INDICE
RIASSUNTO 3
Sezione I: INTRODUZIONE 7
Morfologia e ciclo replicativo di HIV 8
Polimorfismo genetico di HIV 14
Modalità di trasmissione dell’infezione 23
Eventi immunologici e virologici dell’infezione primaria 25
Manifestazioni cliniche dell’infezione primaria 36
Terapia antiretrovirale 41
Sezione II: PAZIENTI E METODI 57
Pazienti 58
Metodi 59
Sezione III: RISULTATI 61
Manifestazioni cliniche 62
Parametri di laboratorio 64
Terapia antiretrovirale 69
Sezione IV: DISCUSSIONE E CONCLUSIONI 71
Discussione e conclusioni 72
Sezione V: TABELLE E FIGURE 80
Sezione VI: RIFERIMENTI BIBLIOGRAFICI 94
RIASSUNTO
Si parla di infezione primaria da HIV per descrivere quella fase dell’infezione che si sviluppa entro un anno dal contagio. L’infezione primaria viene definita acuta entro sei mesi dal contagio o recente da sei mesi ad un anno dopo il contagio.
I dati riportati in questo studio riguardano 78 pazienti ricoverati o seguiti presso gli ambulatori dell’Unità Operativa di Malattie Infettive dell’Ospedale Cisanello di Pisa che sono risultati essere affetti da un’infezione primaria da HIV: 69 tra il 2000 ed il 2007 e 9 tra il 1993 ed il 1999.
Di ogni paziente sono stati valutati: età, sesso, nazionalità, fattori di rischio, segni e sintomi clinici, necessità di ospedalizzazione, necessità di terapia antiretrovirale, tipo di terapia eseguita e parametri di laboratorio. I parametri di laboratorio analizzati sono stati: il profilo sierologico anti-HIV, la carica virale basale e la carica virale a 3 e 6 mesi dall’inizio dell’eventuale terapia antiretrovirale, il numero e la percentuale dei linfociti T CD4+ basali e dopo 3 e 6 mesi dall’inizio dell’eventuale terapia, il pattern di reattività anticorpale contro i diversi antigeni HIV, la presenza di polimorfismi e/o di mutazioni nel gene pol correlabili a resistenza a farmaci antiretrovirali, il sottotipo infettante relativamente alla sequenza del gene pol.
Tutte le analisi virologiche sono state eseguite presso l’Unità Operativa complessa di Virologia dell’Azienda Ospedaliera Universitaria Pisana.
Nel nostro studio i rapporti eterosessuali sono risultati essere il principale fattore di rischio per l’infezione: questo conferma come la malattia, un tempo circoscritta in alcuni gruppi di popolazione definiti a rischio, omosessuali e tossicodipendenti, abbia modificato negli anni le sue caratteristiche epidemiologiche.
Circa i due terzi dei pazienti esaminati presentavano segni e sintomi attribuibili ad una sindrome retrovirale acuta. Le manifestazioni cliniche più comunemente riscontrate erano: febbre, faringodinia, malessere, astenia, rash cutaneo, epatosplenomegalia e linfoadonopatia generalizzata. Meno comuni erano cefalea, tosse, mialgia, sudorazione notturna, nausea, diarrea, vomito, dolori addominali, ulcere genitali. In un quinto dei casi la gravità delle manifestazioni cliniche era stata tale da richiedere la necessità di un ricovero.
L’analogia delle manifestazioni cliniche della sindrome retrovirale acuta con altre patologie infettive, come influenza, mononucleosi infettiva, toxoplasmosi, rosolia, infezione da CMV, sifilide secondaria, pone il problema della diagnosi differenziale, che dovrebbe basarsi su un’attenta valutazione dei fattori di rischio, uno scrupoloso esame obiettivo ed analisi di laboratorio mirate. Due pazienti da noi valutati presentavano segni, sintomi ed anomalie radiologiche attribuibili ad una polmonite; per uno dei due si era reso necessario il ricovero. L’interessamento polmonare nel corso dell’infezione acuta non è un evento molto frequente ed è probabilmente dovuto ad una grave, ma transitoria, immunosoppressione: nei pazienti da noi valutati, infatti, la sintomatologia respiratoria si accompagnava ad un numero particolarmente basso dei CD4+.
Solo due dei pazienti da noi esaminati presentavano un interessamento del sistema nervoso centrale e questo dato contrasta con la frequenza con cui sindromi neurologiche vengono descritte in letteratura. Le patologie neurologiche che solitamente si presentano durante l’infezione primaria sono: meningiti, encefaliti e neuropatie periferiche. Nella nostra casistica un paziente presentava una meningite asettica, risoltasi spontaneamente pochi giorni dopo il ricovero, mentre l’altro presentava una meningomielite, patologia che generalmente compare in una fase tardiva dell’infezione da HIV.
