimersi.
Nella reazione di trascrizione, per la produzione di questi RNA a doppio filamento, sono necessari 500 ng di DNA: questo quantitativo è ottenuto dall’amplificazione di cDNA di planaria mediante PCR, utilizzando i seguenti primers:
⋅ Primer Senso:
5’-TAATACGACTCACTATAGGGAGACAATTACTGATTTATCGTTTTC-3’
(in corsivo: sequenza del promotore T7)
⋅ Primer Antisenso:
5’-TAATACGACTCACTATAGGGAGAAACAACACTACAAAAACTTTCC-3’
Il cDNA e tali primers sono stati utilizzati nella seguente reazione di PCR:
⋅ 1 ciclo a 94°C per 15 minuti per l’attivazione della Hot-start taq-polimerasi (Quiagen);
⋅ 5 cicli da: 30 secondi a 94°C - 45 secondi a 54°C - 45 secondi a 72°C; ⋅ 30 cicli da: 30 secondi a 94°C - 45 secondi a 68°C - 45 secondi a 72°C; ⋅ 1 ciclo di allungamento da 5 minuti a 72°C.
500 ng di amplificato sono stati utilizzati nella seguente reazione di trascrizione (volume finale 20 µl):
DNA x µl
Buffer 10x (Ambion) 2 µl
rNTP 5x 4 µl
RNA pol T7 (Ambion) 2 µl
H2O nuclease-free a volume
La soluzione è stata lasciata per 2 ore e 30 minuti alla temperatura di 37°C, dopo di che è stato aggiunto 1 µl di DNAasi-1 (Ambion) e lasciati per altri 30 minuti a 37°C, quindi overnight a -20°C.
Vengono unite due reazioni di trascrizione, ottenendo un nuovo volume totale di 40 µl, ad ognuno dei quali vengono aggiunti 380 µl di Stop Buffer (NH4OAc 10 M, EDTA 0,5 M, SDS 10%/H2O ribonuclease free), quindi 200 µl di fenolo/cloroformio. Dopo aver agitato con il vortex la soluzione, si effettua una centrifuga a 12.000 rpm per 5 minuti a temperatura ambiente.
Alla fase acquosa prelevata, vengono aggiunti altri 200 µl di fenolo/cloroformio, agitato il tutto con vortex e centrifugato a 12.000 rpm per 5 minuti a temperatura ambiente.
La fase acquosa viene quindi trasferita in eppendorf da PCR (100 µl ciascuna) e messa nella macchina da PCR con il seguente programma:
70°C, 15 minuti – 67°C, 3 minuti – 63°C, 3 minuti – 60°C, 3 minuti – 57°C, 3 minuti – 53°C, 3 minuti – 50°C, 3 minuti – 47°C, 3 minuti – 43°C, 3 minuti – 40°C, 3 minuti – 37°C, 1 ora – 30°C, 10 minuti – 20°C, 10 minuti
Successivamente, il dsRNA viene precipitato aggiungendo 1 ml di EtOH-100%. Dopo aver agitato con il vortex, viene effettuata una centrifugata a 12.000 rpm per 15 minuti a 4°C. Al pellet è, quindi, aggiunto 1 ml di EtOH-80% ed effettuata una centrifuga a 12.000 rpm per 10 minuti a 4°C. Il pellet così ottenuto viene risospeso in 5 µl di H2O ribonuclese-free (RF).
3.9 Protocollo per iniezione
dsDjPHB2
Le planarie sono iniettate nell’intestino, a livello della branca anteriore, utilizzando il microiniettore Nanoject (Drummond Scientific Company).
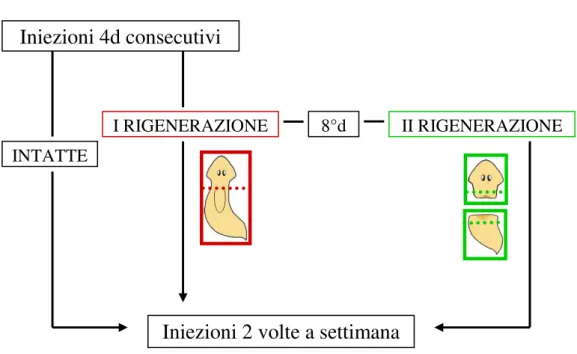
Il protocollo prevede un’iniezione al giorno per 4 giorni consecutivi, a seguito dei quali viene effettuato il primo taglio sopra al faringe per lo studio di planarie in prima rigenerazione, oppure, per lo studio nelle planarie intatte, non si esegue il taglio, ma vengono continuate le iniezioni 2 volte alla settimana, fino alla morte dell’animale.
