Chapter 2
2,3-Unsaturated-O-glycosides from Glycal-derived Allyl Epoxides
and N-mesyl and N-nosyl Aziridines. State of the art
2.1. D-Galactal- and D-allal-derived allyl epoxides and allyl aziridines
As previously described, one of the most common procedures for the synthesis of pseudoglycals is the Ferrier allylic rearrangement of glycals.1 Recently, in the laboratory where I have carried out this thesis, new glycal-derived allyl heterocyclic systems were synthesized and studied: the D -galactal-derived allyl epoxides 2.1β and 2.1β-Tr,2 the D-allal-derived allyl epoxide 2.1α3 and the corresponding allyl N-mesyl- 2.2β4 and 2.2α5 and N-nosyl-aziridines 2.3β and 2.3α.6
Epoxides 2.1α, 2.1β, and 2.1β-Tr and aziridines 2.2α-2.2β and 2.3α-2.3β possess an intrinsic synthetic interest, due to the fact that they are simultaneously glycals and allyl oxiranes or allyl aziridines (Scheme 2.1, for the sake of simplicity only epoxides 2.1β and aziridines 2.2β and 2.3β are shown). As allyl oxiranes and allyl aziridines, they are characterized by a double reactivity when subjected to a nucleophilic addition reaction: the nucleophile can attack (i) at the C(1) vinyl terminus of the “conjugate system” through a typical 1,4-addition pathway (conjugate addition or SN2’ process, route a) to yield α- and/or β-2,3-unsaturated glycosides (pseudoglycals), hereafter
generically called α- and β-1,4-addition products, and (ii) at the allylic C(3) oxirane or aziridine carbon to give substituted glycals through a direct, commonly completely anti-1,2-addition process (SN2 process, route b) to give substituted glycals, hereafter generically called anti-1,2-addition
products. The C(1) of the allyl oxirane and allyl aziridine system also corresponds to the classic reactive site of any glycal system, as epoxides 2.1α-2.1β and aziridines 2.2α-2.2β and 2.3α-2.3β are. O R1O O 2.1!, R1= Bn 2.1!"Tr R1= Tr O BnO O 2.1# O BnO N 2.2!, R2= Ms 2.3!, R2= Ns O BnO N R2 R2 2.2#, R2= Ms 2.3#, R2= Ns
Scheme 2.1. Regioselectivity of nucleophilic addition in the glycal-derived
allyl oxirane 2.1β and aziridines 2.2β and 2.3β
Epoxides 2.1β, 2.1β-Tr and 2.1α and the N-mesyl 2.2α-2.2β and N-nosyl aziridines 2.3α-2.3β turned out to be not sufficiently stable to be isolated, but they could be only prepared in situ by cyclization under alkaline conditions (t-BuOK) of the corresponding ultimate precursor, the trans hydroxy mesylates 2.4β, 2.4β-Tr (Scheme 2.3) and 2.4α (Scheme 2.10) for the epoxides and the trans N,O-dimesylates 2.61α and 2.61β and (Schemes 2.18 and 2.23) and trans N-nosyl-O-mesylates 2.91α and 2.91β (Scheme 2.27) for aziridines 2.2α-2.2β and 2.3α-2.3β spectively, and then left to react immediately with a nucleophile (O-, C-, N-, and S-nucleophiles).
The addition reactions of nucleophiles were carried out making use of two different protocols: - protocol A: the base (t-BuOK, 1 equiv) is added to a solution of the corresponding ultimate precursor of the epoxide or aziridine in the nucleophile (e.g. alcohols) used as the solvent, that is under reaction conditions characterized by a large excess of the nucleophile;
- protocol B; the base (t-BuOK, 1 equiv) is added to a solution of the corresponding ultimate precursor of the epoxide or aziridine in anhydrous solvent (benzene, toluene, THF, MeCN, Et2O)
and the nucleophile is then added (3-4 equiv), that is under reaction conditions characterized by the presence of a small excess of the nucleophile.
Appropriate control experiments indicated that the behavior of 6-OBn substituted epoxide 2.1β was identical to that of 6-O-trityl substituted epoxide Tr. For this reason, epoxides 2.1β and 2.1β-Tr were indifferently used in order to study the behavior of this particular allyl oxirane system in addition reactions with nucleophiles.
Calculations indicated that D-galactal-derived epoxides 2.1β and 2.1β-Tr and aziridines 2.2β and 2.3β exist as the only corresponding conformer β’, whereas D-allal-derived epoxide 2.1α and aziridines 2.2α and 2.3α exist as an almost 65:35 (in the case of epoxide 2.1α) and 1:1 (in the case
O BnO X O BnO HX Nu O BnO HX Nu route b a anti1,2-addition or SN2 process 1 4 3 1,4-addition (conjugate addition) or SN2' process 2 Nu Nu b route a (a and/or b) 2.1!, X = O 2.2!, X = NMs 2.3!, X = NNs 2,3-unsaturated-glycosides substituted glycals
anti-1,2-addition product "- or !-1,4-addition product
Nu Nu
of aziridines 2.2α and 2.3α) equilibrium mixture of the two possible corresponding conformers, α’ and α’’, as shown in Scheme 2.2.4,6-8
Scheme 2.2. Conformers of allyl epoxides 2.1βand 2.1α and allyl aziridines 2.2β , 2.2α, 2.3β and 2.3α
2.2. Synthesis and regio- and stereoselectivity of the addition of O-, C-, and S-nucleophiles to D-galactal-derived allyl epoxides 2.1β and 2.1β-Tr
Trans hydroxy mesylates 2.4β and 2.4β-Tr, the precursors of epoxides 2.1β and 2.1β-Tr, respectively, were prepared from the commercially available tri-O-acetyl-D-glucal (2.5) through a simple protection-deprotection protocol (Scheme 2.3).2
Scheme 2.3. Synthesis of trans hydroxy mesylates 2.4β and 2.4β-Tr and epoxides 2.1β and 2.1β-Tr
2.2.1. O-Nucleophiles
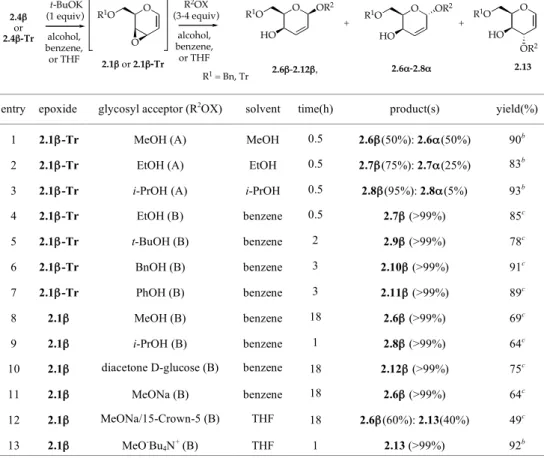
The addition reaction of alcohols (MeOH, EtOH, i-PrOH, t-BuOH) to epoxides 2.1β and 2.1β-Tr (protocol A) afforded the corresponding O-glycosides (addition products) in a completely 1,4-regioselective way, but with a stereoselectivity depending on the type of alcohol used.2 In fact, if in the reaction carried out in MeOH an almost 1:1 mixture of anomeric 2,3-unsaturated methyl α- and β-O-glycosides 2.6α and 2.6β was obtained, the use of more hindered alcohols such as EtOH and i-PrOH led to an increased β-selectivity with an α/β ratio 25:75 and 5:95, respectively (entries 1-3, Table 2.1). Only when a reduced amount of alcohol (3 equiv) was added to the epoxide preformed
O X O X OBn BnO 2.1!', 2.2!', 2.3!' 2.1!", 2.2!", 2.3!" X = O, N-Ms, N-Ns O X OBn O X BnO 2.1"', 2.2"', 2.3"' 2.1"", 2.2"", 2.3"" O OAc AcO AcO O OH R1O MsO 2.5 2.4!, R1=Bn 2.4!-Tr, R1= Tr benzene, Et2O, MeCN, or THF 2.1!, 2.1!-Tr t-BuOK R1O O O
in a benzene solution (protocol B) is a completely 1,4-regio- and β-stereoselective process obtained with the exclusive formation of corresponding β-O-glycosides 2.6β-2.9β. Under these conditions, PhOH and BnOH could also be added in the same regio- and stereoselective fashion to afford β-O-glycosides 2.10β and 2.11β, respectively. In this way, a new, uncatalyzed, completely stereoselective, directly substrate-dependent glycosylation procedure was found, which turned out to be useful also for the synthesis of a disaccharide (2.12β) when diacetone-D-glucose was used as the glycosyl acceptor (entry 10, Table 2.1).2a,b
TABLE 2.1. Regio- and stereoselectivity of the addition reactions of O-Nucleophiles to the in situ
prepared epoxides 2.1β and 2.1β-Tr
entry epoxide glycosyl acceptor (R2OX) (protocol)a
solvent time(h) product(s) yield(%)
1 2.1β-Tr MeOH (A) MeOH 0.5 2.6β(50%): 2.6α(50%) 90b
2 2.1β-Tr EtOH (A) EtOH 0.5 2.7β(75%): 2.7α(25%) 83b
3 2.1β-Tr i-PrOH (A) i-PrOH 0.5 2.8β(95%): 2.8α(5%) 93b
4 2.1β-Tr EtOH (B) benzene 0.5 2.7β (>99%) 85c 5 2.1β-Tr t-BuOH (B) benzene 2 2.9β (>99%) 78c 6 2.1β-Tr BnOH (B) benzene 3 2.10β (>99%) 91c 7 2.1β-Tr PhOH (B) benzene 3 2.11β (>99%) 89c 8 2.1β MeOH (B) benzene 18 2.6β (>99%) 69c 9 2.1β i-PrOH (B) benzene 1 2.8β (>99%) 64c
10 2.1β diacetone D-glucose (B) benzene 18 2.12β (>99%) 75c
11 2.1β MeONa (B) benzene 18 2.6β (>99%) 64c
12 2.1β MeONa/15-Crown-5 (B) THF 18 2.6β(60%): 2.13(40%) 49c
13 2.1β MeO-Bu
4N+ (B) THF 1 2.13 (>99%) 92b
aA=Protocol A; B=Protocol B. bCrude product. cAfter purification by flash chromatography.