Nel nostro studio abbiamo riscontrato che nella maggior parte dei casi la viremia basale era superiore a 105
copie/ml, raggiungendo in otto casi valori superiori a 106
caso di esordio dell’infezione acuta con una viremia al di sotto del limite di rilevabilità del test. Alcuni lavori hanno dimostrato come la carica virale basale sia in grado di condizionare la riduzione dei CD4+ in una fase tardiva dell’infezione, condizionando così la progressione a lungo termine della patologia
Il nostro studio ha rilevato come l’infezione acuta, oltre ad essere caratterizzata da un’elevata carica virale, si associ anche ad una riduzione nel numero e nella percentuale dei linfociti T CD4+ e come la viremia basale sia in grado di condizionare la rapidità di risposta al trattamento antiretrovirale.
Comunque, la maggior parte dei pazienti da noi esaminati raggiungeva valori di viremia inferiori alle 50 copie/ml associati ad una buona ricostituzione immunologica entro 48 settimane dall’inizio del trattamento. I risultati di alcuni studi però suggeriscono che i vantaggi immunologici e virologici di una terapia antiretrovirale iniziata nel periodo dell’infezione primaria potrebbero venir meno entro un anno dalla sospensione del trattamento.
Nonostante la maggior parte degli isolati virali dei pazienti da noi valutati presentasse numerosi polimorfismi a carico del gene pol, soltanto il 5,8% dei pazienti esaminati presentava un’infezione sostenuta da varianti virali con una o più mutazioni correlabili a ridotta risposta alla terapia antiretrovirale. Le mutazioni riscontrate negli isolati dei pazienti da noi esaminati erano associate a resistenza nei confronti degli NRTIs e dei PIs, non si rilevavano, invece, resistenze nei confronti degli NNRTIs.
Alcuni studi mostrano come il tasso di mutazioni associate a farmacoresistenza sia maggiore negli isolati di sottotipo B rispetto ai non-B. Nel nostro studio abbiamo potuto osservare come solo una mutazione fosse presente in un sottotipo non-B, mentre tutte le altre comparivano in isolati di sottotipo B. Queste differenze nel tasso di mutazioni dei sottotipi B e non-B sono probabilmente dovute al fatto che l’esposizione dei sottotipi non-B ai farmaci
antiretrovirali è stata meno prolungata, dal momento che la diffusione di ceppi non-B in Europa ed in Nord America è un fenomeno piuttosto recente.
Infine, il nostro studio mette in evidenza come la prevalenza di sottotipi non- B nella popolazione esaminata sia del 28,4%, dato che conferma l’ampia diffusione dei sottotipi non-B in Italia e nell’Europa occidentale.
Tra i sottotipi non-B quello maggiormente rappresentato nella popolazione da noi esaminata è il sottotipo F; sono risultate essere piuttosto diffuse anche forme ricombinanti URF: ciò indica come la coinfezione e la ricombinazione di diversi sottotipi sia un fenomeno piuttosto frequente. Bisogna segnalare un caso di infezione sostenuta dalla variante CRF02_AG e due casi di infezione sostenuta dal sottotipo C, il sottotipo più rappresentato nel mondo.
La lenta introduzione e diffusione dei sottotipi non-B in Italia ed in Europa potrebbe indurre la necessità di adattare le strategie diagnostiche e terapeutiche alla nuova situazione epidemiologica.
Sezione I
MORFOLOGIA E CICLO REPLICATIVO DI HIV
Il virus dell’immunodeficienza umana (Human Immunodeficiency Virus, HIV) fa parte della famiglia dei Retroviridae, nella quale si possono distinguere tre sottofamiglie: Oncovirinae, Lentivirinae, Spumavirinae.
HIV appartiene al gruppo dei lentivirus, virus che instaurano infezioni persistenti e si associano ad immunosoppressione e a malattie cronico-degenerative con frequente interessamento neurologico (Letvin, 1990).
MORFOLOGIA DI HIV
HIV è un virus ad RNA provvisto di envelope, di forma sferica, con un diametro di 80-120 nm. L’envelope o pericapside è costituito da una membrana a doppio strato lipidico, acquisita dalla membrana citoplasmatica della cellula ospite durante la fase di gemmazione del virus, dove trovano sede due glicoproteine virali, gp120 e gp41. Le due glicoproteine si formano in seguito a taglio proteolitico della poliproteina gp160. La gp120 è una proteina altamente glicosilata, dal peso molecolare di 120 kD, che sporge dalla superficie della particella virale. Questa glicoproteina è responsabile del tropismo tissutale di HIV e viene riconosciuta dagli anticorpi neutralizzanti, anche se la sua antigenicità e specificità recettoriale possono variare nel corso di un’infezione cronoca da HIV. La gp41 è, invece, un glicoproteina transmembrana di 41 kD, capace di promuovere la fusione tra virus e cellula bersaglio.
L’envelope circonda un capside, costituito da una porzione esterna, la matrice, e da una porzione centrale, il core o nucleocapside.
La matrice è formata dalla proteina p17, che si estende tra la porzione interna dell’envelope virale ed il core. La p17 svolge un ruolo fondamentale nel trasportare i precursori strutturali e le glicoproteine virali verso la membrana citoplasmatica della cellula ospite e nel favorirne l’incorporazione nel virione nascente (Zhou et al., 1994).
Il core del virione di HIV ha una forma a tronco di cono ed è costituito principalmente dalla proteina p24.