Anche su animali in prima rigenerazione vengono effettuate iniezioni 2 volte a settimana e, all’ottavo giorno dall’inizio del protocollo, può essere effettuato un ulteriore taglio al fine di eliminare il blastema rigenerativo (per lo
studio di planarie in seconda rigenerazione). Agli animali amputati sono state somministrate dosi di dsDjPHB2 2 volte a settimana, fino alla morte dell’animale (Fig. 3.1)
Figura 3.1: schema del protocollo di iniezione (partenza: “Iniezioni 4d consecutivi”)
Iniezioni 4d consecutivi
Iniezioni 2 volte a settimana
INTATTE3.10 Produzione di sonde a RNA
3.10.1 Template per i geni DjMCM2 e DjPiwi-1
Le sonde ad RNA per geni DjMCM2 e DjPiwi-1 sono state ottenute utilizzando come template DNA plasmidico linearizzato.
I cloni contenenti i geni DjMCM2 e DjPiwi-1 sono stati linearizzati con l’enzima di restrizione ApaI (Promega) per produrre sonde ad RNA marcate con digossigenina (Salvetti et al., 2000; Rossi et al., 2006).
3.10.2 Template per il gene DjPHB2
Le sonde ad RNA per il gene DjPHB2 sono state ottenute utilizzando come template prodotti di PCR.
Dalla sequenza del gene DjPHB2 sono stati disegnati due primers:
⋅ Primer Senso:
5’-CAGAGTACGCGGGGGCATTT-3’
⋅ Primer Antisenso:
5’-TAATACGACTCACTATAGGGAGAGTACTCTCAGAGAAATATTCACC-3’
(in corsivo: sequenza del promotore T7)
Il cDNA e tali primers sono stati utilizzati nella seguente reazione di PCR:
⋅ 1 ciclo a 95°C per 5 minuti per l’attivazione della Hot-start taq-polimerasi (Promega);
⋅ 40 cicli da: 30 secondi a 95°C - 1 minuti a 57°C - 1 minuti a 72°C; ⋅ 1 ciclo di allungamento da 5 minuti a 72°C.
Il prodotto della PCR ottenuto, della lunghezza di 385 bp, è stato analizzato mediante elettroforesi su gel di agarosio, purificato con il kit DNA purification
Wizard (Promega) e utilizzato come stampo per la produzione di sonde a RNA marcate con digossigenina (Dig).
3.10.3 Produzione sonde
1 µg di DNA plasmidico linearizzato o 300 ng di amplificato sono stati utilizzati nella seguente reazione di trascrizione (volume finale 20 µl):
DNA x µl
5x Buffer di trascrizione 4 µl
100 mM DTT 2 µl
10x DIG RNA labelling mix (Roche) 2 µl
RNA pol T7 (Promega) 1 µl
H2O nuclease-free a volume
La soluzione è stata lasciata per 2 h e 30 minuti alla temperatura di 37°C, dopo di che è stato aggiunto 1 µl di DNAasi-1 (Ambion) per 30 minuti a 37°C e successivamente 2 µl di EDTA 200 mM, 2,5 µl di LiCl 4 M e 75 µl di EtOH al 100% RF. Dopo incubazione overnight (O/N) a -20°C la sonda è stata precipitata centrifugando a 12.000 rpm per 30 minuti alla temperatura di 4°C. Il pellet è stato quindi lavato con 300 µl di EtOH al 70% RF, e nuovamente centrifugato a 12.000 rpm per 5 minuti a 4°C. Il pellet ottenuto è stato risospeso in 20 µl di H2O nuclease-free (n.f.) e la sonda è stata quantificata mediante corsa elettroforetica in parallelo con un marcatore di quantità.
3.11 Ibridazione
in situ whole mount
Gli esperimenti di ibridazione in situ whole mount sono stati condotti utilizzando il metodo descritto da Agata et al. (1998). Planarie intatte e rigeneranti sono state trattate con acido cloridrico al 2% diluito in Holtfreter, fissate in Carnoy e depigmentate in acqua ossigenata/metanolo. L’ibridazione è stata condotta utilizzando 200 ng/ml di sonda marcata con digossigenina (Dig) per 72 ore a 55°C. Il segnale è stato rivelato mediante l’utilizzo di anticorpi anti-Dig coniugati con la fosfatasi alcalina e incubando i campioni in AP buffer con 340 µg/ml di NBT e 175 µg/ml di BCIP (Boehringer). Maggiori dettagli possono essere ottenuti consultando la TABELLA A.