The regio- and stereochemical behavior of epoxide 2.1β with O-nucleophiles was examined also with an alcoholate such as MeONa (protocol B). Contrary to expectations, a complete 1,4-regio- and β-stereoselective process was observed, as in the case of alcohols (entry 11, Table 2.1).
O OR2 R1O HO alcohol, benzene, or THF O R1O O t-BuOK (1 equiv) R1O O HO alcohol, benzene, or THF 2.1! or 2.1!-Tr R2OX (3-4 equiv) OR2 2.4! or 2.4!-Tr 2.13 + 2.6!-2.12!, O OR2 R1O HO 2.6"-2.8" + R1 = Bn, Tr
The complete regio- and stereoselective result obtained in the addition reaction of O-nucleophiles (alcohols and alcoholates) to epoxides 2.1β and 2.1β-Tr was rationalized by a coordination between the oxirane oxygen and the nucleophile through a hydrogen bond (alcohols) or the metal (alcoholate), as shown in structures 2.14 and 2.15, respectively (Scheme 2.4). In this way, the nucleophile is efficiently transported on the β-face of the allyl oxirane system and appropriately arranged for an entropically favored β-directed conjugate addition, as experimentally found.
Scheme 2.3. Rationalization of the 1,4-regio- and β-stereoselectivity
Confirmation of this rationalization was obtained in the reaction of epoxide 2.1β with MeONa in the presence of 15-crown-5, the crown ether specific for Na+. In these modified reaction conditions, the corresponding β-1,4-addition product 2.6β was still present in the crude reaction mixture (nearly 60%), but a substantial amount of the corresponding anti-1,2-addition product, the trans methoxy alcohol 2.13 was also obtained (nearly 40%) (entry 12, Table 2.1 and Scheme 2.5). Evidently, as a consequence of the sequestering ability of the crown ether, under these conditions the epoxide is not entirely coordinated with the nucleophile and an equilibrium exists between coordinated and non-coordinated epoxide molecules (structures 2.16 and 2.17, respectively, Scheme 2.5). While in the former, the nucleophilic attack can effectively occur from the coordinated nucleophile to give the β-1,4-addition product (route a), as stated above, the latter can react only with the free, non-coordinated nucleophile. In this case, the nucleophilic attack occurs necessarily at the C(3) allylic oxirane carbon (route b), which, in the absence of any other factors, is the most reactive position in these allyl oxirane system,2a and the corresponding anti-1,2-addition
product (2.13) was obtained in a completely anti fashion in accordance with a classic SN2-type oxirane ring-opening process. These results indicated that in order to have a complete 1,2-addition process with O-nucleophiles, and probably also with other types of nucleophiles, in oxirane system such as 2.1β, it is necessary to use a nucleophile which is not able to coordinate with the oxirane oxygen through a hydrogen bond or by a counterion with a Lewis acid (LA) character.
O OBn O O R H 2.14 2,3-unsaturated-!-O-glycosides O OBn O Na "+ " -1 2.15 O Me "+ " -1
R= Me, Et, i-Pr, t-Bu O BnO
HO
Scheme 2.5. Regio- and stereoselectivity of the addition reaction of alcoholates to epoxide 2.1β
In this framework, it was thought that tetrabutylammonium methoxide (Bu4N+OMe-), simply
prepared by evaporation of commercially available 1M tetrabutylammonium hydroxide (TBAOH) in MeOH, might be an appropriate reagent in order to have a complete 1,2-addition by MeO
-species, because not able to give a hydrogen bond and characterized by the presence of a counterion (Bu4N+) with no LA properties. Actually the reaction of epoxide 2.1β with Bu4N+OMe
-(3 equiv, protocol B) in anhydrous THF resulted in a very clean reaction affording the corresponding anti 1,2- addition product, the trans hydroxy ether 2.13, practically pure, in a completely 1,2-regio- and anti stereoselective fashion (Scheme 2.5 and entry 13, Table 2.1). To date, this is the only protocol available in order to obtain this class of addition products.2a
2.2.2. C-Nucleophiles
As for the reaction of epoxide 2.1β with C-nucleophiles, different results were obtained depending on the type of reagent. Grignard reagents such as MeMgBr and PhMgBr did not react with epoxide 2.1β, generated in situ from hydroxy mesylate 2.4β in the presence of t-BuOK (protocol B).2c
However, when MeMgBr or PhMgBr were added directly to hydroxy mesylate 2.4β, a clear ring contraction reaction occurred with the formation of an almost 1:1 mixture of the diastereoisomeric 4,5-dihydrofurane-derived trans alcohols 2.18 and 2.19 (R2= Me or Ph), as the only reaction products. Alcohols 2.18 and 2.19 derived from a highly stereocontrolled Grob fragmentation process on hydroxy mesylate 2.4β, as shown in 2.20, by the basic Grignard reagent (R2MgBr), initially leading to the trans aldehyde 2.21, then unstereoselectively attacked by the excess of R2MgBr (Scheme 2.6). O BnO O OMe BnO OH HO MsO O BnO O O BnO HO OMe Bu4N+MeO -(4 equiv) THF benzene (or THF) 15-Crown-5 rt t-BuOK (1 equiv) benzene rt MeONa (4 equiv) !-1,4-addition product anti-1,2-addition product route b route a 2.1! 2.6! 2.4! Me O O b O O O 2.13 3 O OBn O O Me 2.16 O OBn O 2.17 "+ " -1 a Na Na+ -O
On their own, cuprates such as Me2CuLi and EtMgBr in the presence of stoichiometric CuCN
afforded only the corresponding anti-1,2-addition product, the trans alcohol 2.22 (R2 = Me, Et,
Scheme 2.6).2c
Only lithium alkyls such as MeLi, BuLi, i-PrLi, t-BuLi and PhLi gave a completely 1,4-regio and β-stereoselective result affording the corresponding β-C-glycosides 2.24β (β-1,4-addition products) as the only reaction products. As previously, a coordination of the reagent (RLi) with the oxirane oxygen through the metal, as shown in 2.23, was considered responsible for the observed regio- and stereoselectivity (Scheme 2.6).2c
Scheme 2.6. Regio- and stereoselectivity of the addition reaction of Grignard reagents, cuprates and
alkyllithium reagents to hydroxy mesylate 2.4β and epoxide 2.1β
As a further C-nucleophile, the cyanide species was examined. The reaction of epoxide 2.1β-Tr with TMSCN, a cyanide species soluble in an organic solvent such as MeCN (protocol B), afforded the corresponding β-C-glycoside, the β-glycosyl cyanide 2.26β, as the only reaction product. Also this result was considered consistent with a coordination between the oxirane oxygen and the
O BnO MsO OH O BnO R2 HO O BnO O O BnO R2 HOH O BnO R2 HOH or EtMgBr/CuCN (3 equiv) 2.4! 2.1! O H BnO O H 2.22, R2= Me,Et Me2CuLi no reaction R2MgBr (3 equiv) O R2 BnO HO 2.19 2.18 + 1) R2MgBr 2) H+ 2.21 R2= Ph, Me 4
R2= Me, Bu, i-Pr, t-Bu, Ph
Et2O 0ºC 2.24! R2Li (3 equiv) 5 4 route a 5 4 O OBn 2.20 O OBn O Li R2 2.23 H H MsO O -"+ " -1 a R2MgBr Et2O Et2O t-BuOK Et2O anti-1,2-addition product !-1,4-addition product
reagent through the TMS- group, as shown in structure 2.25 (Scheme 2.7), followed by a β-directed attack of the coordinated nucleophile on the C(1) of the allyl oxirane system (route a).2a
Scheme 2.7. Addition reaction of TMSCN to epoxide 2.1β-Tr
2.2.3. S-Nucleophiles