All’interno del core si trovano due molecole di RNA genomico a singolo filamento a polarità positiva, covalentemente legate alla trascrittasi inversa ed associate a due proteine basiche, p7 e p9, a formare il cosiddetto complesso nucleoproteico.
Il nucleocapside è anche costituito dalla proteasi, dalla trascrittasi inversa, dall’integrasi e dalla RNasi H, enzimi virali necessari per la replicazione dei virioni.
La proteasi interviene nel clivaggio delle poliproteine virali e nel processamento degli enzimi replicativi.
La trascrittasi inversa (RT) è indispensabile per la sintesi del DNA lineare a doppio filamento a partire dall’RNA genomico. Poiché quest’enzima è privo di attività esonucleasica 3’→5’(Mansky, 1998), durante la trascrizione inversa si osserva un alto tasso di mutazione, pari a 3×10-5
mutazioni per ciclo replicativo. L’alto tasso di mutazione, l’elevato ritmo di replicazione (107
cicli replicativi al giorno) e la frequente ricombinazione tra genomi virali sono responsabili della straordinaria variabilità genetica di HIV (Coffin, 1995; Ho et al., 1995; Wei et al., 1995; Domingo et al., 1999).
L’integrasi è l’enzima necessario per l’integrazione del genoma virale nel DNA della cellula ospite (Masuda et al., 1997; Brown, 1997).
Infine, la RNasi H, che deriva dal clivaggio dell’RT, è un’endonucleasi capace di produrre inneschi e stampi per la trascrittasi inversa (Davies et al., 1991; Hostomsky et al., 1991).
Il genoma di HIV è costituito da tre geni principali, comuni a tutti i retrovirus, e sei geni accessori, presenti solo nell’RNA dei retrovirus complessi, come HIV.
I tre geni principali sono: gag, che codifica le proteine strutturali del capside virale, pol, che codifica la proteasi, la trascrittasi inversa e l’integrasi ed
env, che codifica le glicoproteine dell’envelope (Varmus et al., 1989). I sei geni accessori sono: tat, rev, nef, vif, vpu e vpr (vpx in HIV-2).
Alle due estremità di ciascuna molecola di RNA genomico sono presenti le sequenze LTR (long terminal repeats), fondamentali nella regolazione dell’espressione genica. Queste sequenze contengono geni promotori, la sequenza TAR, a cui si lega la proteina tat determinando l’inizio della trascrizione ed altre sequenze geniche utilizzate per legare diversi fattori di trascrizione cellulari (Schupbach et al., 2000).
CICLO REPLICATIVO DI HIV
La replicazione di HIV inizia con il legame della glicoproteina di superficie del virus, la gp120, al recettore CD4 presente sulla superficie delle cellule bersaglio (linfociti T, magrofagi, cellule dendritiche ed altre cellule capaci di presentare l’antigene). Il legame tra gp120 e CD4 determina cambiamenti conformazionali a carico della glicoproteina virale, rendendo possibile l’interazione della stessa con co-recettori espressi sulla superficie delle cellule bersaglio (Alkhatib et al., 1996; Deng et al., 1996; Feng et al., 1996). I co-recettori sono recettori delle chemochine a sette domini transmembrana; nelle prime fasi della malattia gp120 interagisce principalmente con il co-recettore CCR5, mentre il CXCR4 viene coinvolto successivamente.
Il legame tra la gp120 ed il co-recettore provoca ulteriori cambiamenti conformazionali a carico di gp120, che consentono l’esposizione di gp41, precedentemente nascosta da gp120. Questo permette alle regioni HR1 e HR2 di gp41 di interagire, consentendo l’avvicinamento tra envelope virale e membrana plasmatica della cellula bersaglio. HR2 penetra successivamente nella membrana della cellula ospite, garantendo un contatto stabile che favorisce la fusione tra HIV e cellula bersaglio e la successiva penetrazione (Schupbach et
al., 2000). All’interno della cellula ospite il capside virale viene degradato da enzimi cellulari, liberando l’RNA del virus nel citosol della cellula stessa. La trascrittasi inversa converte l’RNA genomico in DNA a doppio filamento, che rimane associato a proteine virali (p17, vpr, integrasi) capaci di favorire la migrazione del DNA del virus nel nucleo della cellula ospite. Nel nucleo il DNA virale viene integrato nel DNA cromosomico ad opera dell’integrasi virale (Brown, 1990; Brown, 1997). Il genoma di HIV integrato, definito provirus, rimane permanentemente associato al DNA della cellula ospite e può dare inizio ad un’infezione latente, se il DNA virale resta silente, oppure ad un’infezione produttiva, se i geni virali sono espressi attivamente ed il provirus viene trascritto dall’RNA polimerasi II cellulare. Poiché il provirus si comporta come un gene cellulare, la sua replicazione dipende dal grado di metilazione dell’RNA virale, dal ritmo di crescita cellulare e dalla capacità della cellula ospite di utilizzare le squenze promotrici codificate dall’LTR.
