INTRODUZIONE
La Sindrome da Distress Respiratorio (Respiratory Distress sindrome - RDS), nota anche come Hyaline Membrane Disease (HMD), è una sindrome respiratoria dei nati prematuri caratterizzata prevalentemente da un’inadeguata ed insufficiente produzione di surfactante polmonare, correlata, almeno in parte, all’immaturità strutturale delle cellule epiteliali (pneumociti di II tipo) deputate alla sintesi del surfactante.
Lo scopo di questa tesi è quello di riportare i risultati di uno studio sperimentale eseguito su agnelli pretermine come modello sperimentale di RDS. Lo studio ha come obiettivo la valutazione dell’effetto della somministrazione di surfactante, addizionato con superossido dismutasi e catalasi, sullo stress ossidativo a carico polmonare nei nati prematuri affetti da RDS (nati dopo un periodo gestazionale di almeno 25 settimane), così da poter validare un eventuale protocollo terapeutico.
1 RESPIRATORY DISTRESS SYNDROME
1.1 Descrizione della patologia(1)La Sindrome da Distress Respiratorio è una delle principali cause di mortalità nei nati prematuri e rappresenta l’espressione clinica dell’immaturità dello sviluppo polmonare. Nei paesi sviluppati la RDS costituisce a tutt’oggi la principale causa di mortalità perinatale e si verifica quasi sempre nei neonati nati prima delle 37 settimane di gestazione.
Ad oggi la Sindrome da Distress Respiratorio Neonatale (NRDS) è riconosciuta come una sindrome respiratoria dei nati prematuri caratterizzata prevalentemente da un’inadeguata ed insufficiente produzione di surfactante polmonare, correlata, almeno in parte, all’immaturità strutturale delle cellule epiteliali (pneumociti di II tipo) deputate alla sintesi del surfactante.
È stato dimostrato che il rischio di sviluppare RDS aumenta al diminuire del peso alla nascita. L'incidenza di questa malattia è stimato al 90% per i neonati del peso di 750 gr alla nascita ed al 80% per i bambini del peso di 1000 gr(2)
.
L’incidenza e la gravità dell’insufficienza respiratoria risultano essere correlate anche all’età gestazionale (E.G.) (Fig.1)(3): con l’aumentare di questa si assiste infatti ad una
diminuzione lineare della mortalità dalle 26 alle 36 settimane di gestazione, con rari casi oltre le 37 settimane.
Fig. 1: Rapporto tra età gestazionale ed incidenza della RDS(3).
Se alla nascita non è stata raggiunta la piena maturazione del polmone, come avviene in caso di parti pretermine ed in altre forme di gravidanza a rischio, i neonati sviluppano frequentemente un’insufficienza respiratoria nelle prime ore di vita, caratteristica clinica della RDS, e necessitano di ventilazione meccanica.
La possibilità di una ventilazione adeguata dipende infatti dalla stabilità dell’alveolo, intesa come resistenza al collasso durante la fine espirazione, dalla maturità biochimica e morfologica del polmone.
Fisiopatologicamente, la RDS è caratterizzata da una perdita di volume polmonare causata dal collasso diffuso degli spazi aerei, da carenza di surfactante, e da uno sviluppo limitato dei capillari. Si viene così a creare un essudato composto da proteine del plasma, che fuoriescono attraverso il letto capillare danneggiato, e che forma le membrane jaline visibili al microscopio.
Agli inizi del secolo scorso fu descritta per la prima volta la cosiddetta “membrana jalina” in relazione ai decessi per insufficienza respiratoria. Successivamente Von Neergaard nel 1929 descrisse la presenza di un’interfaccia aria-liquido nei polmoni, sostenendo che la tensione superficiale alveolare al confine tra il tessuto umido del polmone e l'aria costituisse una forza che ostacola l’espansione polmonare al primo respiro del neonato.
La ricerca sull’immaturità polmonare però non ha avuto sostanziali progressi fino alle osservazioni prima di Pattle, nel 1955(4), che descrisse la presenza di una sostanza ad attività tensioattiva (surfactante) nella schiuma dell’edema polmonare e poi di Clements(5), nel 1957, che descrisse una sostanza analoga negli estratti polmonari. Furono tuttavia Avery e Mead(6) che nel 1959 correlarono l’insufficienza respiratoria tipica della RDS con le ridotte concentrazioni di surfactante nel liquido di lavaggio broncoalveolare di nati pretermine deceduti.
Una volta stabilita l’associazione fra atelettasia, membrane jaline e diminuita concentrazione di surfactante, la ricerca si è concentrata sullo studio dello sviluppo del “sistema surfactante”.
Un impulso in tal senso fu dato dal notevole finanziamento della ricerca da parte della famiglia Kennedy dopo il decesso del primogenito Patrick di John Fitzgerald Kennedy.
1.2 Eziopatogenesi della RDS
L’eziologia è legata alla presenza di una immaturità strutturale polmonare, caratterizzata da un incompleto sviluppo alveolare con un eccesso di matrice connettivale, associata alla carenza di surfactante, per un ritardo maturativo delle cellule deputate alla sua sintesi.
Nei bambini nati prematuri inoltre l’epitelio alveolare risulta più permeabile alle proteine plasmatiche, la dimensione degli alveoli è minore e si ha una maggior debolezza della parete toracica.
Frequentemente la RDS è complicata da altre patologie quali polmoniti, pervietà del dotto arterioso (PDA), raccolte aeree extra-alveolari, ipertensione polmonare persistente (PPH), emorragia polmonare, displasia broncopolmonare (BPD) ed emorragia intraventricolare (IVH).
La pervietà del dotto arterioso ed i processi infettivi possono rappresentare anche complicazioni della RDS stessa.
Nei nati prematuri si manifestano quindi con maggior facilità asfissia, ipotensione, ipoperfusione, ipotermia ed acidosi.
1.3 Incidenza e fisiopatologia della RDS(1)
Essendo difficile distinguere con certezza l’RDS rispetto ad altre forme, solitamente più lievi, di patologie respiratorie neonatali, non si conosce l’esatta incidenza di tale patologia in popolazioni non selezionate di neonati.
Recenti studi hanno dimostrato una diversa incidenza di RDS in vari Paesi, variabile dallo 0,3% dei nati vivi nei paesi scandinavi, allo 0,8% in Svizzera fino all’1% negli Stati Uniti.
Tra i fattori che sicuramente influiscono sul manifestarsi di questa patologia ricordiamo l’età gestazionale, come già precedentemente esplicitato(3, 8).
Altra variabile rilevante è costituita dal sesso. Diversi studi hanno infatti messo in evidenza che l’RDS si manifesta più frequentemente nei maschi (1.5-2 volte più spesso rispetto alle femmine), in conseguenza di fattori ormonali legati alla differenziazione sessuale. La differenziazione degli pneumociti di tipo II, cellule deputate alla sintesi e secrezione del surfactante polmonare, sarebbe infatti inibita dal testosterone, sia in vivo che in vitro, a causa delle sue proprietà anti-estrogeniche.
Inoltre è stato recentemente dimostrato che le più alte concentrazioni di MIS (Müllerian Inhibiting Substance), glicoproteina prodotta dalle cellule del Sertoli, presenti nei feti
maschi, inibirebbero la produzione polmonare di fosfatidilcolina che costituisce uno dei principali componenti del surfactante.
E’ stata inoltre confermata una predisposizione familiare: uno studio ha mostrato come, in caso di neonati di basso peso, l’incidenza della RDS sia del 90% nel caso in cui il precedente figlio di basso peso era stato anch’esso affetto da RDS e soltanto del 5% nel caso in cui, nella precedente gravidanza, non si era presentata tale sindrome. Sull’incidenza della RDS influiscono anche alcune complicazioni della gravidanza. Nelle gravidanze diabetiche sono state evidenziate significative differenze nel grado di maturazione del polmone fetale in relazione alle differenti classi di diabete gestazionale con un rischio globale di RDS sei volte maggiore rispetto alla popolazione normale. Si è notato infatti un ritardo della maturazione polmonare nel diabete leggero o moderato, mentre, nei casi di diabete più grave, lo sviluppo polmonare appariva accelerato, verosimilmente in relazione alla sofferenza cronica ed all’incremento della produzione endogena di corticosteroidi. Nonostante tali differenze quasi tutti gli autori concordano nel ritenere che nelle gravidanze diabetiche il rischio di RDS sia notevolmente superiore alla norma. Tale maggior rischio è da mettere in relazione all’iperinsulinemia fetale, avendo l’insulina un ruolo antagonista sulla sintesi dei fosfolipidi del surfactante, in particolare della lecitina.