TABELLA A
(Ibridazione in situ whole mount)
SOLUZIONE Temperatura Tempo
2%HCl/5/8 Holtfreter 4°C 5 minuti Carnoy 4°C 2 ore Metanolo -20°C 1 ora 5%H2O2/Metanolo TA 20 ore 70%EtOH/5/8 Holtfreter 4°C -20°C
50%EtOH/5/8 Holtfreter 4°C 30 minuti
30%EtOH/5/8 Holtfreter 4°C 30 minuti
0,1%Triton-X-100/PBS (TPBS) (3 lavaggi) 4°C-TA-37°C 10 minuti
20µg/ml Proteinasi K/TPBS 37°C 6 minuti
5/8 Holtfreter 4°C 1 minuto
4% Formalina/5/8 Holtfreter 4°C 1 ora
5/8 Holtfreter 4°C 1 minuto
5/8 Holtfreter 4°C 1 ora
Trietanolammina in H2O (2 lavaggi) TA 15 minuti
Trietanolammina + 0,25% Anidride Acetica TA 15 minuti
Trietanolammina + 0,50% Anidride Acetica TA 15 minuti
5/8 Holtfreter TA 1 minuto Preibridazione 55°C 1 ora Ibridazione 55°C 56 ore 50%formammide/5XSSC/0,1%Tween20 55°C 5 minuti 50%formammide/5XSSC/0,1%Tween20 55°C 1 ora 50%formammide/5XSSC/0,1%Tween20 55°C 1 ora 50%formammide/5XSSC/0,1%Tween20 55°C 1 ora
MAB 1X/ 0,1%Triton (Buffer I) TA 5 minuti
Buffer I TA 30 minuti
1%blocking/Buffer I (Buffer II) TA 30 minuti
1:2000AntiDIG/Buffer II TA 3 ore Buffer I TA 5 minuti Buffer I TA 1 ora Buffer I TA O/N (4°C) Buffer I TA 1 ora AP buffer TA 5 minuti 340 µg/ml di NBT + 175 µg/ml di BCIP in AP buffer TA or 37°C TA or 37°C TE TA 10 minuti 50%glicerolo/10%metanolo/TE 4°C
5/8 Holtfreter per 1 litro, NaCl 2.188 g, KCl 0.031 g, CaCl2 0.063 g, NaHCO3 0.125 g
Carnoy 60% EtOH, 30% Cloroformio, 10% Acido acetico
PBS per 1 litro, KCl 0.031 g, NaCl 8 g, KH2PO4 0.2 g, Na2HCO3x2H2O 1.44 g
Trietanolammina in 600 ml, 8 ml di trietanolammina, H2O RF a volume, HCl per pH a 7.6 (ca. 3 ml)
Preibridazione 50% formammide, 5xSSC, 0,1 mg/ml RNA di lievito, 0,1 mg/ml eparina, 0,1% Tween 20, 10 mM DTT
Ibridazione
50% formammide, 5xSSC, 0,1 mg/ml RNA di lievito, 0,1 mg/ml eparina, 10% Dextran-solfato, 0,1% Tween 20, 10 mM DTT, 200 ng/ml Sonda a RNA marcata
SSC 20x per 1 litro, NaCl 175.3 g, Na-citrato 88.2 g
MAB 5x per 1 litro, Acido maleico 58 g, NaCl 43.85 g, pH 7.5
AP buffer 0.1M Tris HCl, 0.1 M NaCl
TE 10 mM Tris/HCl, 1 mM EDTA
3.12 Trattamento con colchicina
Al fine di bloccare le mitosi, gli animali sono stati immersi in una soluzione di colchicina al 3‰ in acqua per 6 ore, dopo di che è stata effettuata la macerazione. In particolare, animali intatti di controllo e trattati con dsDjPHB2 sono stati trattati in colchicina 20 e 35 giorni dalla prima iniezione e poi processati per l’analisi dell’indice mitotico e per esperimenti di immunolocalizzazione con anticorpo contro H3 fosforilato.
Inoltre, sono stati trattati in colchicina animali di controllo e iniettati con dsDjPHB2 20 giorni dopo il taglio (prima rigenerazione) e dopo 3 e 7 giorni dal taglio per animali in seconda rigenerazione.