La trascrizione del genoma virale dà origine a tre classi di mRNA: mRNA di lunghezza completa (9 kb), mRNA di dimensioni intermedie (4 e 5 kb) ed mRNA di piccole dimensioni (2 kb); questi ultimi due originano dall’mRNA di lunghezza completa in seguito fenomeni di taglio post-trascrizionale (splicing). I trascritti di 2 kb codificano le proteine tat, rev e nef, prodotte in elevata quantità durante le prime fasi del ciclo replicativo di HIV, mentre i trascritti di lunghezza completa ed intermedia codificano le proteine gag, pol, env, vif, vpr e vpu e sono prodotti nelle fasi tardive della replicazione virale (Pavlakis et al., 1992). Inoltre, i trascritti a piena lunghezza possono essere anche incorporati all’interno di nuovi virioni.
Inizialmente il genoma di HIV viene trascritto in mRNA di lunghezza completa, che viene sottoposto a numerosi eventi di splicing, dando origine a trascritti di 2 kb, codificanti le proteine tat, rev e nef.
La proteina tat è un attivatore della trascrizione ed agisce legandosi alla sequenza TAR del genoma virale. Questo processo favorisce la produzione di trascritti di 9 kb (Zhou et al., 1995).
La proteina rev si lega alla sequenza RRE (rev responsive element) dell’mRNA virale, promuovendo il trasporto dell’mRNA di HIV dal nucleo al citoplasma della cellula ospite e prevenendo ulteriori eventi di splicing (Rosen et al., 1988; Malim et al., 1989; Pollard et al., 1998). Nella fase tardiva dell’infezione rev porta selettivamente all’aumento dell’espressione dei geni strutturali che subiscono un solo processo di splicing, mentre viene inibita l’espressione dei geni regolatori (Cullen, 1998; Pongoski et al., 2002).
La proteina nef si origina dopo numerosi fenomeni di splicing e riduce l’espressione del recettore CD4 sulla superficie della cellula ospite favorendo l’endocitosi del recettore stesso in vescicole rivestite da clatrina (Aiken et al., 1994). Inoltre, nef favorisce la degradazione delle molecole del complesso maggiore di istocompatibilità di classe prima (MHC I) riducendo la capacità dei linfociti T citotossici di riconoscere ed eliminare le cellule infettate dal virus (Collins et al., 1998). La proteina nef è quindi in grado di regolare la citotossicità di HIV, di favorire il mantenimento di un’elevata carica virale e sembra essere essenziale per determinare la progressione dell’infezione ad AIDS.
Le proteine codificate dai geni gag, gag-pol ed env vengono sintetizzate sotto forma di poliproteine e successivamente processate nelle proteine funzionali.
Le glicoproteine virali, codificate dal gene env, vengono sintetizzate a partire da un unico precursore poliproteico gp160, che viene glicosilato e processato nel reticolo endoplasmatico della cellula ospite da una proteasi cellulare per dare origine alla gp120 e alla gp41. Queste due glicoproteine vengono trasportate alla membrana della cellula ospite, dove interagiscono formando trimeri (Freed et al., 1995).
Le poliproteine gag e gag-pol migrano verso la membrana citoplasmatica della cellula ospite e si ancorano alla membrana stessa in prossimità delle glicoproteine dell’envelope virale. Inoltre, la poliproteina gag favorisce l’incorporazione dell’RNA genomico nel virione nascente e stimola la gemmazione della particella virale dalla cellula ospite.
Dopo il rilascio del virione dalla cellula ospite, la proteasi virale determina il taglio proteolitico delle poliproteine gag e gag-pol.
La poliproteina gag dà origine alle proteine strutturali del capside virale p17, p24, p7 e p6, mentre dal precursore gag-pol originano i tre enzimi virali proteasi, trascrittasi inversa ed integrasi.
I geni accesori vpr, vpu e vif non sono essenziali per il ciclo replicativo virale in vitro, ma contribuiscono all’infettività del virus ed alla progressione della malattia.
La proteina vpr (vpx in HIV-2) è in grado di favorire la replicazione di HIV determinando l’arresto delle cellule infettate in fase G2, stadio del ciclo
cellulare in cui il processo di trascrizione è più intenso (He et al., 1995; Chowdhury et al. ,2003). Inoltre, vpr favorisce la migrazione del DNA virale nel nucleo della cellula ospite (Agostani et al., 2002; Sherman et al., 2002) ed è coinvolta nell’attivazione di promotori virali e cellulari (Cohen et al., 1990).
La proteina vpu favorisce la degradazione del recettore CD4 espresso sulla superficie della cellula infettata (Margotti net al., 1998) ed aumenta il rilascio delle particelle virali (Klimkait et al., 1990; Paul et al., 1998; Deora et al., 2001).
Infine, la proteina vif promuove l’assemblaggio e la maturazione dei virioni e sembra intervenga nel difendere il virus dalla mutagenesi letale indotta dal sistema delle citidina deaminasi della cellula infettata (Harris et al., 2004).
POLIMORFISMO GENETICO DI HIV
Sulla base dell’analisi filogenetica di numerosi isolati virali, HIV può essere suddiviso in tipi, gruppi, sottotipi, ceppi, forme ricombinanti circolanti (CRF) e ricombinanti unici (URF).