Un altro fattore di rischio per l’RDS è rappresentato dall’espletamento del parto tramite intervento cesareo, effettuato prima del termine di gravidanza. Ciò sembra dovuto alla mancata increzione di catecolamine fetali (in particolare l’adrenalina), come avviene invece normalmente durante il travaglio di parto senza anestesia. E’ noto che essa svolge un ruolo chiave nella diffusione del surfactante polmonare negli alveoli(9,10). La gravidanza gemellare è stata indicata come un fattore protettivo per l’insorgenza della NRDS. Tuttavia la gemellarità di per sé non rappresenta un fattore che accelera la maturazione polmonare fetale, a meno che non siano presenti alcune complicazioni ostetriche, quali l’ipertensione e/o la rottura prematura delle membrane che,
determinando sofferenza fetale, accelerano la maturazione polmonare fetale nella gravidanza gemellare così come nella gravidanza singola.
Un effetto analogo è rilevabile in tutte quelle condizioni che possono determinare un distress fetale cronico, come l’ipertensione indotta dalla gravidanza(11)
.
È stata inoltre dimostrata una base genetica legata alla possibilità di sviluppo della RDS. Le varianti alleliche dei geni che codificano per le quattro proteine del surfactante sono state infatti associate con l’RDS, in particolare per quanto riguarda la SP-A e la SP-B. Le mutazioni genetiche dei geni codificanti per queste proteine sono state correlate con una maggiore suscettibilità allo sviluppo della Sindrome da Distress Respiratorio. I fattori di suscettibilità genetica dipendono dal grado di prematurità alla nascita, coerentemente con lo sviluppo sequenziale del polmone e le differenze nella presentazione clinica, legata alla gestazione(12,13).
A partire dal 1959, a seguito degli studi di Avery e Mead(6), il concetto di base della fisiopatologia dell’RDS risulta essere la “mancanza del fattore tensioattivo polmonare”, cioè del surfactante (surface active substance). Tale fattore riduce la tensione superficiale (propria dell’interfaccia aria-ipofase, liquido che bagna le pareti alveolari), regolata dalla legge di Laplace (Fig. 2), che è alla base della meccanica respiratoria. Avvicinandosi ad una tensione superficiale pari a 0 dyne/cm, il surfactante evita la tendenza al collasso degli alveoli durante l’espirazione, cosicché è necessaria una pressione minore per mantenere gli alveoli distesi.
La legge di Laplace afferma che P = 2T/r con
P = pressione,
T = tensione superficiale,
r = raggio.
Secondo tale legge, la pressione risulta essere inversamente proporzionale alla dimensione degli alveoli.
Il primo respiro del neonato richiede normalmente un notevole sforzo inspiratorio, legato alla notevole pressione di apertura delle vie aeree. Con livelli normali di surfactante, che riduce la tensione superficiale, sono favoriti l’espansione polmonare iniziale ed il reclutamento sequenziale delle piccole vie aeree (reclutamento alveolare). È altresì impedito il collasso degli alveoli a fine espirazione. Nei polmoni viene infatti mantenuto più del 40% del volume di aria residuo dopo il primo atto respiratorio; da ciò deriva che la pressione inspiratoria necessaria per gli atti respiratori successivi è inferiore.
Se il polmone presenta un deficit di surfactante ciò non avviene poiché gli alveoli collassano ad ogni atto respiratorio, forzando il neonato a compiere per ogni atto respiratorio un lavoro maggiore, pari a quello compiuto durante il primo respiro.
L’atelettasia costituisce infatti la principale manifestazione anatomo-patologica della RDS. Gli alveoli polmonari, ancora immaturi, collabiscono a fine espirazione, ritornando ad uno stato simil-fetale, in cui gli spazi aerei sono ripieni di liquido polmonare fetale (FPL) e non rimangono areati, come accade nei neonati maturi. È richiesta inoltre una maggior pressione di apertura delle piccole vie aeree durante l’insufflazione, soprattutto per il loro reclutamento(7).
La ridotta compliance polmonare e la progressiva atelettasia, con aree polmonari perfuse ma non ventilate, portano ad una alterazione del rapporto ventilazione/perfusione con ipoventilazione alveolare e conseguente ipossiemia ed ipercapnia. Si instaura quindi uno stato di acidosi respiratoria. L’ipossiemia e l’acidosi possono compromettere ulteriormente l’ossigenazione causando vasocostrizione polmonare con ipoperfusione polmonare, shunt destro-sinistro a livello del forame ovale e del dotto arterioso e danno endoteliale dei capillari. Ne deriva un’essudazione ricca di proteine e di fibrina negli spazi alveolari con formazione delle membrane jaline. Tali membrane sono costituite da cellule epiteliali degenerate, cellule ematiche, fibrina e componenti dello strato di rivestimento alveolare(14). Le membrane jaline
rappresentano così una barriera allo scambio gassoso, aggravando l’ipossiemia, l’ipercapnia, quindi lo stato di acidosi e gli eventi successivi.
Negli ultimi decenni, numerosi studi hanno confermato che i polmoni di neonati con RDS sono caratterizzati da un’elevata tensione superficiale e tendono perciò a collabire; le curve pressione-volume nei polmoni di neonati con RDS presentano, infatti, quadri anormali, in quanto sono necessarie elevate pressioni di apertura per distendere e reclutare gli alveoli mentre la capacità di mantenere il volume residuo a fine espirazione è ridotta.
Nei neonati inoltre la minor rigidità della parete toracica fa si che le forze che si oppongono al collasso alveolare siano di minore entità, rendendo ancor più di fondamentale importanza la necessità della riduzione della tensione superficiale a livello alveolare.
In definitiva, la fisiopatologia della RDS è caratterizzata da alterazioni della meccanica polmonare determinate dal deficit di surfactante, quali una ridotta compliance e capacità funzionale residua (CFR) con instabilità alveolare e tendenza degli alveoli più piccoli a collassare, uno shunt D-S intra ed extrapolmonare secondario alla formazione di atelettasie, all’ipossia ed all’acidosi.
Il lavoro respiratorio inoltre è aggravato dalla riduzione del volume corrente per l’ipoespansione polmonare e dall’aumento relativo dello spazio morto.
1.4 Sintomi, quadro radiologico e complicanze(15,16)
I segni di RDS appaiono subito dopo la nascita o entro 4 ore. La sintomatologia è rappresentata da tachipnea (FR >60 atti/minuto con Ti spontaneo di 0,25-0,35”)(17)
, gemito espiratorio, agitamento delle pinne nasali, retrazioni intercostali e subcostali e cianosi; la tachipnea può alternarsi a bradipnea o ad episodi di apnea.
La tachipnea è dovuta al tentativo di aumentare la ventilazione minuto per compensare la diminuzione del volume tidalico e l’aumento dello spazio morto. Le retrazioni si
per espandere i polmoni, a causa della scarsa compliance. Gli atti respiratori risultano essere quindi frequenti, difficoltosi e rumorosi.
L'entità dell'atelettasia e la severità dell'insufficienza respiratoria tendono a peggiorare con il tempo. Nelle RDS gravi si verifica un esaurimento del diaframma e dei muscoli intercostali con ritenzione di CO2 e conseguente acidosi respiratoria. Si viene da ultimo
a creare anche uno stato di acidosi metabolica in quanto il sangue che perfonde la porzione atelettasica del polmone non viene ossigenato ed il bambino diventa ipossiemico.
Il polso periferico può presentarsi debole, con edema delle estremità e ridotta produzione di urina.
I neonati con peso molto basso alla nascita (< 1000 g) possono non presentare i segni della sindrome respiratoria, e addirittura non essere in grado di dare inizio all'attività respiratoria già in sala parto, data la rigidità dei loro polmoni(16).
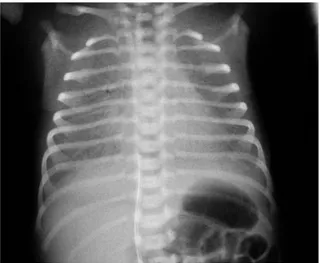
Il tipico quadro radiologico del torace mostra una ridotta espansione polmonare ed un modello reticolare granulare bilaterale, in rapporto a fenomeni di atelettasia alveolare, con sovrapposto broncogramma aereo per l’aria presente nelle vie aeree. Nei casi più gravi, vi è un completo "sbiancamento" dei campi polmonari, con un’opacità così marcata da nascondere la silhouette cardiaca(15).