3.13 Preparazione macerati e colorazione con Hoescht per conta delle
cellule in mitosi
Per ottenere cellule dissociate del corpo degli animali, è stata effettuata una macerazione dei campioni trattati con colchicina: ogni planaria è stata lasciata 20 ore a 4°C in 250 µl di una soluzione contenente glicerolo, acido acetico e acqua sterile con un rapporto di 1:1:13. Dopo le 20 ore, è stato aggiunto colorante Hoescht ad una concentrazione di 20 µg/ml.
20 µl di macerato con Hoescht sono stati deposti su vetrini poli-D-lisinati, lasciati asciugare per circa 2 ore a temperatura ambiente ed infine montati per analisi al microscopio (5 minuti in PBS e chiusura dei vetrini in glicerolo/PBS (2:1)).
L’indice mitotico è stato calcolato analizzando i preparati al microscopio a fluorescenza Axioplan (Zeiss) e il numero di cellule totali è stato contato mediante l’utilizzo di un ematometro.
3.14 Inclusione in paraffina ed immunolocalizzazione su sezione di
cellule in mitosi, con anticorpi anti-istone H3 fosforilato
Animali intatti trattati con dsDjPHB2 a 20 e a 35 giorni dall’inizio dei trattamenti, in prima rigenerazione a 20 giorni dal primo taglio e in seconda rigenerazione a 7 giorni dal secondo taglio, con i rispettivi controlli, sono stati sottoposti a trattamenti per l’immunolocalizzazione su sezione di cellule in
TABELLA B
(Inclusione in paraffina e immunolocalizzazione su sezione)
SOLUZIONE Temperatura Tempo
2% HCl/5/8 Holtfreter 4°C 5 minuti
Soluzione rilassante di Kobayashi 4°C O/N
EtOH/5/8 Holtfreter 30% 4°C 5 minuti
EtOH/5/8 Holtfreter 50% 4°C 5 minuti
EtOH/5/8 Holtfreter 70% 4°C 5 minuti
EtOH/5/8 Holtfreter 80% 4°C 5 minuti
EtOH/5/8 Holtfreter 90% 4°C 5 minuti
EtOH/5/8 Holtfreter 100% 4°C 5 minuti
2 ml EtOH 100% TA 5 minuti
Aggiunta xilolo graduale: 0.5, 0.5, 0.5, 1 e 3 TA 15 minuti ciascuno
Xilolo assoluto TA 15 minuti
Xilolo caldo 60°C 15 minuti
50% Xilolo – 50% Paraffina 60°C 20 minuti
Paraffina (3 lavaggi) 60°C 20 minuti ciascuno
Inclusione in petri TA 4°C
Preparazione di sezioni con 8 µm di spessore.
Xilolo (2 lavaggi) TA 15 minuti ciascuno
EtOH 100% 4°C 5 minuti
EtOH/5/8 Holtfreter 95% 4°C 5 minuti
EtOH/5/8 Holtfreter 70% 4°C 5 minuti
EtOH/5/8 Holtfreter 50% 4°C 5 minuti
EtOH/5/8 Holtfreter 30% 4°C 5 minuti
PBS 1x TA 10 minuti
Blocking solution (B.Sol.) TA 2 ore
Anticorpo I/B.Sol. O/N
PBS 1x + 0,3% TRITON (4 lavaggi) TA 15 minuti ciacuno
B.Sol. TA 30 minuti
Anticorpo II TA 2 ore
PBS 1x + 0,3% TRITON (3 lavaggi) TA 15 minuti ciascuno
PBS 1x TA 5 minuti
H2O
Chiusura vetrini con glicerolo/PBS (2:1)
Soluzione rilassante di Kobayashi
1,6% Formaldeide-37%, 20 µM MgSO4·7H2O 1 M,
1% HNO3 (65%)/5/8 Holtfreter
Blocking solution 1% BSA, PBS 1x, 0,3% TRITON
Anticorpo I anti-istone H3 fosforilato (Upstate), 1:250
3.15 Colorazione preparati istologici con ematossilina ed eosina
Come coloranti dei preparati istologici sono stati utilizzati l’ematossilina e l’eosina: il primo un colorante basico di colore blu-viola che va ad evidenziare i nuclei, il secondo un colorante acido di colore rosa-fucsia che, invece, mette in evidenza i citoplasmi.
I campioni sottoposti a questa analisi istologica derivano da planarie intatte, trattate con dsDjPHB2 e relativi controlli, a 35 giorni dall’inizio delle iniezioni e da animali rigeneranti, trattati e di controllo, a 7 giorni dal secondo taglio.