Lo straordinario polimorfismo di HIV è dovuto principalmente a tre fattori:
• l’attività della trascrittasi inversa, che favorisce l’accumularsi di errori di trascrizione che l’enzima non è in grado di correggere perché privo di attività esonucleasica 3’→5’(Mansky, 1998);
• l’elevato tasso di replicazione virale;
• la propensione di HIV alla ricombinazione genetica. Poiché il virione neoformato contiene due molecole di RNA a singolo filamento, la coinfezione di una stessa cellula da parte di più varianti virali può determinare lo sviluppo di un genoma ricombinante costituito da regioni appartenenti a ceppi diversi. Tale processo di ricombinazione genetica è alla base della formazione dei cosiddetti ceppi “mosaico” (Peeters et al., 2000). Alcuni di questi ricombinanti sono piuttosto diffusi e vengono definiti
circulating recombinant forms (CRF), altri, definiti unique recombinant
forms (URF), costituiscono reperti isolati.
La variabilità genetica di HIV è in grado di influenzare le modalità di trasmissione dell’infezione e di interazione tra virus e cellula bersaglio, l’andamento clinico dell’infezione stessa e la risposta al trattamento. Questa variabilità non soltanto coinvolge la popolazione virale di individui differenti (variabilità interhost), ma anche quella dello stesso individuo (variabilità
intrahost) (Lukashov, 1998).
Esistono due tipi di HIV, HIV-1 e HIV-2. L’amplificazione ed il sequenziamento di frammenti genomici ha consentito il confronto tra diverse sequenze di isolati HIV-1 e HIV-2.
HIV-1 è il più aggressivo dei due ed il principale responsabile della pandemia. Sulla base del sequenziamento dell’intero genoma virale è possibile distinguere tre gruppi di HIV-1: M (major), O (outlier), N (non-M/non-O). Il gruppo M comprende più del 95% dei ceppi isolati e può essere ulteriormente suddiviso in nove sottotipi. I sottotipi identificati sono A, B, C, D, F, G, H, J e K. Sono stati distinti alcuni sottotipi anche nei gruppi O e N.
Le sequenze genomiche che codificano per le proteine dell’envelope e per le regioni gag dei diversi gruppi di HIV-1 differiscono tra loro per più del 25%. I diversi sottotipi appartenenti al gruppo M presentano differenze nella sequenza aminoacidica di almeno il 20% nella regione del rivestimento esterno e del 15% nella regione gag (Gao et al., 1994; Myers et al., 1995; Gao et al., 1998).
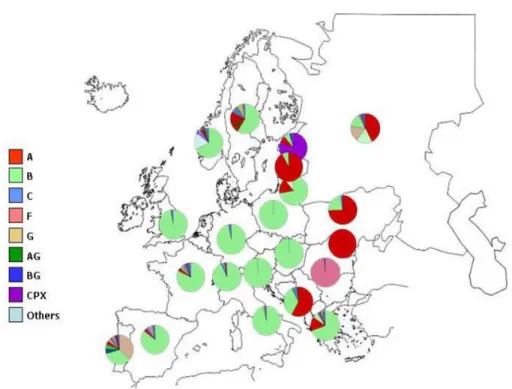
In Europa, Nord America ed Australia il sottotipo predominante è il sottotipo B, anche se più del 40% delle nuove infezioni in Europa sono sostenute da varianti virali non-B (Figura I.1).
Il sottotipo A, ulteriormente suddiviso in A1 e A2, prevale nell’Africa centrale ed occidentale (Piot et al., 2002).
Il sottotipo C è il sottotipo predominante nell’Africa meridionale ed orientale, in India, Nepal e Cina (Weniger et al., 1994; Janssens et al., 1997; Novitsky et al., 1999; Oelrichs et al., 2000; Chen et al., 2000).
Il sottotipo D è limitato a casi sporadici nell’Africa centrale ed orientale (Piot et al., 2002).
Il sottotipo F è stato osservato nell’ Africa centrale, Sud America ed Europa orientale, soprattutto in Romania. Alcuni ricercatori suggeriscono di suddividere il sottotipo F in due ceppi: F1, isolato in Brasile, Romania e Finlandia e F2, identificato in Cameroon e Repubblica Democratica del Congo (Peeters, 2000).
Il sottotipo G è presente nell’Africa orientale ed occidentale, in Russia ed in Europa centrale (Piot et al.,2002).
La presenza del sottotipo H è stata documentata solo nell’Africa centrale (Janssens et al., 1997; Piot et al., 2002).
Il sottotipo J è stato osservato in America centrale e più recentemente anche in Spagna.
Infine, il sottotipo K è stato identificato nella Repubblica Democratica del Congo ed in Cameroon (Triques et al., 2000).
I virus ricombinanti emergono quando sottotipi diversi si diffondono simultaneamente nella popolazione. Le CRFs sono forme ricombinanti epidemiologicamente rilevanti. Virus ricombinanti che coinvolgono quattro o più sottotipi sono denominati complessi ed indicati con la sigla cpx. Attualmente sono riconosciute 34 diverse CRFs (Figura I.2) derivate da sottotipi appartenenti al gruppo M (http://hiv-web.lanl.gov).