Fig. 3: Radiografia del torace di un neonato con RDS: Polmoni scarsamente areati con il modello reticolo-nodulare “a vetro smerigliato”.
Il prematuro con RDS è ad alto rischio di emorragia intraventricolare (IVH), con sanguinamento all'interno degli spazi ventricolari del SNC e conseguente morte neonatale, nonché di lesioni della sostanza bianca periventricolare. Le complicazioni intracraniche sono state associate ad ipossiemia, ipercapnia, ipotensione, oscillazioni nella pressione arteriosa e bassa perfusione cerebrale(16).
1.5 Prognosi della RDS(16)
Il rischio dello sviluppo della RDS può essere stimato nel periodo prenatale mediante la valutazione della maturità polmonare fetale, così da poter determinare il momento ottimale per espletare il parto. Viene misurata la quantità di surfactante presente nel liquido amniotico, ottenuto tramite amniocentesi o raccolto dalla vagina (se le membrane sono rotte).
La maturazione polmonare può essere considerata buona, e di conseguenza il rischio di RDS basso, quando il rapporto lecitina/sfingomielina è >2 ed è presente fosfatidilglicerolo.
Se la RDS viene adeguatamente trattata la prognosi è eccellente, con mortalità <10%. Anche soltanto con un adeguato supporto ventilatorio, la produzione di surfactante inizierà e una volta che la produzione comincia si ha risoluzione della RDS entro 4 o 5 giorni. Tuttavia, se non si interviene, l’ipossiemia grave può causare un’insufficienza d’organo multipla e morte del neonato.
2 ANATOMIA E FISIOLOGIA FETALE E NEONATALE
2.1 Circolazione fetale(16)La circolazione fetale è caratterizzata da resistenze arteriolari polmonari molto elevate e da un flusso ematico polmonare ridotto (soltanto il 5-10% della gittata cardiaca); al contrario, la resistenza al flusso ematico nella circolazione sistemica, compresa quella placentare, è bassa.
Sono presenti inoltre due shunts destra-sinistra. Uno è costituito dal dotto arterioso che collega l'arteria polmonare all'aorta e l’altro dal forame ovale che collega l’atrio destro col sinistro.
Circa il 90-95% del sangue proveniente dal cuore destro bypassa i polmoni e entra direttamente nella circolazione sistemica. Il dotto arterioso fetale è mantenuto pervio dalla presenza di una bassa PaO2 sistemica fetale (circa 25 mmHg), accompagnata da
una produzione locale di prostaglandine.
La pressione atriale sinistra è relativamente bassa, dato il ridotto ritorno ematico polmonare, mentre la pressione atriale destra è relativamente elevata a causa dei grandi volumi di ritorno sanguigno dalla placenta; la differenza di pressione nei due atri, fa sì che il forame ovale rimanga pervio.
Dopo i primi respiri si assiste ad un profondo cambiamento di questo sistema, che esita nell’aumento del flusso sanguigno polmonare e nella chiusura del forame ovale. Le resistenze polmonari arteriolari diminuiscono improvvisamente come conseguenza della vasodilatazione causata dall’espansione polmonare, dall’aumento della PaO2 e
riduzione della PaCO2.
Le forze elastiche delle coste e della parete toracica riducono, inoltre, la pressione interstiziale polmonare, migliorando ulteriormente il flusso di sangue attraverso i capillari polmonari. Si ha quindi un aumento del ritorno venoso dai polmoni e, di conseguenza, aumenta anche la pressione a livello atriale sinistro, riducendo così il differenziale di pressione tra i due atri, effetto che contribuisce alla chiusura del forame ovale.
Con la respirazione si ha un aumento della PaO2 che porta al restringimento delle
arterie ombelicali. Il flusso ematico placentare si riduce o si arresta del tutto, con minor ritorno sanguigno all'atrio destro. Si verifica così una diminuzione della pressione atriale destra con contemporaneo aumento di quella atriale sinistra, che porta alla definitiva chiusura del forame ovale.
Alla nascita si ha quindi un’inversione della situazione fetale, con le resistenze sistemiche che superano quelle polmonari. Si viene a verificare, pertanto, uno shunt ematico sinistro-destro, chiamato circolazione transitoria, con un’inversione della direzione del flusso sanguigno attraverso il dotto arterioso. Questo stato può durare da poco dopo la nascita (quando si verifica l'aumento del flusso ematico polmonare e la chiusura funzionale del forame ovale) fino a circa 24-72 ore di vita, quando di norma il dotto arterioso dovrebbe chiudersi.
La costrizione e chiusura del dotto arterioso avviene per due motivi principali: per l’elevata pO2 del sangue proveniente dall'aorta che entra nel dotto arterioso e nei suoi vasa vasorum e per le modificazioni che si verificano nel metabolismo delle prostaglandine.
Una volta che il dotto arterioso si chiude, si instaura una circolazione di tipo adulto. I due ventricoli ora pompano in serie e non esistono shunt tra le circolazioni polmonare e sistemica.
In caso di distress neonatale, nei giorni seguenti la nascita, si può osservare un ritorno alla circolazione di tipo fetale. L'asfissia infatti, accompagnata da ipossia e ipercapnia, provoca la vasocostrizione delle arteriole polmonari e porta alla dilatazione del dotto arterioso, invertendo i processi precedentemente descritti, con conseguente shunt destro-sinistro attraverso il dotto arterioso ormai pervio e/o il forame ovale riaperto od entrambi. Di conseguenza il neonato presenterà la cosiddetta ipertensione polmonare persistente o circolazione fetale persistente, caratterizzata da grave ipossiemia.
2.2 Polmone(18)
Nonostante lo sviluppo polmonare abbia inizio durante la gestazione, a partire dalle 6 settimane, intorno alle 25 settimane di età gestazionale si ritrova una elevato numero di alveoli ben sviluppati.
Lo sviluppo del polmone prosegue fino al secondo anno di vita. A carico degli alveoli polmonari si susseguono due processi principali: la formazione delle unità di scambio gassoso e la loro dilatazione. Si ha una deposizione di collagene ed elastina ed una riorganizzazione dei capillari, che portano alla definitiva maturazione alveolare.
Durante lo sviluppo intrauterino, nei polmoni si ha una produzione continua del cosiddetto liquido polmonare fetale (FPL), trasudato derivante dai capillari polmonari, che contiene anche il surfactante prodotto e secreto dagli pneumociti di tipo II. La produzione di liquido polmonare, fondamentale nello sviluppo dell’organo, è fortemente legata a quella di liquido amniotico, tanto che condizioni che interferiscono con la normale produzione di liquido amniotico, quali ad esempio la compressione uterina da oligoidramnios, la presenza di alcuni tumori o l’occlusione di un’arteria polmonare, alterano gravemente lo sviluppo polmonare(19).
Affinché si possa avere un corretto scambio di gas alla nascita, il liquido polmonare fetale deve essere adeguatamente rimosso tramite la compressione del torace del feto durante il parto e l’assorbimento dello stesso da parte delle cellule polmonari. Nel caso la clearance di questo liquido sia ritardata si può avere una tachipnea transitoria nel neonato.
Per quanto riguarda la maturazione polmonare del feto umano intorno alla 20a settimana di gestazione avviene la differenziazione dell’epitelio polmonare in pneumociti di tipo I e II ed intorno alla 24a settimana di gestazione si ha la formazione dell’unità alveolo-capillare. A partire dalla 20a
settimana, fino circa alla 36a, abbiamo la maturazione del sistema enzimatico per la sintesi del surfactante.
Lo sviluppo polmonare può essere inoltre descritto attraverso 4 stadi:
- stadio embrionale, con comparsa della gemma polmonare nel ventiseiesimo giorno di gestazione, come estroflessione dell’intestino primitivo;
- stadio pseudo-ghiandolare, dalla sesta alla diciassettesima settimana di gestazione, si assiste alla suddivisione dei bronchi, fino alla formazione dei bronchioli terminali e delle primitive strutture acinari;
- stadio canalicolare, dalla diciassettesima alla ventiseiesima settimana: si sviluppano quindi le vere e proprie unità funzionali polmonari, gli acini, che vengono invasi da capillari polmonari;
- stadio alveolare o sacculare, a partire dalla venticinquesima settimana, con una spiccata dilatazione delle vie aeree terminali, con notevole incremento del volume polmonare e della superficie di scambio, con un ulteriore assottigliamento della barriera alveolo-capillare. Si ha la maturazione degli pneumociti di tipo II, deputati alla produzione del surfattante, a partire da cellule totipotenti.