Thiols such as PhSH and EtSH were considered as typical S-nucleophiles. If compared with the corresponding reaction with alcohols, the addition reaction of PhSH and EtSH to epoxide 2.1β-Tr (protocol B) led to the obtainment of the corresponding anti-1,2-addition product (hydroxy thioethers 2.29 and 2.30, respectively, Scheme 2.8), as largely the main (in the case of PhSH) or the sole reaction product (in the case of EtSH). Only in the case of PhSH, a slight, even if significant, amount (15%) of the corresponding β-1,4-addition product, the β-phenyl thioglycoside 2.31β, was observed. In the framework of the rationalization previously used for the corresponding reaction with alcohols, the reduced ability of thiols, with respect to alcohols, to coordinate with the oxirane oxygen considerably reduces the amount, in the reaction medium, of a coordinated species such as 2.28, the only one which can lead to the β-1,4-addition products 2.31β (route a, Scheme 2.8), to the point that this type of addition is not experimentally observed with the less acidic EtSH (pKa=
10.78) or observed only at a slight extent with the more acidic PhSH (pKa= 6.61). As a
consequence, under these conditions most of the epoxide is not coordinated with the nucleophile, and the reaction with the free thiol can occur only at the C(3) allylic oxirane carbon (route b, Scheme 2.8), to yield the corresponding anti-1,2-addition product, as experimentally observed.2a
O TrO OH MsO O TrO O O TrO TMSO 2.26! t-BuOK (1 equiv) benzene benzene 2.1!-Tr TMSCN (3 equiv) 2.4! CN C N route a Si Me MeMe O OTr O 2.25 "+ a " -!-1,4-addition product
Scheme 2.8. Regio- and stereoselectivity of the addition reaction of thiols to epoxide 2.1β-Tr
2.2.4. Coordination and non-coordination products
Taken as a whole, the results obtained in the addition reaction of O-, C-, and S- nucleophiles to D -galactal-derived epoxide 2.1β allowed to make some general considerations about the behavior of these simple and interesting allyl oxirane systems. In particular: i) when the nucleophile is able to coordinate with the oxirane oxygen of 2.1β through a hydrogen bond (ROH), a "protonation" process (TMSCN) or a metal having LA properties (RONa, RLi), a nucleophilic attack by the coordinated nucleophile is highly favored and necessarily occurs, for structural reasons, only at the C(1) carbon from the same side (β) as the oxirane system (structures 2.33 and 2.34, Scheme 2.9) affording the corresponding β-1,4-addition product, as the only reaction product. On the basis of the rationalization proposed for their formation, the configuration of the glycosides arising from coordination processes necessarily corresponds to that of the starting epoxide. As a useful generalization, glycosides having the same configuration of the starting epoxide (or aziridine, as we will see in the following Chapters), because arising, in our opinion, from a coordination process, could be simply recognized and identified from the other reaction products as “coordination product”; ii) if the nucleophile is not able to coordinate with the oxirane oxygen of 2.1β, the opening reaction pathway necessarily involves the reaction between a free, non-coordinated, epoxide with the free nucleophile. In this framework, the nucleophilic attack can occur only at the C(3) oxirane carbon which, in the absence of any other factors such as coordination, is the most reactive position in these glycal-derived oxirane systems (structure 2.35, Scheme 2.9). The anti 1,2-addition product is, in this way, selectively and exclusively obtained. Products of this type, because
O TrO O SR1 TrO OH HO MsO O TrO O benzene 2.31!, R1= Ph (15%) 2.32!, R1= Et (not observed) benzene O TrO HO SR1 2.4! 2.1!-Tr route a route b 2.29, R1=Ph (85%) 2.30, R1=Et (>99%) t-BuOK (1 equiv) 2.27 b H R1 (3 equiv) 3 O OTr O O OTr O S R1 H 2.28 1 a S R1SH "+ "- !-1,4-addition product anti-1,2-addition product
formed through a non-coordinated process, in an anti fashion, could be simply recognized and identified from the other reaction product as “non-coordination product”.2a
Scheme 2.9. Addition reactions of O-, C-, and S-nucleophiles to epoxide 2.1β
2.3. Synthesis and regio- and stereoselectivity of the addition of O-, C-, and S-nucleophiles to D-allal derived allyl epoxide 2.1α
The observation that the β-stereoselectivity obtained in the nucleophilic addition of alcohols (protocol B) to epoxides 2.1β and 2.1β-Tr corresponds to the β-configuration of the oxirane ring led our group to consider it interesting to prepare the allyl epoxide 2.1α, the α-diastereoisomer of the previously examined allyl epoxide 2.1β and to study the corresponding regio- and stereochemical behavior in nucleophilic addition reactions.3
The synthesis of trans hydroxy mesylate 2.4α, the stable precursor of epoxide 2.1α, started from epoxide 2.1β and proceeded through a simple protection-deprotection protocol applied to trans diol 2.36 (Scheme 10).
Scheme 2.10. Synthesis of trans hydroxy mesylate 2.4α and epoxide 2.1α O OBn O H Nu 2.33 O O O BnO HO Nu O BnO HO Nu BnO 1 NuH coordination product 3 2.1! NuM NuH or Nu -anti-1,2-addition product non-coordination product NuH or Nu-X+ !-1,4-addition product 2.35 M= Li+, Na+, TMS- O OBn O M Nu 2.34 O OBn O "+ " -1 "+ " -1 3 H+ Bu4N+Me 3SiO -2.1! THF O O BnO O OH BnO HO O OH BnO MsO O BnO O 2.4" 2.1" 2.36 benzene or Et2O t-BuOK
To enable a direct comparison with the diastereoisomeric epoxide 2.1β under the same conditions, the regio- and stereoselectivity of epoxide 2.1α in opening reactions with nucleophiles was examined in the addition reaction of simple O-, C-, and S-nucleophiles.3,9
2.3.1. O-Nucleophiles
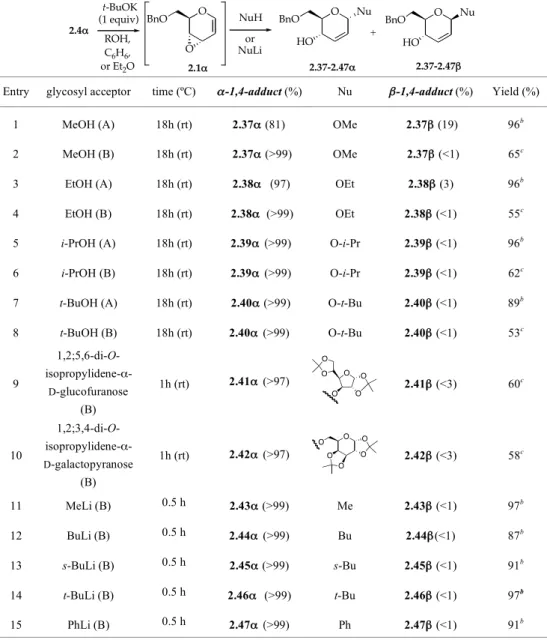
The results obtained in the reaction of epoxide 2.1α with alcohols (O-nucleophiles) under protocol A indicated that the addition reaction is completely 1,4-regioselective, but with an α/β stereoselectivity which depends on the type of alcohol used: with MeOH and EtOH an 81:19 and a 97:3 mixture of the corresponding α- and β-glycosides, 2.37α,β and 2.38α,β, was obtained, respectively, whereas with i-PrOH and t-BuOH the corresponding α-glycosides 2.39α and 2.40α were practically the only reaction products (entries 1, 3, 5, and 7, Table 2.2). In the alternative protocol B, a completely 1,4-regio- and α-stereoselective result was observed with the obtainment of the corresponding α-glycosides 2.37-2.40α as the only addition products (α-1,4-addition products and coordination products), with all the alcohols examined (Table 2.2, entries 2, 4, 6, and 8). The use of 1,2;5,6-di-O-isopropylidene-α-D-glucofuranose (diacetone-D-glucose) and 1,2;3,4-di-O-isopropylidene-α-D-galactopyranose, as the glycosyl acceptors, showed that this new protocol was also useful for the construction of disaccharides (entries 9 and 10, Table 2.2).3
The complete 1,4-regio- and α-stereoselectivity observed in the reaction of epoxide 2.1α with alcohols and di-O-isopropylidene monosaccharides (the glycosyl acceptors) could be rationalized by a possible coordination between the oxirane oxygen and the nucleophile through a hydrogen bond, as shown in structure 2.48 in Scheme 2.11.