Il sottotipo ricombinante AE è stato rinominato CRF-01AE ed è particolarmente diffuso in Thailandia.
Il sottotipo I, inizialmente isolato a Cipro, è considerato un ricombinante e viene attualmente designato come CRF-04cpx.
Il ricombinante AG, CRF-02AG, è prevalente in alcune parti dell’Africa. I sottotipi A, B, C, D ed i ricombinanti CRF-01AE e CRF-02AG sono responsabili di più del 90% delle infezioni nel mondo. Circa i tre quarti delle nuove infezioni che si manifestano nel mondo sono sostenute dai sottotipi A, C e CRF-02AG. Il sottotipo C si sta diffondendo nell’Africa centrale ed in Sud Africa, sta diventando prevalente in Cina, India ed Etiopia e potrebbe essere oggi responsabile di più della metà delle infezioni da HIV nel mondo (http://www.unaids.org).
Circa il 25% degli isolati virali del Cameroon appartiene al gruppo O, che viene suddiviso da alcuni ricercatori in cinque sottotipi: I, II, III, IV, V.
Infine, sempre in Cameroon, è stata rilevata la presenza di isolati appartenenti al gruppo N. I gruppo N di HIV-1 sembra essere filogeneticamente più vicino a SIV che non ai gruppi M ed O di HIV-1.
Sono stati distinti otto gruppi di HIV-2: A, B, C, D, E, F, G, H. I gruppi A, isolato in Senegal e Guinea Bassau e B, identificato nella Costa d’Avorio, sono i più diffusi. Il gruppo G è stato isolato nella Costa d’Avorio, i gruppi C, D, E, F nelle aree rurali della Sierra Leone e della Liberia. Non è stato ancora identificato alcun sottotipo di HIV-2.
E’ evidente che l’Africa rappresenta il maggior serbatoio dell’infezione; infatti nel continente africano non solo sono stati individuati tutti i sottotipi del gruppo M, ma anche le rare varianti dei gruppi N ed O ed il tipo 2 di HIV (Peeters, 2003).
I diversi sottotipi di HIV presentano delle differenze nell’uso dei co-recettori e nella capacità della variante virale di indurre la formazione di sincizi. E’ stato osservato come il principale co-recettore utilizzato dai virus capaci di indurre la formazione di sincizi sia il CXCR4, mentre il CCR5 viene utilizzato dai ceppi virali che non tendono ad indurre la formazione di sincizi (Cocchi et
al., 1995; Deng et al., 1996; Doranz et al., 1996; Tersmette et al., 1998;). Mentre la maggior parte dei sottotipi di HIV tende ad utilizzare il co-recettore CCR5 nelle fasi iniziali dell’infezione ed il CXCR4 negli ultimi stadi della malattia (Keet et al.,1993; Cornelissen et al., 1995), i sottotipi A, C e D tendono a comportarsi diversamente. I virus appartenenti ai sottotipi A e C utilizzano prevalentemente il co-recettore CCR5 in ogni stadio della patologia, mentre le varianti D presentano lo stesso tropismo per CCR5 e CXCR4 in ogni fase dell’infezione (Zhang et al., 1996; Tscherning et al., 1998; Chen et al., 2000). Infine, è stato osservato come molti isolati HIV-2 utilizzino un ampio spettro di co-recettori alternativi per infettare le cellule bersaglio.
Inoltre, studi in vitro suggeriscono che gli isolati del gruppo M abbiano una capacità replicativa fino a 100 volte superiore rispetto agli isolati del gruppo O e di HIV-2. Sempre in vitro si è visto come il sottotipo B sia in grado di replicarsi più velocemente del sottotipo C in cellule T CD4+ ed in macrofagi di donatori diversi, ma non in cellule di Langherans umane: questo potrebbe essere associato a minor progressione di malattia. In realtà gli studi in vitro non correlano con le osservazioni cliniche sui sottotipi.
Numerosi studi dimostrano come le caratteristiche cliniche della patologia HIV relata differiscano a seconda del sottotipo virale responsabile dell’infezione. E’ stato osservato che le infezioni sostenute dai sottotipi A e G tendono a rimanere asintomatiche più a lungo e a progredire più lentamente, mentre il sottotipo C è associato ad una progressione della patologia particolarmente rapida (Kanki et al., 1999).
Oltre alle caratteristiche cliniche anche le modalità di trasmissione dell’infezione tendono a variare nei diversi sottotipi. I rapporti omosessuali e l’abuso di droghe per via endovenosa sono le principali modalità di trasmissione osservate per il sottotipo B. Al contrario, per i sottotipi A, C, D ed E la principale modalità di trasmissione dell’infezione sono i rapporti eterosessuali (Soto-Ramirez et al., 1996; van Harmelen et al., 1997; Hu et al., 1999).