2.3 Movimenti respiratori fetali (FBM)(1)
Fisiologicamente, in utero, il feto può presentare episodi di respirazione spontanea alternati a momenti di apnea. I movimenti respiratori sono rapidi, con una frequenza di circa 40-70 movimenti/minuto, intermittenti e per lo più irregolari e superficiali, costituendo circa il 90% di tutta l’attività respiratoria fetale. Non si hanno ancora però informazioni precise riguardo l’inizio e il mantenimento di tali movimenti respiratori. La loro presentazione ciclica è associabile ai cambiamenti dello stato di sonno del feto, osservandosi soprattutto durante la fase del sonno non-REM. Esistendo, più in particolare, una correlazione tra i FBM e lo stato comportamentale del feto, è plausibile pensare che essi possano, in associazione con i movimenti corporei, condizionare non soltanto lo sviluppo e la crescita del polmone, ma possibilmente anche lo sviluppo del cervello fetale.
E’ stato inoltre dimostrato un altro “pattern” di movimenti respiratori, conosciuto con il nome di «gasp». Questi sono meno frequenti dei precedenti (1-4 mov/min) e si presentano periodicamente come sforzi inspiratori profondi o come sforzi espiratori o
È utile ricordare che il feto occasionalmente può tossire, mostrando brevi sforzi espiratori e può avere accessi di respirazione ad alta frequenza in risposta ad ipertermia.
A partire dal 1970 i movimenti respiratori del feto umano sono stai approfonditi mediante l’utilizzo di tecniche ultrasonografiche, dell’esame bidimensionale in “real-time” e successivamente del color Doppler.
Normalmente si è osservato che il liquido polmonare fetale viene riversato nella cavità amniotica in quantità maggiore durante i FBM rispetto ai periodi di apnea. Tale fenomeno non è stato osservato nel feto umano fino al 1985, quando Chiba et al.(20), per primi, elaborarono un metodo ultrasonografico per lo studio del flusso fetale intratracheale mentre lo scambio di fluido amniotico associato con i FBM era invece già stato ampiamente dimostrato in feti di pecora.
Nel 1993 Badalian et al.(21) hanno dimostrato inoltre come l’attività respiratoria fetale possa essere determinata attraverso la valutazione dei movimenti respiratori fetali correlati alla velocità del flusso del fluido nasale evidenziato mediante color Doppler. Questo studio ha permesso di confermare che i movimenti respiratori fetali favoriscono, a loro volta, un movimento di liquido tra la cavità amniotica e le vie aeree respiratorie fetali.
Sono vari i fattori che possono influenzare i FBM, fra questi possiamo riconoscere: travaglio di parto: in diversi studi è stato riportato un significativo decremento
degli FBM durante il travaglio del parto spontaneo, a seguito sia della liberazione di prostaglandine, sia della graduale ipossemia fetale durante le contrazioni uterine;
età gestazionale: i movimenti respiratori fetali sono osservabili a partire dal secondo trimestre di gravidanza. In particolare l’intervallo respiro-respiro e la durata della fase inspiratoria durante i movimenti respiratori fetali aumentano tra la 22a e la 35a settimana, per poi decrescere successivamente;
stato acido-base, pH, ed ipossia: il feto risponde a stati di ipossia all’opposto di quanto si verifica nell’adulto. Riducendo infatti l’ossigenazione materna, fino ad ottenere un notevole decremento della pO2 fetale (a valori di 10-12 mmHg), si
verifica una diminuzione dei FBM, come dei movimenti del corpo e degli arti fetali e dell’attività REM. In caso di ipercapnia, un aumento della pCO2 materna
è stato correlato ad un incremento dei FBM sia nei feti di pecora che in quelli umani. In condizioni di asfissia, dati ad esempio da una prolungata occlusione del cordone ombelicale, si ha inizialmente una cessazione dei FBM quindi si hanno “gasps” fetali, consistenti in sforzi inspiratori, isolati, profondi e prolungati. Stati di acidosi, sia respiratoria che metabolica;
ormoni: un incremento dei FBM si è osservato in seguito alla somministrazione di estrogeni coniugati alla madre, così come di betametasone o di desametasone;
aminofillina: sfruttando la sua capacità di stimolare i centri respiratori bulbari è stata utilizzata nella prevenzione dell’apnea della prematurità. Cosmi et al.(125)
hanno visto che la somministrazione per via endovenosa di aminofillina alla madre è seguita dalla comparsa di movimenti della parete toracica ed addominale fetale, accompagnati da un flusso di liquido a livello delle narici fetali e formazione di veri e propri vortici;
fumo di sigaretta ed alcool, con azione inibente i FBM.
2.4 Fisiopatologia del sistema respiratorio del neonato(18)
La compliance polmonare ridotta, le maggiori resistenze delle vie aeree e la compliance della gabbia toracica aumentata, rappresentano degli svantaggi per il lavoro respiratorio del neonato, in particolare se pretermine.
1. Ridotta compliance polmonare: essendo il polmone un tessuto elastico, è necessario prendere in considerazione i parametri di elastanza e compliance.
L’elastanza è una misura di funzionalità polmonare che si definisce come il risultato delle forze di retrazione elastica del polmone e del torace, esprime la tendenza di una struttura a ritornare allo stato di partenza a seguito dell’applicazione di una forza che ne abbia determinato una deformazione rispetto alle condizioni iniziali. La compliance è invece indice della distensibilità o cedevolezza di una struttura, indicata come variazione di volume che si crea in seguito ad una variazione di pressione (ΔV/ΔP), e può quindi essere considerata l’inverso dell’elastanza.
Il polmone dei neonati prematuri presenta una ridotta compliance polmonare poiché, data la scarsa produzione di surfactante, si ha un aumento della tensione superficiale polmonare e una riduzione della capacità funzionale residua (CFR), chiamata anche volume polmonare di fine espirazione.
Inoltre nel nato prematuro, soprattutto in seguito a parto cesareo, si ha una clearance del liquido polmonare più lenta che nel nato a termine, con accumulo di questo negli alveoli, aspetto che costituisce un ostacolo alla normale espansione polmonare.
2. Maggiori resistenze delle vie aeree: per permettere una corretta ventilazione del sistema respiratorio devono essere vinte, oltre alla forza di retrazione elastica della gabbia toracica, anche le resistenze opposte dalle vie aeree al flusso d’aria. Nel sistema respiratorio circa il 30-40% delle resistenze è localizzato a livello del naso, dell’oro-faringe e del faringe, mentre a livello dell’albero tracheobronchiale le strutture che offrono la maggiore resistenza al flusso sono i bronchi di media grandezza e non le vie aeree di piccolo calibro, poiché nonostante singolarmente siano più piccole, sono in numero maggiore e quindi la loro sezione complessiva risulta più ampia.
Nel neonato prematuro, peraltro, non è presente la fascia di tessuto superficiale adiposo del collo, che di norma tende a stabilizzare e quindi a mantenere pervie le prime vie aeree(22,23)
,
ed inoltre il muscolo genioglosso, che ha la funzione di stabilizzare il faringe durante la fase inspiratoria, presenta una ridotta attività(24).3. Aumentata compliance della gabbia toracica: nell’adulto, il rapporto esistente fra la compliance della gabbia toracica e quella del polmone è di 1:1. Nel neonato a termine è circa 3:1, mentre nel nato prematuro è circa 5:1. La compliance della gabbia toracica è quindi inversamente proporzionale all’età gestazionale (E.G.).
L’aumentata compliance della gabbia toracica nel neonato prematuro costituisce senza dubbio una delle cause della bassa capacità funzionale residua del polmone e può contribuire allo sviluppo di un’insufficienza polmonare cronica.
La gabbia toracica del neonato prematuro è infatti più cedevole e flessibile, non riuscendo a mantenere un’adeguata pressione transpolmonare a fine espirazione. In conseguenza di ciò abbiamo il collasso alveolare, accompagnato da uno squilibrio nel rapporto ventilazione/perfusione, con conseguente aumento del gradiente di O2
alveolo-arterioso e della concentrazione di CO2, che porta all’ipossiemia del soggetto.
La riduzione dell’ossigenazione, inoltre, altera la funzione dei muscoli respiratori. Si ha quindi una limitazione all’espansione toracica, dovuta anche all’andamento orizzontale delle coste ed al diaframma che si presenta più piatto.