TABLE 2.2. Glycosylation of alcohols and lithium alkyls by epoxide 2.1α
Entry glycosyl acceptor (protocol)a
time (ºC) α-1,4-adduct (%) Nu β-1,4-adduct (%) Yield (%)
1 MeOH (A) 18h (rt) 2.37α (81) OMe 2.37β (19) 96b
2 MeOH (B) 18h (rt) 2.37α (>99) OMe 2.37β (<1) 65c
3 EtOH (A) 18h (rt) 2.38α (97) OEt 2.38β (3) 96b
4 EtOH (B) 18h (rt) 2.38α (>99) OEt 2.38β (<1) 55c
5 i-PrOH (A) 18h (rt) 2.39α (>99) O-i-Pr 2.39β (<1) 96b
6 i-PrOH (B) 18h (rt) 2.39α (>99) O-i-Pr 2.39β (<1) 62c
7 t-BuOH (A) 18h (rt) 2.40α (>99) O-t-Bu 2.40β (<1) 89b
8 t-BuOH (B) 18h (rt) 2.40α (>99) O-t-Bu 2.40β (<1) 53c 9 1,2;5,6-di-O- isopropylidene-α-D-glucofuranose (B) 1h (rt) 2.41α (>97) 2.41β (<3) 60c 10 1,2;3,4-di-O- isopropylidene-α-D-galactopyranose (B) 1h (rt) 2.42α (>97) 2.42β (<3) 58c 11 MeLi (B) 0.5 h (0°C-rt) 2.43α (>99) Me 2.43β (<1) 97b 12 BuLi (B) 0.5 h (0°C-rt) 2.44α (>99) Bu 2.44β(<1) 87b 13 s-BuLi (B) 0.5 h (0°C-rt) 2.45α (>99) s-Bu 2.45β (<1) 91b 14 t-BuLi (B) 0.5 h (0°C-rt) 2.46α (>99) t-Bu 2.46β (<1) 97b 15 PhLi (B) 0.5 h (0°C-rt) 2.47α (>99) Ph 2.47β (<1) 91b
aA=protocol A; B=protocol B. bCrude product. cPurified product (flash chromatography or preparative TLC).
In this way, the nucleophile could be effectively transported onto the α-face of the allyl oxirane system and appropriately disposed for a α-direct attack on the C(1) carbon to give the corresponding α-1,4-addition product (the corresponding α-glycoside), as actually found. In this addition process, epoxide 2.1α should reasonably react through conformer 2.1α’, in which the
O O O O O O O O O O O O O BnO BnO O Nu HO O Nu BnO HO O NuH t-BuOK (1 equiv) + 2.1! 2.4! 2.37-2.47! ROH, C6H6, or Et2O 2.37-2.47" or NuLi
conjugate pathway proceeds by a more favorable pseudoaxial nucleophilic attack on C(1) carbon (route a, Scheme 2.11).3
Scheme 2.11. Rationalization of the 1,4-regio- and α-stereoselectivity in the glycosylation of alcohols by
epoxide 2.1α
The complete 1,4-regio and α-stereoselectivity observed in the reaction of epoxide 2.1α with O-nucleophiles was not only limited to alcohols and partially protected monosaccharides. In fact, also an alcoholate as MeONa when added to epoxide 2.1α led to the methyl α-O-glycoside 2.37α, the coordination product, as the only reaction product in a completely conjugated addition process occurring from the same side of the oxirane functionality (Scheme 2.12).
Scheme 2.12. Reaction of epoxide 2.1α with MeONa and Bu4N+MeO
-Evidently, as previously admitted, the reagent-epoxide coordination through the metal (Na+), as
shown in 2.49, was responsible of the observed complete regio- and stereoselectivity, no trace of the corresponding anti-1,2-addition product having been found. Once again, tetrabutylammonium methoxide (Bu4N+MeO-) turned out to be the only reagent able to afford a complete
anti-1,2-addition process with the formation of the trans methoxy alcohol 2.50, the non-coordination product, as the only reaction product, in a completely regio- and stereoselective fashion.
O BnO O O OR BnO HO 2.37-2.42! 2.1! H O R 2.48
ROH= MeOH, EtOH, i-PrOH, t-BuOH, di-O-isopropylidene-!-D-monosaccharide benzene ROH O O OBn O O BnO a route a coordination product !-1,4-addition product 2.1!' 2.1!" O
BnO BnO O OMe
HO O t-BuOK (1 equiv) 2.1! 2.4! 2.37! benzene 2.1!' O O OBn MeONa Na O Me O O OBn 2.49 Bu4N+MeO -BnO O O 2.1!'' MeO- Bu 4N+ O BnO HO 2.50 OMe "+ " -coordination product non-coordination product BnO O O 2.1!'' 2.1!'
2.3.2. C-Nucleophiles
As for C-nucleophiles, lithium alkyls such as MeLi, BuLi, s-BuLi, t-BuLi and PhLi (3 equiv) were added to epoxide 2.1α, previously prepared in Et2O from hydroxy mesylate 2.4α (protocol B). In
all cases, a complete 1,4-regioselective and α-stereoselective addition of the alkyl group occurred with the exclusive formation of the corresponding α-1,4-addition products, the α-C-glycosides 2.43-2.47α (entries 11-15, Table 2.2). As previously observed for the reactions with O-nucleophiles, the complete 1,4-regio- and α-stereoselectivity observed in the reaction of epoxide 2.1α with RLi was rationalized by the possible occurrence of a coordination between the oxirane oxygen and the nucleophile through the metal, as shown in structure 2.51 in Scheme 2.13.3
Scheme 2.13. Rationalization of the 1,4-regio- and α-stereoselectivity in the addition of lithium alkyls to
epoxide 2.1α
The reaction of epoxide 2.1α with trimethylsilyl cyanide (TMSCN) turned out to be completely 1,4-regio- and stereoselective with the exclusive obtainment of α-glycosyl cyanide 2.52α, the corresponding α-1,4-addition product (coordination product). As usual, a coordination between the oxirane oxygen and the “acidic” portion, the trimethylsilyl group, of the reagent, was considered responsible of the observed 1,4-regio- and α-stereoselectivity, as tentatively shown in structure 2.53 (Scheme 2.14).9
Scheme 2.14. Addition reaction of TMSCN to epoxide 2.1α
The reaction of epoxide 2.1α with Me2CuLi was not regio- and stereoselective and an almost 1:1:1
mixture of all the reasonable addition products was obtained: the anti 1,2-addition product (the
route a O BnO O O R BnO HO !+ 2.1" !+ 2.51 ! -Li R 2.43-2.47"
RLi= MeLi, BuLi, s-BuLi, t-BuLi, PhLi Et2O RLi O O OBn O O BnO a coordination product "-1,4-addition product O BnO BnO O CN TMSO O t-BuOK (1 equiv) 2.1! 2.4! 2.52! TMSCN Si O O OBn 2.53 Me MeMe C N "+ " -BnO O O Si Me MeMe C N "+ "
-trans methyl alcohol 2.54, 30%), the α-1,4-addition product (the α-glycoside 2.55α, 38%) and the β-1,4-addition product (the β-glycoside 2.55β, 32%) (Scheme 2.15).