Le planarie sono state fissate, incluse in paraffina, tagliate al microtomo e poste su vetrini gelatinati (vedi paragrafo successivo), quindi le fette ottenute sono state deparaffinate e colorate, seguendo il protocollo descritto in TABELLA C.
TABELLA C
(Inclusione in paraffina e colorazione in ematossilina ed eosina)
SOLUZIONE Temperatura Tempo
HCl/Holtfreter 4°C 5 minuti
Liquido di Bouin 4°C 15 ore
EtOH/5/8 Holtfreter 50% (5 lavaggi) 4°C 30 minuti ciascuno
EtOH/5/8 Holtfreter 70% 4°C 30 minuti
EtOH/5/8 Holtfreter 80% 4°C 30 minuti
EtOH/5/8 Holtfreter 90% 4°C 30 minuti
EtOH/Holt 100% 4°C 30 minuti
2 ml EtOH 100% TA 5 minuti
Aggiunta xilolo graduale: 0.5, 0.5, 0.5, 1 e 3
ml TA 15 minuti ciascuno
Xilolo assoluto TA 15 minuti
Xilolo caldo 60°C 15 minuti
50% Xilolo – 50% Paraffina 60°C 20 minuti
Paraffina (x3) 60°C 20 minuti ciascuno
Inclusione in petri TA 4°C
Preparazione di sezioni con 6 µm di spessore. Sono stati utilizzati vetrini gelatinati
Xilolo (x2) TA 15 minuti ciascuno
EtOH 100% TA 5 minuti
EtOH/5/8 Holtfreter 95% TA 5 minuti
EtOH/5/8 Holtfreter 70% TA 5 minuti
EtOH/5/8 Holtfreter 50% TA 5 minuti
EtOH/5/8 Holtfreter 30% TA 5 minuti
Ematossilina 1:10 TA 3 minuti
Acqua distillata TA 1 minuto
Eosina TA 3 minuti
Acqua distillata TA 30 secondi
EtOH 95%/Acqua distillata TA 30 secondi
EtOH 100% TA 30 secondi
Xilolo TA 5 minuti
Montare con BIOMOUNT Mounting Medium (British BioCell International)
Liquido di
3.16 Preparazione vetrini gelatinati
La gelatina utilizzata per fare vetrini gelatinati è stata preparata aggiungendo, per litro di H2O distillata, 0,1 g di allume di cromo (CrK(SO4)2·2H2O) e 1 g di gelatina.
Dopo aver lavato accuratamente con detergente e risciacquato i vetrini sotto acqua corrente per 30 minuti, i vetrini sono stati lavati in H2O distillata e, quindi, fatti asciugare in apposito porta vetrini.
I vetrini così puliti, possono essere passati due o tre volte nella gelatina, precedentemente preparata, per 5 minuti, fatti asciugare in stufa a 37°C e conservati in frigorifero.
3.17 Analisi ultrastrutturali - microscopia elettronica a trasmissione
(TEM)
Le analisi al TEM sono state condotte su planarie trattate con dsDjPHB2 a 3 giorni dall’inizio della seconda rigenerazione e i relativi controlli.
Sono stati fissati piccoli frammenti ricavati dalla zona in cui era stato effettuato il taglio, seguendo il protocollo, descritto in TABELLA D.
TABELLA D
(Fissazione in ultrastruttura)
SOLUZIONE Temperatura Tempo
Tampone cacodilato 0.2 M, pH 7.4 4°C 5 minuti
2.5% Glutaraldeide/Tampone cacodilato 0.1M 4°C 1 ora
Tampone cacodilato 0.1M (2 lavaggi) 10 minuti ciascuno
1% Osmio/Tampone cacodilato 0,2M 2 ore
Tampone cacodilato 0,1M (2 lavaggi) 10 minuti ciascuno
EtOH: 30%, 50%, 70%, 90%, 95%, 100% (x3) 10 minuti ciascuno
OxPropilene (3 lavaggi) 10 minuti ciascuno
EPON/OxPropilene 1:1 O/N
EPON/OxPropilene 2:1 5 ore
EPON 60°C 48 ore
Le sezioni ultrafini, ottenute mediante l’utilizzo di una lama diamante (Micro Star) e di un ultramicrotomo (Ultracut Reichert-Jung ultramicrotome), vengono poste sopra dei retini di rame, e colorate con acetato di uranile e citrato di piombo. L’osservazione dei preparati è stata condotta con un microscopio elettronico a trasmissione Jeol 100 SX.