Il neonato prematuro, affetto da RDS, può mettere in atto alcuni meccanismi per mantenere un’adeguata CFR. Uno di questi consiste nel mantenere parzialmente attivi i muscoli inspiratori anche durante l’inizio dell’espirazione, o iniziare la fase inspiratoria prima del termine della fase espiratoria precedente. Anche la chiusura della glottide, con restringimento laringeo, in espirazione sembra avere un ruolo in tal senso, per aumentare la capacità funzionale residua del polmone.
Questi meccanismi prendono il nome di aumento dinamico della CFR e sono messi in atto per facilitare lo scambio gassoso, anche se risultano svantaggiosi per quanto riguarda il dispendio energetico(25). In caso di affaticamento o momenti di apnea nel neonato prematuro, si assiste all’aumento delle aree di atelettasia polmonare, con conseguente insufficienza respiratoria(26).
Il collasso polmonare comporta un danno epiteliale con formazione di essudato a base proteica e consumo di surfactante.
2.5 Equilibrio Acido-Base(27)
L’equilibrio acido-base è mantenuto attraverso una varietà di processi fisiologici e la sua regolazione deriva dall’integrazione di differenti sistemi di controllo.
Il normale metabolismo esita nella produzione di acidi: un acido è un donatore di protoni o ioni idrogeno (H+), mentre una base è un accettore di protoni o ioni idrogeno. Il termine pH esprime il logaritmo negativo della concentrazione di H+ liberi. Il rapporto è inversamente proporzionale, in quanto all’aumentare della concentrazione di H+
liberi il pH diminuisce, e viceversa. Per mantenere il pH entro il suo normale intervallo di neutralità (7,35-7,45), gli acidi devono essere tamponati od escreti.
Il principale meccanismo di regolazione della concentrazione di H+ è il sistema tampone, distinto in sistema tampone extracellulare ed intracellulare. Quando la capacità di tali complessi regolatori viene superata, e non può essere assicurata una corretta omeostasi, il polmone ed il rene intervengono attraverso un’attività di compensazione.
2.6 I sistemi tampone
Le sostanze che costituiscono i diversi sistemi tampone, hanno la caratteristica, se presenti in soluzione, di far aumentare la quantità di acidi o di basi che devono essere aggiunti alla soluzione stessa per modificarne il pH.
- Sistema tampone extracellulare: la maggiore quantità di acido che ritroviamo nel corpo è in forma di acido carbonico (H2CO3), che è un acido volatile. L’enzima anidrasi
carbonica catalizza le seguenti reazioni: CO2 + H2O < - > H2CO3 < - > HCO3- + H+
Questo sistema tampone è quindi costituito dalla coppia acido carbonico-bicarbonato (H2CO3- HCO3-). Il primo è un acido debole e si attiva in caso di alterazioni del pH
dovute ad una base forte, il bicarbonato è invece una base debole e si attiva in caso di acidi forti. Ogni volta che si attiva il sistema tampone, la concentrazione di un membro della coppia aumenta mentre l'altra diminuisce.
Tale sistema è il più efficace e rapido dell’organismo ed è presente anche durante la vita fetale, quando il compito di rimuovere la CO2, prodotta dalla reazione, è affidata
alla placenta.
Il sistema tampone extracellulare è il più diffuso e rappresenta il 65% dei sistemi tampone, lo si ritrova nel fluido interstiziale, nel plasma, negli eritrociti, nelle cellule e nelle ossa. E’ inoltre l’unico sistema che presenta due vie di eliminazione: una rapida (entro pochi minuti) attraverso i polmoni, tramite la CO2, ed una lenta (in alcune ore o
giorni), tramite il bicarbonato e gli idrogenioni che vengono eliminati per via renale. - Sistema tampone intracellulare: è costituito principalmente dall’emoglobina e dall’ossiemoglobina, e secondariamente dalle proteine e dai fosfati. Le proteine, in particolare, essendo composte da aminoacidi, contengono almeno un gruppo amminico ed uno carbossilico. Il gruppo carbossilico tende a funzionare come un acido, mentre quello amminico tende ad agire come una base. Le proteine possono quindi comportarsi come tamponi sia per gli acidi che per le basi.
I fosfati, che agiscono ugualmente alla coppia tampone acido carbonico-bicarbonato, costituiscono un importante sistema regolatore del pH, sia negli eritrociti che a livello tubulare renale.
Il sistema tampone intracellulare ha una risposta più lenta rispetto a quello extracellulare, tanto che richiede alcune ore per raggiungere la sua massima capacità. Nonostante ciò, è attivo già durante la vita fetale, ma la sua capacità tamponante migliora dopo la nascita, grazie alla progressiva espansione del compartimento extracellulare.
2.7 I meccanismi di compenso
- Polmone: la ventilazione assume un ruolo importante nel mantenimento di un corretto pH. Il sistema respiratorio è infatti in grado di modificare il pH, entro 1-3 minuti, eliminando o conservando CO2, in modo più rapido ed efficiente rispetto a tutti gli altri
sistemi tampone, anche combinati fra loro.
Come già discusso, quando un acido forte è presente nel corpo, la coppia bicarbonato-acido carbonico viene attivata per tamponare l'bicarbonato-acido. Questo si traduce in un incremento netto di acido carbonico, che è rapidamente dissociato in CO2 e H2O.
L’eccesso di anidride carbonica viene poi eliminato attraverso i polmoni.
Un aumento della concentrazione di H+ nel sangue, od una diminuzione di HCO3-,
sollecita i chemorecettori dell’arco aortico e del glomo carotideo e stimola il centro respiratorio midollare, portando ad un aumento della frequenza respiratoria, con maggior eliminazione di CO2
.
Se, invece, il pH è elevato, secondariamente ad un aumento del bicarbonato, il centro respiratorio è inibito e la frequenza respiratoria diminuisce. Ciò esita in una ritenzione di CO2 che diventa quindi disponibile per formare acido carbonico, tamponando
l’eccesso di bicarbonato.
Il sistema respiratorio, tuttavia, non può agire sugli ioni idrogeno, ma esclusivamente sulla CO2
.
La compensazione respiratoria, nonostante riesca ad attivarsi in pochi minuti, è di solito completamente funzionale entro 1-2 giorni.
- Rene: il controllo renale dell'equilibrio acido-base sfrutta diversi processi di trasporto attivo altamente sviluppati. La compensazione renale, però, è un processo abbastanza lento. È necessario fino ad un giorno per la sua completa attivazione in caso di alcalosi respiratoria, ed anche di più in corso di acidosi.
I reni reagiscono alle variazioni di pH regolando l'escrezione o la conservazione di H+ e HCO3-
.
Situazioni di acidosi, con basso pH, stimolano l’escrezione di H+
nelle urine. Una volta nelle urine lo ione idrogeno spiazza un altro ione positivo, generalmente Na+. Allo stesso tempo viene riassorbito bicarbonato (HCO3-) in cambio degli H+. lo ione sodio
viene poi riassorbito nelle cellule del tubulo dove si combina con HCO3- per formare
NaHCO3 che va a tamponare gli H+ nel sangue. Il tasso dell’escrezione degli H+, e
quindi il tasso di riassorbimento di HCO3-, è proporzionato alla pCO2 arteriosa.
Data l’escrezione di H+ l’urina tende ad avere pH acido, con valori intorno a 6.
In ambito clinico, il controllo del pH può essere un indicatore utile del grado di compensazione renale, nel valutare lo stato acido-base.
Tutte le reazione precedentemente descritte saranno invertite in caso invece aumenti il pH.
I sistemi che regolano e mantengono l'equilibrio acido-base diventano pienamente efficienti in diversi periodi di sviluppo. I sistemi tampone sono già funzionali in utero. Il controllo respiratorio del sistema di equilibrio acido-base è maturo nei neonati, purché la loro funzione polmonare sia adeguata. Infatti qualsiasi disturbo respiratorio, neuromuscolare, o neurologico che altera l’eliminazione della CO2 può risultare in
un’alterazione dell’equilibrio acido-base.
Il sistema di compensazione renale non è pienamente funzionale alla nascita. I neonati hanno infatti una limitata capacità di espellere gli ioni idrogeno, ma questa diventa pienamente efficiente entro i 2 mesi di età, sia nei neonati a termine che in quelli pretermine.
2.8 Alterazioni dell’equilibrio acido-base
I sistemi tampone di compenso, sia respiratorio che renale, necessitano, come abbiamo visto, di tempi specifici per ripristinare l'equilibrio acido-base. Il significato clinico dei disturbi dello stato acido-base, nonché i segni ed i sintomi di questi, sono
direttamente correlati alla velocità con cui si verificano le variazioni di pH. Processi cronici permettono all’organismo di mettere in atto tutti i meccanismi di compenso e sono quindi accompagnati di minime variazioni di pH. Insulti rapidi e progressivi esitano, invece, in profonde alterazioni nel pH, che posso risultare fatali se non si interviene in modo pronto ed efficace.