Scheme 2.15. Regio- and stereoselectivity of the addition reaction of Me2CuLi to epoxide 2.1α
2.3.3. S-Nucleophiles
The reaction with tiophenol (PhSH) and ethanthiol (EtSH) was examined. The reaction of epoxide 2.1α with EtSH led to a 74:26 mixture of the corresponding anti-1,2 addition product (the trans ethylthio alcohol 2.56, non-coordination product) and α-1,4-addition product (α-ethylthio glycoside 2.57α, coordination product, Scheme 2.16).9
Scheme 2.16. Addition reactions of EtSH and PhSH to epoxide 2.1α
The rationalization of this result derived from the observation that the only 1,4-addition product present in this reaction has the same configuration (α) of the epoxide (α) and that the only 1,2-addition product had a relative trans configuration. In this framework, the anti-1,2-1,2-addition product (the non-coordination product) derives from a nucleophilic attack on the allylic C(3) carbon from a free, non-coordinated nucleophile on a free, non-coordinated epoxide which is reasonably largely present in the reaction mixture as a consequence of the reduced ability of thiols to coordination. In this process, the epoxide reacts through its conformer 2.1α’’, as shown in Scheme 2.17, in order to allow a more favorable trans diaxial opening pathway (route b), even if it determines a certain steric and/or torsional strain between the incoming nucleophile and the C(5)-C(6) bond of the side chain. As for the α-1,4-addition product, its formation comes from the
O BnO O t-BuOK 2.1! 2.4! O Me BnO HO 2.55! (38%) O BnO HO Me 2.54 (30%) + O Me BnO HO 2.55" (32%) + Me2CuLi non-coordination
product coordinationproduct
O BnO O t-BuOK 2.1! 2.4! O SR BnO HO 2.57! (26%) 2.59! (43%) O BnO HO SR EtSH or PhSH R= Et R= Ph 2.56 (74%) 2.58 (47%) +
coordination product non-coordination product O BnO HO 2.59" (10%) + SPh
occurrence, even if reduced, of a coordination of the nucleophile to the epoxide, in the form of an hydrogen bond. As usual, the coordinated nucleophile is appropriately disposed for an entropically favored α-directed attack on C(1) with complete facial selectivity and with the epoxide reacting through its conformer 2.1α” (Scheme 2.17).
Scheme 2.17. Regio- and stereoselectivity of the addition reaction of EtSH and PhSH to epoxide 2.1α
In the framework of this rationalization and as a consequence of the increased acidity of the PhSH and related ability to hydrogen bonding, an increased amount of the corresponding α-1,4-addition product (the α-phenylthio glycoside 2.59α, 43%, coordination product) was observed in the reaction of epoxide 2.1α with PhSH. The α-1,4-addition product was correctly accompanied by a similar amount of the corresponding anti-1,2-addition product (the trans phenylthio alcohol 2.58, non-coordination product, 47%) and unexpectedly by a certain amount (10%) of the corresponding β-phenylthio glycoside 2.59β (Schemes 2.16 and 2.17).9
2.3.4. Comparison of the results obtained with epoxides 2.1β and 2.1α
The comparison of the results obtained with α epoxide 2.1α3,9 and with the previously studied β epoxides 2.1β and 2.1β-Tr2 in their reactions with O- and C-nucleophiles indicated that, in these
glycal-derived allyl oxirane systems, the configuration α or β of the oxirane ring and the related coordination or chelation effects could be responsible for the complete, substrate-dependent, α- orβ-stereoselectivity, respectively observed in the completely regioselective conjugate addition of O- and C-nucleophiles. In this way, α- (from 2.1α) and β-O- and C-glycosides (from 2.1β) can be stereospecifically obtained by a simple and efficient protocol which does not need a catalyst, but only smoothly basic conditions in order to generate epoxides 2.1α and 2.1β from the corresponding hydroxy mesylates 2.4α and 2.4β, respectively, in a new, uncatalyzed, directly substrate-dependent, stereoselective, glycosylation process.
BnO O O O BnO HO SR O SR BnO HO X S R O O OBn 2.1!' "+ " -S R H b route b a route a 2.1!" R= Et, Ph
!-1,4-addition product anti-1,2-addition product
2.4. D-Allal- and D-galactal-derived allyl N-(mesyl)- and N-(nosyl)-aziridines Alkyl O-glycosides having differently functionalized amino groups in different positions (aminosugars) are an important category of modified carbohydrate units present in several oligosaccharides and glycoconjugates.10 Furthermore, aminosugars are important as essential components of bacterial capsular polysaccharides and as structural elements of aminoglycoside antibiotics with antiviral and antitumor activity.11 In consideration of the biological importance of natural products containing aminosugars,12 the development of efficient synthetic routes to these
carbohydrates is an attractive goal.
In this framework, the interest of our group was directed towards the stereoselective introduction of a nitrogen functionality at the C(4) carbon of a glycal system with simultaneous glycosylation to give 2,3,4-trideoxy-4-N-(substituted-amino)-hex-2-enopyranosides as valuable, nitrogen-containing, synthetic intermediates since the unsaturation allows further functionalization.
Few methods were reported to date for the synthesis of these synthetically useful compounds: the most convenient of these involves an allyl cyanate-to-isocyanate rearrangement of 3-enopyranosides and a palladium-catalyzed allylic substitution by secondary amines of suitable hex-2-enopyranosides.13
2.4.1. Stereoselective synthesis of 4-(N-mesylamino)-2,3-unsaturated-α-O-glycosides via the new
D-allal-derived N-(mesyl)-aziridine
The observation that in the stereospecific α- and β-O-glycosylation of alcohols and phenol and C-glycosidation of lithium alkyls by epoxides 2.1α and 2.1β,2,3 the C(4)-OH group present in the addition products comes from the epoxide led our group to pursue the prospect of achieving an analogous nitrogen transfer to the C(4) position via a corresponding activated aziridine intermediate.
As a first preliminary approach to the chemistry of glycal-derived aziridines, the readily available D-allal-derived N-mesyl-aziridine 2.2α (Scheme 2.18) turned out to be appropriate in order to check the chemical behavior of this new class of activated aziridines.5
Scheme 2.18. Synthesis of N-mesyl aziridine 2.2α O BnO MsO NHMs 2.61! O BnO O 2.1" O BnO N Ms t-BuOK (1 equiv) 2.2! benzene or MeCN
Actually, the N-acetyl aziridine 2.2α-Ac (obtained by base-catalyzed cyclization of N-acetyl-O-mesylate 2.62, Scheme 2.19), corresponding to aziridine 2.2α, was initially prepared and examined in addition reaction with alcohols.
Scheme 2.19. N-Acetyl aziridine 2.2α-Ac
Unfortunately, N-acetyl aziridine 2.2α-Ac turned out to be completely unreactive with alcohols under protocol A reaction conditions. Corresponding 1,4-Addition products were obtained, even if in an unsatisfactory yield, only when 2.2α-Ac was left to react under protocol B only with MeOH and EtOH.
The synthesis of trans N,O-dimesylate 2.61α, the stable precursor of N-mesyl-aziridine 2.2α, was achieved starting from epoxide 2.1β of inverted configuration (Scheme 2.18).
2.4.1.1. O-Nucleophiles
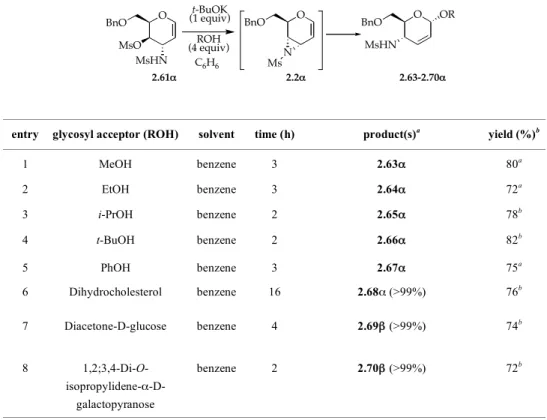
To verify the regio- and stereochemical behavior of aziridine 2.2α in glycosylation reactions, the usual alcohols and partially protected monosaccharides were employed. Under protocol B, MeOH, EtOH, i-PrOH, phenol as well as more hindered O-nucleophiles such as t-BuOH, (+)-dihydrocholesterol, diacetone D-glucose and 1,2;3,4-di-O-isopropylidene-α-D-galactopyranose were glycosylated with good yields and the corresponding 4-N-(mesylamino)-2,3-unsaturated-α-glycosides (2.63-2.68α) and disaccharides (2.69α and 2.70α) were obtained with complete 1,4-regio- and high (93-95%, in the case of 2.64α, 2.65α and 2.67α) or complete α-stereoselectivity (in the case of 2.66α and 2.68-2.70α) (Table 2.3).
O BnO MsO NHAc 2.62 O BnO N Ac t-BuOK (1 equiv) 2.2!-Ac
TABLE 2.3. Regio- and stereoselectivity of the addition reaction of O-nucleophiles to the in situ formed N-mesyl-aziridine 2.2α under protocol B.
entry glycosyl acceptor (ROH) solvent time (h) product(s)a yield (%)b
1 MeOH benzene 3 2.63α 80a 2 EtOH benzene 3 2.64α 72a 3 i-PrOH benzene 2 2.65α 78b 4 t-BuOH benzene 2 2.66α 82b 5 PhOH benzene 3 2.67α 75a 6 Dihydrocholesterol benzene 16 2.68α (>99%) 76b 7 Diacetone-D-glucose benzene 4 2.69β (>99%) 74b 8 1,2;3,4-Di-O- isopropylidene-α-D-galactopyranose benzene 2 2.70β (>99%) 72b
aIn all cases, the corresponding β-anomer was detected (1H NMR): entries 1-3 (7%), entry 5 (5%), entries 4 and 6-8 (less than 1%). bPurified product (flash chromatography or preparative TLC).