Infine, la diversità genetica che si riscontra nei diversi sottotipi di HIV può avere delle conseguenze importanti anche nel condizionare la sensibilità del virus alla terapia antiretrovirale. Infatti, è stato osservato come i sottotipi non-B non solo presentino una minore sensibilità all’HAART, ma siano anche in grado di sviluppare più rapidamente resistenze multiple nei confronti delle diverse classi di farmaci antiretrovirali rispetto al sottotipo B (Caride et al., 2000; Pillay et al., 2000).
Figura I.2. CRFs di HIV-1 fino ad oggi caratterizzate CRF01_AE CRF02_AG CRF03_AB CRF04_cpx CRF05_DF CRF06_cpx CRF07_BC CRF08_BC CRF09_cpx CRF10_CD
CRF11_cpx CRF12_BF CRF13_cpx CRF14_BG CRF15_01B CRF16_A2D CRF18_cpx CRF19_cpx CRF20_BG CRF21_A2D CRF23_BG
CRF24_BG CRF28_BF CRF29_BF CRF31_BC CRF32_06A1 CRF33_01B CRF34_01B
MODALITA’ DI TRASMISSIONE DELL’INFEZIONE DA HIV
I rapporti sessuali, l’esposizione a sangue infetto e la trasmissione perinatale materno-fetale sono le tre principali modalità di trasmissione dell’infezione da HIV.
I rapporti sessuali sono il più importante fattore di rischio. La probabilità di trasmettere o acquisire l’infezione con un unico rapporto sessuale dipende da numerosi fattori, come, ad esempio, il numero di partners, il tipo di rapporto sessuale, lo stadio della malattia del partner infetto, la coesistenza di altre infezioni a trasmissione sessuale.
La carica virale è il più importante fattore predittivo di trasmissione dell’infezione: la probabilità di contagio è massima nella fase acuta dell’infezione, si riduce gradualmente nei mesi successivi per aumentare di nuovo negli stadi terminali della malattia. (Wawer et al., 2003). La terapia antiretrovirale, abbassando la viremia, può ridurre in modo significativo, ma non annullare, il rischio di contagio per via sessuale (Musicco et al., 1994; Hosseinipour et al., 2002).
Poiché HIV è stato rinvenuto in minime concentrazioni nella saliva di soggetti infetti, la possibilità di trasmissione dell’infezione attraverso rapporti orali è estremamente bassa; inoltre, è difficile attribuire la trasmissione dell’infezione al contatto con la saliva poiché spesso quest’ultima è commista a sangue (Levy et al., 1988; Goto et al., 1991).
Dati epidemiologici e di laboratorio indicano che la confezione con altri agenti patogeni a trasmissione sessuale e la presenza di lesioni genitali aumentano il rischio di trasmissione dell’infezione (Piot et al., 1989). Infatti, lesioni e processi infiammatori a carico del tratto genitale favoriscono la distruzione della barriera mucosale ed aumentano il numero di cellule bersaglio di HIV, favorendo la penetrazione del virus (Stamm et al., 1988; Plummer et al.,1991; Dyer et al., 1998).
Infine, poiché i rapporti sessuali per via anale provocano più facilmente delle lesioni rispetto a quelli per via vaginale i rapporti omosessuali siano correlati ad un maggior rischio di trasmissione dell’infezione rispetto a quelli eterosessuali.
Tra i tossicodipendenti la trasmissione dell’infezione avviene per via parenterale, attraverso la condivisione di aghi infetti. Basse condizioni socioeconomiche, vagabondaggio e appartenenza a minoranze etniche sono associati ad un aumentato rischio di contagio (McCusker et al., 1990).
La probabilità di contrarre l’infezione da HIV dopo aver ricevuto una trasfusione di sangue o di emoderivati da un sieropositivo si aggira intorno al 100% (Ward et al., 1987; Donegan et al., 1990). Il test di screening anticorpale per HIV venne introdotto nel 1985, con conseguente drastica riduzione del rischio di trasmissione dell’infezione tramite trasfusioni. Inoltre, sono stati riportati casi di trasmissione dell’infezione da HIV in seguito a trapianto d’organo solido (Erice et al., 1991).
La trasmissione perinatale dell’infezione da HIV può avvenire durante la gestazione, al momento del parto o durante l’allattamento. Il rischio di contagio perinatale dipende dalla carica virale materna (Mofenson et al., 1999; Jamieson et al., 2003): madri con un’infezione primaria (Ehrnst et al.,1991) o ad uno stadio avanzato della malattia (Ryder et al., 1989) hanno una maggiore probabilità di trasmettere l’infezione ai figli. Oltre alla viremia materna, anche lesioni della barriera emato-placentare possono aumentare il rischio di trasmissione verticale dell’infezione. Altri fattori di rischio sono: parto pretermine, prematura rottura delle membrane, abuso di stupefacenti durante la gravidanza, basso peso alla nascita (Landsman et al., 1996; Kuhn et al., 1999).
Infine, modalità meno comuni di trasmissione dell’infezione sono: l’esposizione professionale a materiale biologico infetto e l’inseminazione artificiale con liquido seminale contaminato.