Le alterazioni della rimozione polmonare di anidride carbonica, in difetto od in eccesso, prendono il nome di acidosi e alcalosi respiratoria. Tutte le circostanze in cui si abbia un innalzamento od un abbassamento del pH ematico a seguito di disordini di altro tipo costituiscono l’acidosi e l’alcalosi metabolica
.
L’emogasanalisi su sangue arterioso è lo strumento diagnostico più utile nel determinare gli squilibri acido-base nel contesto clinico. In tabella sono riportati i valori fisiologici di alcuni parametri:
PARAMETRI
RANGE
pH
7.35-7.45
pO
2100 mmHg
SpO
298-100%
pCO
235-45 mmHg
HCO
3-22-26 mmol/L
BE
+2 -2 mEq/L
SpO2: è la saturazione dell’ossigeno periferico e corrisponde alla saturazione
d’ossigeno dell’emoglobina. Se la perfusione tissutale è adeguata, il valore di SpO2 è
approssimativamente simile a quello di saturazione dell’emoglobina arteriosa (SaO2).
BE: è l’eccesso di basi (o Base Excess). Esso quantifica la presenza di basi nel sangue (per lo più HCO3-), viene misurato in mEq/L e normalmente assume valore pari
a zero. Le modificazioni delle basi ematiche sono dette “eccesso” o “deficit di basi”. L'eccesso di basi può essere utilizzato per calcolare la quantità di trattamento (neutralizzazione) richiesta per contrastare l'acidosi (o l'alcalosi) metabolica.
I soggetti affetti da RDS, a seguito della difficoltà respiratoria, saranno soggetti più frequentemente a stati di acidosi respiratoria, condizione caratterizzata da un eccesso di acido carbonico nel liquido extracellulare, conseguente all’ipoventilazione ed alla ritenzione di CO2. Questa potrà essere risolta, almeno in parte, assistendo il paziente
con la ventilazione artificiale.
Può instaurarsi anche uno stato di acidosi metabolica che riconosce come causa più comune nella popolazione pediatrica un’insufficiente perfusione tissutale. Lo stato acidosico è caratterizzato da un deficit di basi (determinabile attraverso il parametro BE dell’emogasanalisi) che può essere trattato con la somministrazione di bicarbonato, seguendo la formula:
Dose (mEq) = 0.3 x peso(kg) x BE (mEq/L)
Ciò che si ottiene dalla formula è la quantità di bicarbonato necessario a portare a zero il valore di BE. Di solito non viene somministrata la dose totale, ma o la metà, oppure una limitata dose standard, valutando poi le modificazioni cliniche nel tempo. Nei neonati, infatti, una rapida infusione di bicarbonato può essere causa di emorragia intracranica. Inoltre il bicarbonato è accompagnato dagli ioni sodio, che portano ad un aumento dell'osmolarità del liquido extracellulare, che in combinazione con altre terapie, come glucosio endovenoso, può diventare critica e causare coma.
Una buona parte del bicarbonato iniettato viene per di più convertita in anidride carbonica ed eliminata(28).
3 LO STRESS OSSIDATIVO
3.1 Fisiopatologia dello stress ossidativo(29)
Il termine stress ossidativo indica l’insieme delle alterazioni che si manifestano a livello di tessuti, cellule e macromolecole biologiche quando queste vengono esposte ad un eccesso di agenti ossidanti. Identifica, quindi, una modificazione del normale equilibrio intracellulare, esistente tra sostanze ossidanti, prodotte fisiologicamente dalle cellule durante i processi metabolici, ed efficienza dei sistemi di difesa antiossidanti.
Quando le sostanze ossidanti, tra cui le specie reattive dell’ossigeno (ROS), prevalgono e/o le sostanze antiossidanti si riducono, si instaura una condizione di stress ossidativo.
I vari tessuti presentano una diversa suscettibilità a questo fenomeno; il sistema nervoso centrale è estremamente sensibile a questo tipo di danno, a causa del basso livello di enzimi antiossidanti, dell’elevato contenuto di substrati ossidabili e della gran quantità di ROS prodotte durante le reazioni neurochimiche.
Nel comparto intracellulare è presente, di norma, un ambiente riducente che viene preservato da enzimi che mantengono lo stato ridotto attraverso un costante apporto di energia metabolica. Disturbi del normale stato redox possono provocare effetti tossici attraverso la produzione di specie chimiche reattive che danneggiano le componenti della cellula, inclusi proteine, lipidi ed acidi nucleici, e sono in grado di condurre la stessa all’apoptosi.
Le ROS ed altre specie reattive vengono continuamente prodotte dal nostro organismo attraverso numerosi processi biochimici; determinate quantità di sostanze ossidanti sono infatti indispensabili per mantenere il corretto funzionamento cellulare, regolando i meccanismi propri dell’omeostasi.
A seconda dell’atomo responsabile della loro reattività, si ritrovano specie chimiche reattive dell’ossigeno (ROS), dell’azoto (RNS) o del carbonio (RCS).
In particolare i radicali liberi sono definiti come specie chimiche reattive aventi un singolo elettrone spaiato nell’orbitale esterno. Questa caratteristica conferisce loro una configurazione instabile tale da renderle capaci di reagire con diverse molecole quali proteine, lipidi, carboidrati e acidi nucleici, dalle quali sottraggono un elettrone, ossidandole, nel tentativo di acquisire stabilità. In tal modo vengono prodotti altri radicali liberi secondo reazioni autocatalitiche che si propagano a catena.
Gli effetti dannosi si esplicano, ad esempio, con la perdita della compartimentazione cellulare e dei trasporti selettivi, in caso siano attaccati i fosfolipidi di membrana; con accumulo di mutazioni ed alterazioni dell’espressione genica, per quanto riguarda gli acidi nucleici; con modificazioni della struttura e perdita della funzione enzimatica, recettoriale e di trasporto, a seguito dell’ossidazione di alcuni gruppi amminoacidici delle proteine.
I radicali liberi centrati sull’ossigeno sono i principali sottoprodotti che si formano nelle cellule degli organismi aerobi. Le forme più comuni sono il radicale ossidrile (OH
-
), l’anione superossido (·O2
-) e l’ossido nitrico (NO
-
). Anche le forme non radicaliche come il perossido di idrogeno (H2O2) ed il perossinitrito (ONOO
-) possono, in molti casi, indurre danno cellulare generando radicali attraverso varie reazioni chimiche(30). Possono essere identificate due fonti di produzione di specie chimiche reattive:
- endogena: a livello dei mitocondri, nel metabolismo del citocromo P450, nei perossisomi ed a seguito dell’attivazione delle cellule infiammatorie;
- esogena: gli agenti ambientali possono generare direttamente, o indirettamente, le ROS. È stata osservata un’induzione di stress e danno ossidativo dopo esposizione a diversi tipi di xenobiotici, quali metalli (ridotti e non ridotti), ioni, radiazioni (UV, raggi gamma, raggi X), farmaci (barbiturici), contaminanti ambientali ed agenti cancerogeni(31).
In particolare, nei neonati può verificarsi un’aumentata generazione di ROS come risultato di molte condizioni, quali l’iperossia, la riperfusione post-ischemica, e/o la presenza di stati di infiammazione.
Il neonato prematuro è particolarmente suscettibile al danno indotto dalle specie chimiche reattive dell’ossigeno per alcune ragioni principali:
- assenza di un'adeguata concentrazione di antiossidanti alla nascita;
- ridotta capacità antiossidante, poiché l’aumento di questa si verifica nell’ultima parte della gestazione, in preparazione alla vita extrauterina;
- capacità di aumentare la sintesi di sostanze antiossidanti, in risposta all’iperossia o ad altre condizioni ossidanti, relativamente compromessa.
Tutto ciò può portare ad un aumentato rischio, per il nato prematuro, di sviluppare patologie ROS-indotte, quali la Sindrome da Distress Respiratorio (RDS), in forma acuta, o la displasia broncopolmonare (BPD), esito della cronicizzazione della RDS(32). Inoltre fattori quali il ritardo di crescita intrauterina e la prematurità, possono influenzare lo squilibrio tra sostanze ossidanti ed antiossidanti e, quindi, i danni indotti dai radicali liberi(33).