The great tendency of aziridine 2.2α, and in general of three-membered heterocycles (epoxides and aziridines) derived from glycals, to give 1,4-addition products with O-nucleophiles was furtherly demonstrated by the reaction of aziridine 2.2α with AcONa in DMF, that is to say, under conditions which should reasonably favor a typical SN2 process on the allylic C(3) carbon. Actually, the corresponding α-acetyl glycoside, the acetate 2.72α, turned out to be clearly the main addition reaction product (91%) with only a small amount of the expected 1,2 addition product, the trans mesylamino acetate 2.71 (9%) (Scheme 2.20).
A selective 1,2-addition process with O-nucleophiles was obtained only by making use of reagents in which the appropriate O-nucleophile is the counter ion of the non-coordinating tetrabutylammonium cation (Bu4N+). In this way, the treatment of a solution of aziridine 2.2α with
tetrabutylammonium methoxide (TBAOMe) and tetrabutylammonium trimethylsilanolate (TBAOSiMe3) afforded exclusively the corresponding trans methoxy- and trans
O BnO MsO MsHN 2.61! O BnO N Ms O BnO MsHN OR t-BuOK (1 equiv) ROH (4 equiv) 2.2! 2.63-2.70! C6H6
hydroxymesylamino derivatives 2.73 and 2.74, respectively, in a complete 1,2-regio- and anti-stereoselective fashion (Scheme 2.20).
Scheme 2.20. Regio- and stereoselectivity of the addition reaction of alcoholates to aziridine 2.2α
In the case of MeOH, EtOH, i-PrOH and t-BuOH, the addition reactions were repeated by using the alcohol itself as the solvent (protocol A). Under these conditions, the glycosylation reactions were still completely 1,4-regioselective, but the α/β anomeric ratio turned out to depend on the alcohol used (Scheme 2.21).
Scheme 2.21. Regio- and stereoselectivity of addition of alcohols to aziridine 2.2α (protocol A) O BnO N O OAc BnO MsHN O BnO NHMs MsO 2.2! 2.61! Ms + O BnO MsHN OAc 2.72a (91%) 2.71 (9%) [2.2!] O BnO MsHN OH DMF AcONa Ac2O t-BuOK TBAOSiMe3 2.74 O BnO MsHN OMe [2.2!] t-BuOKTBAOMe 2.73 anti-1,2-addition product anti-1,2-addition
product !-1,4-additionproduct
anti-1,2-addition product O BnO N Ms 2.2! O OR BnO MsHN t-BuOK (1 equiv) ROH + MeOH EtOH i-PrOH t-BuOH R=Me R=Et R=i-Pr R= t-Bu 2.63! 2.64! 2.65! 2.66! (60) (75) (93) (>99) (40) (25) (7) (<1) 2.63" 2.64" 2.65" 2.66" O OR BnO MsHN ROH (solvent/nucleophile) 2.63-2.66! 2.63-2.66" O BnO MsO NHMs 2.61! !-glycoside "-glycoside
The results obtained indicated that in the case of aziridine 2.2α there is a close relationship between the configuration (α) of the three-membered heterocycle (the aziridine ring) and the largely predominant or exclusive direction (α) of the O-glycosylation process, as previously observed for the corresponding epoxide 2.1α in related addition reactions.
The occurrence of an effective coordination (hydrogen bond) between the aziridine nitrogen and the O-nucleophile (ROH), as shown in structure 2.75 (Scheme 2.22) with the aziridine reacting through conform 2.2α’, can reasonably rationalize the results. In this way the nucleophile alcohol (ROH) is brought onto the α-face of the aziridine system and is suitably arranged for an entropically favored α-directed nucleophilic attack on the C(1) carbon of the unsaturated system, via pseudoaxial attack (route a, Scheme 2.22) to give the corresponding α-1,4-addition products (coordination products). Because not assisted by the aziridine nitrogen in its addition process, a β-directed attack by a free non-coordinated O-nucleophile can occur through conformer 2.2a’ (pseudoequatorial attack) or through conformer 2.2a” (pseudoaxial attack) and should correctly be less active under protocol A and absent under protocol B reaction conditions in which only a small amount of nucleophiles is present, as experimentally observed. Moreover, a non-coordinated attack along the more favorable pseudoaxial attack (route b) suffers from an unfavourable 1,3-diaxial interaction (torsional and/or steric strain) with the axial -CH2OBn side chain (Scheme 2.22).5
Scheme
Scheme 2.22. Rationalization of the regio- and α-stereoselective addition of alcohols to aziridine 2.2α
2.4.2. Stereoselective uncatalyzed synthesis of 2,3-unsaturated-4-N-substituted-β-O-glycosides by means of a new D-galactal-derived N-(mesyl)-aziridine
In order to evaluate the synthetic utility of this new class of activated allylic aziridines, and to check whether the stereoselectivity observed in the glycosylation of alcohols and monosaccharides was substrate-dependent, the diastereoisomeric activated D-galactal-derived allyl aziridine 2.2β was synthesized and its regio- and stereochemical behavior in nucleophilic addition reactions with O-nucleophiles was examined.4
1 O OBn N Ms O R 2.2!' a b route b route a O OR BnO MsHN O OR BnO MsHN "-1,4-addition product !-1,4-addition product (coordination product) H O R -H+ -H+ H ROH benzene O BnO N Ms 2.2! O N BnO Ms 1 b H O R 2.2!'' 2.75
O BnO O 2.1! O NHMs BnO MsO 2.61" O BnO 2.2" N Ms t-BuOK benzene or MeCN
The stereoselective synthesis of aziridine 2.2β started from the previously described glycal-derived allyl epoxide 2.1α of opposite configuration and led to the trans-N,O-dimesylate 2.61β, the stable precursor of allyl aziridine 2.2β (Scheme 2.23).
Scheme 2.23. Synthesis of N-mesyl aziridine 2.2β
The glycosylation of simple alcohols (MeOH, EtOH, i-PrOH, t-BuOH) by the in situ-formed allyl aziridine 2.2β, carried out in the alcohol itself as the solvent (protocol A), turned out to be completely 1,4-regioselective, with a stereochemical behavior (the α/β anomeric ratio) not only largely dependent on the alcohol used, but also decidedly different from that observed with aziridine 2.2α and related allylic oxiranes 2.1α and 2.1β in the same experimental conditions.In fact, both with MeOH and EtOH, a practically identical 31:69 β/α selectivity was observed, indicating, for the first time, the same stereochemical behavior for these two alcohols and, more notably, a stereoselectivity (α) opposite to the configuration (β) of the starting heterocycle, the aziridine 2.2β, in the present case.Only in the case of the more sterically demanding i-PrOH was the usual selectivity, even if inferior to expectations (60%), favoring the anomer (β) configurationally homogeneous with the configuration (β) of the aziridine intermediate, to the point that with t-BuOH a complete β-stereoselectivity was observed (Scheme 2.24).
Scheme 2.24. Regio- and stereoselectivity of the addition reaction of alcohols to aziridine 2.2β (protocol A)
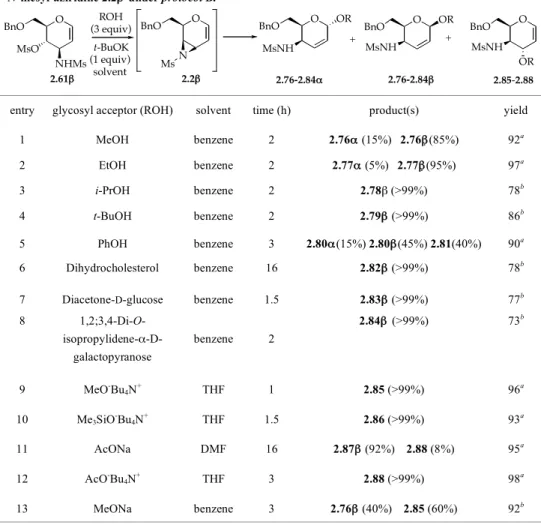
The addition reactions were repeated by treating a benzene solution of aziridine 2.2β with only a small amount of nucleophile (3-4 equiv) (protocol B, Table 2.4). Under these conditions, with the only exclusion of phenol, the addition reactions were completely 1,4-regioselective and showed a stereoselectivity drastically driven towards that anomer (β) having the same configuration (β) as the
O BnO N O OR BnO MsHN O BnO NHMs MsO 2.2! 2.61! t-BuOK (1 equiv) Ms + O OR BnO MsHN 2.76" (69%) 2.77" (69%) 2.78" (40%) 2.79" (<1%) 2.76! (31%) 2.77! (31%) 2.78! (60%) 2.79! (>99%) R= Me R=Et R=i-Pr R=t-Bu "-glycoside !-glycoside
ROH= MeOH, EtOH, i-PrOH, t-BuOH ROH
starting aziridine. The nature of the O-nucleophile determines only the extent of the generally observed stereoselectivity. In this framework, whereas MeOH showed only a moderate β-stereoselectivity (β-anomer:α-anomer = 85:15), showing an inverted behavior with respect to the same reaction carried out under protocol A, EtOH led to an almost exclusive β-stereoselectivity (95%), and a complete β-stereoselectivity (>99% of the corresponding β−anomer turned out to be present in the crude reaction product) was observed with those O-nucleophiles such as i-PrOH, t-BuOH, dihydrocholesterol, diacetone-D-glucose, and 1,2;3,4-diisopropyliden-D-galactopyranose which are characterized by an increasing steric demand around the nucleophilic center (entries 1-8, Table 2.4).