3.2 Indici di stress ossidativo: NPBI, TH e AOPP
I neonati ed in particolare i nati prematuri sono ad alto rischio di stress ossidativo e sono molto sensibili al danno ossidativo indotto dai radicali liberi. È stato messo in evidenza uno squilibrio tra i sistemi antiossidanti dell’organismo e quelli che portano alla creazione di sostanze ossidanti che sono causa di danno ossidativi.
L’encefalo, in particolare, è più a rischio di lesioni mediate dai radicali liberi poiché le membrane neuronali sono ricche di acidi grassi polinsaturi ed il neonato ha una relativa deficienza di agenti antiossidanti, quali la superossido dismutasi e la glutatione perossidasi, a livello encefalico(34).
Alcuni studi hanno dimostrato che le lipoproteine plasmatiche sono un target comune di stress ossidativo indotto dai radicali liberi in neonati ipossiemici.
Ci sono diversi studi che hanno mostrato come la presenza di ferro libero (NPBI) nel plasma e nei globuli rossi, gli idroperossidi totali (TH) ed i prodotti di degradazione avanzata delle proteine (AOPP) possano essere usati come markers per valutare lo stress ossidativo indotto da radicali liberi in neonati ipossiemici.
Il parametro TH è una misura dello stress ossidativo in generale, dato che misura i prodotti intermedi ossidativi di lipidi, peptidi e aminoacidi, mentre la determinazione di AOPP fornisce informazioni per quanto riguarda il coinvolgimento delle proteine nelle reazioni che portano alla formazione dei radicali liberi.
L’AOPP, indicando i prodotti di ossidazione avanzata delle proteine, costituisce un marker che identifica la quantità di proteine modificate da processi di ossidazione, a livello di specifici residui amminoacidici, ad opera di specie chimiche reattive.
In contrasto con i lipidi, la reazione delle proteine con vari ossidanti durante l'ipossia non è stata studiata estesamente fino al 2000, quando Buonocore et al.(35) ipotizzarono che l’ipossia dei tessuti portasse ad un aumento dell’ossidazione delle proteine nel sangue di neonati prematuri. Lo studio si è basato sull’analisi di campioni di sangue ottenuti dalla vena ombelicale di 39 neonati pretermine ipossici e 16 controlli.
Nei campioni eparinizzati sono stati misurati i seguenti parametri plasmatici: idroperossidi totali (TH), prodotti di ossidazione avanzata delle proteine (AOPP) ed anche ipoxantina (Hx), xantina (Xa) ed acido urico (UA). Sono stati ritrovati valori più alti di tutti questi parametri nel plasma dei neonati pretermine ipossici rispetto a quelli di controllo, dimostrando una correlazione fra questi ed il danno causato dall’ipossia: più l’ipossia risulta grave, maggiore è il danno indotto dai radicali liberi a lipidi e proteine. L’NPBI (non-protein-bound iron) indica una forma di ferro a basso peso molecolare, privo dell’affinità di legame per la transferrina, che si ritrova nel plasma, complessato a citrato, lattato, o fosfato o legato blandamente all'albumina od ad altre proteine. Questa
provoca il rilascio del radicale ossidrile (OH
·
), a partire dal superossido o dal perossido di idrogeno, eventualmente attraverso complessi ferro-ossigeno. Il radicale ossidrile è una specie estremamente ossidante ed attacca tutte le classi di macromolecole biologiche, depolimerizzando i polisaccaridi, rompendo i filamenti di DNA, inattivando gli enzimi, e determinando la perossidazione dei lipidi. NPBI viene rilasciato dall'emoglobina quando gli eritrociti si trovano in condizione di stress ossidativo.Il neonato è molto suscettibile allo stress ossidativo indotto da NPBI. Studi recenti hanno riportato che NPBI è rilasciato dagli eritrociti in concentrazioni più elevate nei neonati pretermine, in condizioni ipossiche, rispetto ai neonati a termine(36).
L’asfissia potrebbe quindi influenzare il metabolismo del ferro e condurre ad un aumento significativo di NPBI e di perossidazione lipidica nel plasma dei neonati con encefalopatia ipossico-ischemica, indicando che la delocalizzazione di ferro indotta da asfissia gioca un ruolo nel danno cerebrale di neonati asfittici(37).
Buonocore et al.(38), nel 2003, dimostrarono come NPBI fosse un marcatore precoce affidabile, indicatore di stress ossidativo intrauterino, e conseguentemente dei problemi nello sviluppo cerebrale.
Nel 2008 Shouman et al.(39), valutando le concentrazioni di NPBI e malondialdeide (MDA) nel siero e nel fluido cerebrospinale (CSF) di otto neonati sani e di nove invece affetti da encefalopatia ipossico-ischemica (HIE) moderatamente grave, conclusero che l'ipossia/ischemia altera il metabolismo del ferro e provoca perossidazione lipidica, portando ad un aumento dei valori di questi due parametri nel CSF nei neonati con HIE. Questo studio supporta, quindi, un ruolo del ferro nel danno ossidativo al sistema nervoso centrale, a seguito di insulti ipossico-ischemici.
4 TRATTAMENTI DELLA RDS
4.1 Surfactante(1)Il surfactante polmonare è un complesso di natura lipoproteica, con alto potere tensioattivo, che si trova al di sopra del film alveolare (ipofase acquosa basale) delle vie aeree terminali e degli alveoli polmonari.
Il surfactante va quindi a determinare quella che è l’interfaccia aria-ipofase, con i gruppi polari (idrofili) della molecola orientati verso la fase liquida ed i gruppi non polari (idrofobi) verso la fase gassosa. Così disposto il surfactante ha come funzione fisiologica principale quella di ridurre la tensione superficiale a livello dell’interfaccia aria-liquido, stabilizzando gli alveoli e prevenendone il collasso al termine dell’espirazione.
Le cellule deputate alla sintesi ed alla secrezione del surfactante sono gli pneumociti di tipo II, cellule voluminose, cuboidali, dotate di microvilli e di corpi lamellari, organelli specifici contenenti inclusioni citoplasmatiche granulari, costituite da un presecreto dello stesso surfactante.
Gli pneumociti di tipo II costituiscono solo una piccola percentuale delle cellule alveolari, l’85% delle cellule epiteliali alveolari è infatti rappresentato dagli pneumociti di tipo I, contenenti prevalentemente materiale lipidico.
Dal punto di vista chimico, il surfactante è composto per il 90% da lipidi, di cui il 70-80% è costituito da fosfolipidi o lecitine ed il 10% da lipidi neutri, per l’8% da proteine e per il restante 2% da carboidrati.
Il maggior costituente dei fosfolipidi è la fosfatidilcolina satura (dipalmitoilfosfatidilcolina, DPPC) che rappresenta il 50% del contenuto lipidico del surfactante. Il fosfatidilglicerolo (PG) rappresenta il 5-10% dei lipidi, mentre fosfatidilinositolo (PI), la fosfatidilserina (PS) e la fosfatidiletonolamina (PE) generalmente sono presenti in quantità inferiore al 10%. Il colesterolo e le sue forme esterificate rappresentano meno del 5% della massa lipidica del surfactante. Il loro
ruolo non è stato ancora chiarito a fondo, ma con certezza il colesterolo influisce sulla fluidità e sull’organizzazione delle membrane lipidiche. La sfingomielina rappresenta meno del 2% dei lipidi.
In alcuni studi clinici è stata dimostrata l’importanza della DPPC, in quanto in neonati pretermine, nei quali il surfactante contiene scarse quantità di tale lipide, presentano un rischio più elevato di RDS con esito infausto.
Mentre il ruolo della componente fosfolipidica nel conferimento delle proprietà di superficie al surfactante fu stabilito immediatamente dopo l’isolamento di tali composti, soltanto agli inizi degli anni ‘70 furono riconosciute, come parte integrante del sistema surfactante, anche alcune proteine specifiche, che vennero purificate e parzialmente caratterizzate, rendendo così possibile lo studio delle loro proprietà e degli effetti esercitati sulla struttura, sulla funzione e sul metabolismo del surfactante.
Nel 1988 Possmayer(40) propose una nomenclatura, tuttora accettata, per queste proteine, chiamate da allora SP-A (surfactant-associated protein A), SP-B, SP-C ed SP-D.
Le proteine SP-A e SP-D sono idrofile in natura e consistono in assemblaggi macromolecolari di grandi dimensioni, appartenenti alla famiglia delle collectine.
La SP-B e la SP-C costituiscono, invece, piccole proteine idrofobiche derivanti dal clivaggio di precursori proteici di dimensioni maggiori.
Nel metabolismo del surfactante le proteine hanno funzioni diverse e talvolta sovrapponibili. La SP-A e la SP-B, in particolare, partecipano alla trasformazione calcio-dipendente dei corpi lamellari in mielina tubulare, fenomeno molto importante nella secrezione del surfactante.
Surfactant protein A (SP-A)
(41):
La SP-A è una proteina idrofilica, di 34-36 kDa, che facilita la proprietà dei fosfolipidi di ridurre la tensione superficiale negli alveoli, regola la sintesi dei fosfolipidi tensioattivi, la loro secrezione e ricaptazione negli pneumociti di tipo II, contrastando gli effetti
inibitori sulle funzioni del surfactante esercitati dalle proteine plasmatiche rilasciate durante il danno polmonare.
Appartiene alla famiglia delle collectine, delle lectine di tipo C. Questa è caratterizzata dall’avere un dominio collagene-simile e dalla capacità di legare i carboidrati.
La SP-A non è rilevabile nel tessuto polmonare fetale umano durante la prima parte del secondo trimestre, prima della differenziazione dell'epitelio alveolare. Pneumociti differenziati di tipo II contenenti corpi lamellari sono osservati nel polmone fetale umano a partire da 22 settimane di gestazione e la secrezione di tensioattivo attivo nel liquido amniotico si verifica dopo le 30 settimane di gestazione. Questa proteina può essere quindi rilevata nel liquido amniotico a partire da 30-32 settimane di gestazione.
Surfactant protein D (SP-D)
(42):
La SP-D è un membro della famiglia delle collectine di 43-kDa ed è espressa dagli pneumociti di tipo II e dalle cellule epiteliali polmonari.
Isolata di recente sembra avere molte analogie funzionali con la SP-A nella regolazione selettiva dell’omeostasi dei fosfolipidi del surfactante e nella sua composizione.
Non è ancora chiaro se l'SP-D agisca attraverso cambiamenti intracellulari od extracellulari, e, specificatamente, se regoli l’omeostasi del surfactante a livello di sintesi e/o catabolismo da parte delle cellule epiteliali di tipo II, o di catabolismo da parte dei macrofagi alveolari.
Oltre alla funzione regolatrice sull’omeostasi del surfactante, SP-A e SP-D, svolgono un’importante funzione immunomodulatoria a livello polmonare.
La SP-A e la SP-D aumentano la captazione di batteri e virus da parte dei macrofagi alveolari e dei neutrofili e possono influenzare la funzione macrofagica sfruttando i recettori per le collectine presenti sulle cellule bersaglio(42)
.
La SP-A stimola una varietà di risposte macrofagiche quali la chemiotassi, la polimerizzazione dell’actina e la fagocitosi, svolgendo un ruolo importante nella regolazione della funzione immunitaria innata(43)
.
Surfactant protein B (SP-B)
(44):
La SP-B è espressa dagli pneumociti di tipo II e dalle cellule Clara. È una proteina idrofoba e potenzia la velocità di adsorbimento di fosfolipidi al monostrato superficiale ed aiuta a mantenere la bassa tensione superficiale durante la compressione dinamica dell’alveolo; esperimenti di ricostituzione in vitro hanno suggerito che la SP-B è necessaria per la formazione della mielina tubulare.
Il deficit congenito di SP-B porta allo sviluppo di distress respiratorio, nel neonato, entro il primo giorno di vita. Il tasso di progressione della malattia e dell’insufficienza respiratoria è variabile, ma porta a morte il neonato entro pochi mesi di vita.
Surfactant protein C (SP-C)
(45):
E’ una proteina estremamente idrofobica espressa esclusivamente dagli pneumociti alveolari di tipo II verso la fine del secondo terzo della gravidanza e costituisce circa il 4% del surfactante totale. La SP-C accelera la formazione del film multistrato che ricopre gli alveoli; in vitro essa migliora anche la ricaptazione dei fosfolipidi del surfactante e sembra avere un ruolo nel catabolismo di questo. La SP-C può essere, essa stessa, ricaptata dallo spazio alveolare e incorporata nei corpi lamellari(46).
Il deficit di SP-C può causare una pneumopatia interstiziale che esordisce nei primi anni di vita.
Entrambe le proteine idrofobiche (SP-B e SP-C) facilitano l’adsorbimento del film fosfolipidico all’interfaccia aria-liquido, contribuendo al mantenimento delle proprietà tensioattive del surfactante stesso; la SP-B partecipa inoltre alla formazione della mielina tubulare(47)
. La SP-B e la SP-C aumentano inoltre la resistenza del surfactante
all’inattivazione da parte di varie proteine, quali ad esempio fibrinogeno, fibrina,albumina, ed emoglobina, eventualmente presenti all’interno degli alveoli in corso di RDS(48)
.
Il surfactante viene prodotto nel reticolo endosplasmatico degli pneumociti di tipo II, dalla 25a alla 30a settimana di gestazione e raggiunge la sua completa capacità di stabilizzare gli alveoli intorno alla 33a -36a settimana.
Step iniziale è quindi la differenziazione degli pneumociti di tipo II (inizio alla 20a-24a settimana di gestazione), per la quale è necessario uno stretto contatto fra queste cellule ed i fibroblasti che producono un peptide conosciuto come fibroblast pneumocyte factor, che favorisce la sintesi dei composti tensioattivi e la cui funzionalità è controllata dai glucocorticoidi.
All’interno degli pneumociti di tipo II sono sintetizzate sia la frazione lipidica che quella proteica, che vengono poi accumulate, prima della secrezione, nei corpi lamellari attraverso un meccanismo di trasporto intracellulare. La secrezione avviene poi per esocitosi mediante la fusione della loro membrana esterna con quella plasmatica apicale della cellula (secrezione merocrina) e conseguente diffusione del loro contenuto nello spazio alveolare.
Il surfactante, una volta secreto negli spazi alveolari, può essere riciclato, ovvero, a seguito della ricaptazione da parte degli pneumociti di tipo II, i componenti vengono riutilizzati. Può anche essere degradato ed i suoi componenti riutilizzati per la sintesi di nuovi lipidi e proteine senza uscire dagli pneumociti, oppure può essere rimosso dal sistema in forma di molecole intatte o di prodotti di degradazione, quali gli acidi grassi. Nel feto, una volta secreto, il surfactante viene riversato nel liquido polmonare fetale (FPL) che riempie gli alveoli in concentrazioni crescenti con il progredire dell’età gestazionale. Il FPL rappresenta un ultra-filtrato del plasma fetale, avendo una composizione simile, e costituisce oltre il 60% del peso del polmone fetale. Tale liquido, alla nascita, viene sostituito iso-volumetricamente da aria, per un volume
attraverso due meccanismi: per fuoriuscita dalla trachea a causa della compressione del torace durante il passaggio del feto in vagina, ma soprattutto per riassorbimento nel circolo ematico attraverso i vasi linfatici.
Il riassorbimento completo del FPL coincide con lo stabilirsi di un normale strato di rivestimento alveolare, costituito dal surfactante, il quale deriva in parte dall’accelerata attività secretoria delle cellule di tipo II con l’inizio del respiro, ed in parte dal surfactante presente nel FPL ancora non riassorbito.
Esistono diverse condizioni in grado di alterare il sistema di produzione del surfactante, quali ad esempio un’immaturità dei processi metabolici e degli enzimi deputati alla sintesi del surfactante, presenti in bassa concentrazione, come spesso si può osservare in caso di acidosi; mancanza dei precursori del surfactante (per ischemia polmonare o basse concentrazioni ematiche); inadeguata secrezione delle cellule di tipo II, degenerate ad esempio per asfissia fetale acuta; inibizione del surfactante da fibrinogeno, trasudato plasmatico, etc.; scarso riassorbimento alla nascita del FPL, per insufficiente circolazione ematica e linfatica, con conseguente mancata formazione dello strato delimitante dell’alveolo.
4.1.1 Impiego del surfactante(49)
L'insufficienza respiratoria secondaria a carenza di surfactante è una delle principali cause di morbilità e di mortalità nei neonati pretermine, che possono essere ridotte sostanzialmente con la somministrazione di surfactante esogeno.
A partire dagli anni ‘70 sono stati condotti numerosi studi che hanno dimostrato l’efficacia della somministrazione di surfactante naturale in modelli animali di RDS. Durante gli anni ‘80 numerosi studi effettuati utilizzando sia surfactanti naturali che sintetici, dimostrarono una riduzione della mortalità neonatale, a seguito della somministrazione del tensioattivo.