TABLE 2.4. Regio- and stereoselectivity of the addition reaction of O-nucleophiles to the in situ formed N-mesyl-aziridine 2.2β under protocol B.
entry glycosyl acceptor (ROH) solvent time (h) product(s) yield (%) 1 MeOH benzene 2 2.76α (15%) 2.76β(85%) 92a 2 EtOH benzene 2 2.77α (5%) 2.77β(95%) 97a 3 i-PrOH benzene 2 2.78β (>99%) 78b 4 t-BuOH benzene 2 2.79β (>99%) 86b 5 PhOH benzene 3 2.80α(15%) 2.80β(45%) 2.81(40%) 90a 6 Dihydrocholesterol benzene 16 2.82β (>99%) 78b
7 Diacetone-D-glucose benzene 1.5 2.83β (>99%) 77b
8 1,2;3,4-Di-O- isopropylidene-α-D-galactopyranose benzene 2 2.84β (>99%) 73b 9 MeO-Bu 4N+ THF 1 2.85 (>99%) 96a 10 Me3SiO-Bu4N+ THF 1.5 2.86 (>99%) 93a 11 AcONa DMF 16 2.87β (92%) 2.88 (8%) 95a 12 AcO-Bu 4N+ THF 3 2.88 (>99%) 98a 13 MeONa benzene 3 2.76β (40%) 2.85 (60%) 92b
a Crude product. b Purified product (flash chromatography or preparative TLC).
O BnO BnO O OR MsNH O OR BnO MsNH N + 2.2! Ms + O BnO MsNH OR 2.76-2.84" 2.76-2.84! 2.85-2.88 ROH (3 equiv) t-BuOK (1 equiv) solvent O NHMs BnO MsO 2.61!
As previously observed with aziridine 2.2α, the reaction of aziridine 2.2β with AcONa in DMF, that is under typical SN2 conditions, does not lead to the expected anti-1,2-addition product 2.88, but the corresponding β-acetyl glycoside 2.87β was the main addition reaction product (92%). The expected anti-1,2-addition product, the trans mesylamino acetate 2.88 was present only in a small amount (8%) (entry 11, Table 2.4 and Scheme 2.25).
Scheme 2.25. Regio- and stereoselectivity of the addition reaction of alcoholates to aziridine 2.2β
In the same way, the reaction of aziridine 2.2β with a nucleophile such as MeONa afforded a crude reaction mixture containing the corresponding anti-1,2 and β-1,4-addition products in a 60:40 ratio (entry 13, Table 2.4). A selective 1,2-addition process was obtained only by treatment of aziridine 2.2β with tetrabutylammonium methoxide (TBAOMe), tetrabutylammonium trimethylsilanolate (TBAOSiMe3) and tetrabutylammonium acetate (TBAOAc), which afforded exclusively the
corresponding trans methoxy-, trans hydroxy-mesylamino and trans acetoxy-mesylamino derivative 2.85, 2.86 and 2.88, respectively, in a complete 1,2-regio- and anti-stereoselective fashion (entries 9, 10 and 12, Table 2.4, and Scheme 2.25).4
With the only exception of these three last reactions, which are completely 1,2-regio- and anti-stereoselective, the addition reactions of O-nucleophiles to aziridine 2.2β, at least in conditions of a reduced amount of nucleophile present (protocol B, Table 2.4), showed a complete 1,4-regioselectivity and a stereoselectivity (β), which appeared to be driven by the configuration of the
O BnO N O OAc BnO MsHN O BnO NHMs MsO 2.2! 2.61! Ms + O BnO MsHN anti-1,2-addition product OAc !-1,4-addition product 2.87! (92%) 2.88 (8%) O BnO MsHN OMe 2.85 [2.2!] [2.2!] O BnO MsHN OH 2.86 DMF AcONa Ac2O t-BuOK t-BuOK TBAOMe TBAOSiMe3 2.88 [2.2!] TBAOAc t-BuOK anti-1,2-addition product anti-1,2-addition product anti-1,2-addition product
aziridine. As in the case of the other previously examined aziridines and epoxides derived from the glycal system, the occurence of a suitable coordination, in the form of a hydrogen bond between the reactive substrate, the aziridine 2.2β reacting in the only stable conformation 2.2β’, and the O-nucleophile, appears to be adequate in order to rationalize the observed results (Scheme 2.26).
Scheme 2.26. Rationalization of the regio- and stereoselective addition of alcohols to aziridine 2.2β
Even if favored by the coordination, the pseudoequatorial nature of this type of nucleophilic attack (route b), together with the reduced ability of the aziridine nitrogen to give rise to hydrogen bonds and the impossibility for aziridine 2.2β to adopt the alternative conformation 2.2β”, makes the attack on the C(1) from an external nucleophile through a pseudoaxial attack (route a), decidedly competitive particularly in the presence of a large excess of nucleophile (protocol A), to the point that with the more nucleophilic MeOH and EtOH, a stereoselectivity in favor of the α-anomer is observed under these conditions (Schemes 2.24 and 2.26).4
A comparison of the results obtained with the D-galactal-derived aziridine 2.2β with the previously studied D-allal-derived aziridine 2.2α in their reactions with O-nucleophiles, under protocol B, indicated that, in these glycal-derived vinyl aziridine systems, the configuration β or α of the aziridine ring and the related coordination effects are responsible for the complete or almost complete β- or α-stereoselectivity, respectively observed in the completely regioselective conjugate addition of O-nucleophiles. In accordance with the pseudoaxial or pseudoequatorial nature of the corresponding coordination-driven nucleophilic attack on C(1), the α-stereoselectivity observed with aziridine 2.2α under protocol A is larger than the β-stereoselectivity obtained with aziridine 2.2β, under the same conditions.
2.5. D-Galactal- and D-allal-derived N-(nosyl)-aziridines 2.3α and 2.3β
The use of N-mesyl aziridines 2.2α and 2.2β in the glycosylation process made the regio- and stereoselective insertion of a N-mesylamino functionality at C(4) of a pseudoglycal system possible.4,5 Actually, limited to this application, the N-mesylamino group is not the best choice to
have a free amino group by deprotection procedures. As a consequence, it appeared necessary to
O OBn N O R H Ms O H O N Ms BnO 2.2!' 2.2!" 1 "+ b a route b !-1,4-addition product (coordination product) #-1,4-addition product " -R route a
introduce on the starting aziridine a different N-activating group, which could easily be removed after the glycosylation process had taken place. Considering that the simple N-acetyl group could not be used because the corresponding N-acetyl aziridine 2.2α-Ac had proved to be not sufficiently reactive (see paragraph 2.4.1 and Schemes 2.19), the choice fell on aziridines 2.3α and 2.3β which bear the N-(o-nitrobenzenesulfonyl) [N-(nosyl)] protecting/activating group.6 Actually, the N-nosyl group could easily be removed from the addition products by the PhSH/K2CO3 protocol, in
accordance with a SNAr reaction mechanism,14 in order to have free amino group-containing products.
The synthesis of the N-nosyl aziridines starts from trans 3-amino alcohols 2.89 and 2.90obtained from epoxides 2.1β and 2.1α, respectively, leading to trans N-nosyl-O-mesylates 2.91α and 2.91β respectively, the stable ultimate precursors of aziridines 2.3α and 2.3β. Base-catalyzed cyclization (K2CO3/MeCN) of 2.91α and 2.91β afforded the desired aziridines 2.3α and 2.3β, respectively
(Scheme 2.27)
Scheme 2.27. Synthesis of N-nosyl aziridines 2.3α and 2.3β
To check the efficiency of aziridines 2.3α and 2.3β as glycosyl donors, the possible influence of the N-nosyl group on the regio- and stereoselectivity and the applicability of the deprotection procedure on the N-(nosylamino)-substituted products deriving from the glycosylation process, the regio- and stereochemical behavior of aziridines 2.3α and 2.3β in the reaction with alcohols, partially protected monosaccharides and phenol (O-nucleophiles) was examined.
The results obtained indicated that the regio- (only the corresponding 1,4-addition products were observed in each case) and stereoselectivity (α-1,4-addition product/β-1,4-addition product ratio) of N-nosyl aziridines 2.3α and 2.3β under protocols A and B susbsantially resembled that of the corresponding N-mesyl aziridines 2.2α and 2.2β, respectively. The only slight difference is found under protocol B in the sense that the glycosylation reactions of aziridines 2.3α and 2.3β are completely stereoselective towards that anomer (2.92α and 2.92β, respectively) having the same
O NH2 BnO HO 2.89 O BnO 2.3! N Ns O NHNs BnO MsO 2.91! O NH2 BnO HO 2.90 O BnO 2.3" N Ns O NHNs BnO MsO 2.91" O BnO 2.1! O O BnO 2.1" O K2CO3 K2CO3 MeCN MeCN
configuration (α or β, respectively) of the starting aziridine also in those cases in which N-mesyl aziridines 2.2α and 2.2β were not (Scheme 2.28).
The complete 1,4-regioselectivity and aziridine ring configuration-related stereoselectivity, observed in all the reactions of the N-nosyl aziridines 2.3α and 2.3β with O-nucleophiles, could be rationalized, as previously admitted for 2.2α and 2.2β, by the occurrence of an effective coordination (hydrogen bond) of the O-nucleophile with the aziridine nitrogen of 2.3α and 2.3β, followed by a nucleophilic attack on the nearby C(1) carbon of the allyl aziridine system (routes a and b for 2.3α and 2.3β, respectively, Scheme 2.28). In the present case, the metal ion (K+ from
K2CO3 used in the cyclization process) and/or an intrinsically higher ability to coordination by the
aziridine nitrogen of the N-nosyl group probably plays a role in determining the complete stereoselectivity observed.6
Scheme 2.28. Rationalization of the regio- and stereoselective addition of alcohols (ROH)
to N-nosyl aziridines 2.3α and 2.3β
The 4-N-(nosylamino)-O-glycosides 2.92α (R=Me, Et, i-Pr, Bn, menthyl, respectively) and 2.92β (R= t-Bu, allyl, diacetone-D-glucopyranolsyl) were chosen in order to check the applicability of these pseudoglycals to the deprotection procedure which makes use of the PhSH/K2CO protocol.
In this way, a solution of the methyl 4-N-(nosylamino)-4-deoxy-α-O-glycoside 2.92α (R= Me), taken as an example, in MeCN was treated with PhSH (3 equiv) in the presence of K2CO3 (4 equiv)
(solution phase conditions): the deprotection reaction was very fast and was completed in 3h (conversion >99%, TLC and 1H NMR) and the corresponding methyl 4-amino-4-deoxy-α-O-glycoside 2.93α (R= Me) was obtained in pure form (65% yield) after simple preparative TLC or flash chromatography (Scheme 2.29). Application of the same protocol to all the other O-glycosides 2.92α and 2.92β available (Scheme 2.28) afforded the corresponding free-amino group containing O-glicosydes 2.93α and 2.93β with good yield. An alternative procedure carried out by means of a PhSH-supported resin (PS-thiophenol) (solid phase conditions) turned out to be unsatisfactory, as for yield and reaction time.6
O OBn N O R H Ns 2.3!' 1 "+ b route a !-1,4-addition product " -1 O OBn N Ns H O R a route b BnO O OR NsHN #-1,4-addition product "+ 2.3#' O OR BnO NsHN " -2.92! 2.92#
coordination product coordination product
Scheme 2.29. Deprotection reaction of 4-N-(nosylamino)-O-glycosides 2.92α and 2.92β.
In conclusion, the original glycosylation protocol of alcohols, partially protected monosaccharides and phenol by the diastereoisomeric D-allal and D-galactal-derived allyl N-mesyl aziridines 2.2α and 2.2β was substantially improved by the use of the corresponding N-nosyl aziridines 2.3α and 2.3β. On passing from the N-mesyl to the N-nosyl protecting/activating group, the stereoselectivity of all the O-glycosylation reactions increases, and the N-(nosylamino) functionality, regio- and stereoselectively introduced at C(4) carbon of O-glycosides 2.92α and 2.92β, could be easily deprotected by the simple PhSH/K2CO3 protocol and transformed into corresponding O-glycosides
bearing a free amino group in the same position. As a consequence, these results indicated that the use of our O-glycosylation process by means of N-nosyl aziridines 2.3α and 2.3β, followed by the deprotection protocol, may constitute a simple and valid tool for the completely C(4)-regioselective and stereospecific synthesis of 2,3-unsaturated-4-deoxy-4-amino-O-glycosides as 2.93α and 2.93β, bearing a free amino group at C(4) carbon, with an added value to the final product and the glycosylation process itself.6
References
1. Ferrier, R. J.; Zubkov, O. A. In Organic Reactions; Overman, L. E., Ed, John Wiley; Hoboken, NJ (USA), 2003; Vol. 62, pp 569-736.
2. a) Di Bussolo, V.; Caselli, M.; Romano, M. R.; Pineschi, M.; Crotti, P. J. Org. Chem.. 2004, 69, 8702. b) Di Bussolo, V.; Caselli, M.; Pineschi, M.; Crotti, P. Org. Lett. 2002, 4, 3695. c) Di Bussolo, V.; Caselli, M.; Pineschi, M.; Crotti, P. Org. Lett. 2003, 5, 2173.
3. Di Bussolo, V.; Caselli, M.; Romano, M. R.; Pineschi, M.; Crotti, P. J. Org. Chem. 2004, 69, 7383. 4. Di Bussolo, V.; Romano, M. R.; Favero, L.; Pineschi, M.; Crotti, P. J. Org. Chem. 2006, 71, 1696. 5. Di Bussolo, V.; Romano, M. R.; Pineschi, M.; Crotti, P. Org. Lett. 2005, 7, 1299.
O OR BnO NsHN 2.92! PhSH K2CO3/MeCN O OR BnO NsHN 2.92" O OR BnO H2N 2.93" PhSH K2CO3/MeCN O OR BnO H2N 2.93!
6. Di Bussolo, V.; Romano, M. R.; Pineschi, M.; Crotti, P. Tetrahedron 2007, 63, 2482. 7. Crotti, P.; Di Bussolo, V.; Pomelli, C. S.; Favero, L. Theor. Chem. Account 2009, 122, 245. 8. Di Bussolo, V,; Checchia, L.; Romano, M. R.; Pineschi, M.; Crotti, P. Tetrahedron 2010, 66, 689. 9. Di Bussolo, V.; Favero, L.; Romano, M. R.; Pineschi, M.; Crotti, P. Tetrahedron 2008, 64, 8188.
10. a) Banoub, J.; Boullanger, P.; Lafont, D. Chem. Rev. 1992, 92, 1167. b) van den Bos, L.J.; Codée, J. D. C.; van Boom, J. H.; Overkleeft, H. S.; van der Marel, G. A. Org. Biomol. Chem. 2003, 1, 4160.
11. a) Wang, Y.; Kalka-Moll, W. M.; Roehrl, M. H.; Kasper, D.L. Proc. Natl. Acad. Sci. USA 2000, 97, 13478. b) Choi, Y.-H; Roehrl, M. H.; Kasper, D.L.; Wang, Y. Biochemestry 2002, 41, 15144. c) Sears, P.; Wong, C.-H. Angew. Chem. Int. Ed. 1999, 38, 2300.
12. Knapp, S Chem. Soc. Rev. 1999, 28, 61.
13. a) Ichikawa, Y.; Kobayashi, C.; Isobe, M. J. Chem. Soc., Perkin Trans. 1 (Org. Bioorg. Chem.) 1996, 377. b) Ichikawa, Y.; Kobayashi, C.; Isobe, M. Synlett 1994, 919 and references therein. c) de Brito, T. M. B.; da Silva, L. P.; Siqueira, V. L.; Srivastava, R. M. J. Carbohydr. Chem. 1999, 18, 609.