EVENTI IMMUNOLOGICI E VIROLOGICI DELL’INFEZIONE PRIMARIA
Le modalità con cui si realizzano le interazioni tra HIV e sistema immunitario umano sono molto complesse e variabili. Tale variabilità è dovuta sia a fattori virali, come ad esempio il sottotipo infettante, che a fattori propri dell’organismo ospite, come ad esempio i livelli di co-recettore espressi dalle cellule bersaglio e si traduce in un’elevata variabilità di progressione della malattia.
Immunopatogenesi dell’infezione primaria da HIV
L’ingresso di HIV nelle cellule bersaglio è mediato dall’interazione tra le glicoproteine dell’envelope virale gp120 e gp41 ed il recettore CD4 espresso sulla superficie dei bersagli cellulari (Doms, 2001), ma, affinché il processo si completi ed il virus penetri nella cellula bersaglio, occorre che le glicoproteine dell’envelope interagiscano anche con altre molecole, definite co-recettori, presenti sulla cellula bersaglio (Broder et al., 1995).
I principali co-recettori utilizzati da HIV sono il CCR5 ed il CXCR4, recettori chemochinici a sette domini transmembrana. Il primo co-recettore ad essere stato identificato è il CXCR4, il cui ligando naturale è SDF-1 (stromal cell-derivated factor) (Bleul et al., 1996; Oberlin et al., 1996). Successivamente è stato scoperto il CCR5, i cui ligandi naturali sono le chemochine MIP-1α, MIP-1β e RANTES (Samson et al., 1996; Combadiere et al., 1996; Raport et al., 1996). Altri recettori delle chemochine, come il CCR1, il CCR2 ed il CCR3, sono stati recentemente identificati come possibili co-recettori utilizzati da alcuni ceppi virali per entrare nelle cellule bersaglio (Choe et al., 1996; Doranz et al., 1996; Liao et al., 1997).
Ancora non si sa con certezza quali siano le cellule che vengono infettate da HIV nelle fasi precoci dell’infezione primaria, né si conoscono i meccanismi
attraverso i quali il virus riesca ad oltrepassare le mucose nel corso di un’infezione contratta per via sessuale. Sicuramente un processo infiammatorio preesistente o una lesione di continuo a livello della porta di ingresso dell’infezione favoriscono il superamento della barriera mucosale (Royce et al., 1997; Fleming et al., 1999; Wang et al., 2005).
In studi condotti su macachi infettati con SIV per via intravaginale è stato osservato che la prime cellule a contenere DNA virale, due giorni dopo l’esposizione al virus, sono le cellule dendritiche dei tessuti periferici (Spira et al., 1996). Le cellule dendritiche, che sono in grado di trattenere il virus infettante sulla loro superficie per prolungati periodi di tempo, si legano ad antigeni virali, li processano in peptidi associati all’MHC, migrano nelle regioni paracorticali degli organi linfatici, dove interagiscono con le cellule T.
La precisa natura degli eventi che si susseguono nelle fasi precoci di un’infezione primaria da HIV e che conducono alla replicazione virale nei tessuti linfatici, non è stata ben chiarita. La capacità di HIV di infettare le cellule dendeitiche tissutali è limitata, dal momento che queste cellule esprimono bassi livelli di CD4 (Mc Ilroy et al., 1995). Studi in vitro hanno dimostrato che le cellule dendritiche ed i linfociti T CD4+ sono in grado di formare dei sincizi in cui HIV è capace di replicare attivamente (Pope et al., 1996).
Simili sincizi contenenti antigeni virali sono stati identificati in vivo nel tessuto tonsillare (Frankel et al., 1996), nel sangue periferico (Weissman et al., 1995) e nella sottomucosa vaginale di soggetti che presentavano un’infezione da HIV.
Inoltre, recentemente è stata dimostrata l’importanza di una molecola di adesione cellulare, l’ICAM-3, espressa dalle cellule dendritiche dell’area T-cellulare di tonsille, linfonodi e milza, nel favorire l’interazione tra HIV e cellule dendritiche e probabilmente nel consentire la trasmissione di HIV dalle cellule dendritiche ai linfociti T (Geijtenbeek et al., 2000).
La replicazione di HIV inizia a livello della porta d’entrata del virus, in modo tale da amplificare la carica virale iniziale (Miller et al., 2005; Haase, 2005). Successivamente le cellule dendritiche trasportano HIV ai linfonodi loco-regionali, dove le cellule stesse interagicono con i linfociti T che vengono così infettati. L’infezione a carico del sito d’ingresso del virus e dei linfonodi loco-regionali si realizza entro 72 ore dall’esposizione ad HIV. A questo punto incomincia una nuova replicazione virale e l’infezione diviene sistemica entro la fine della prima settimana, quando ha inizio l’infezione massiva a carico delle cellule T CD4+ (Picker, 2006).
Durante l’infezione primaria HIV utilizza come principale co-recettore il CCR5, espresso da un’ampia gamma di cellule T CD4+, presenti in ogni sito extra-linfonodale dell’organismo, inclusi polmoni, fegato, mucosa intestinale, mucosa genitale e cute (Douek et al., 2003; Vazey et al., 2003).
Dopo dieci giorni dall’inizio dell’infezione, la maggior parte dei linfociti T CD4+ presenti nei tessuti extra-linfonodali è stata infettata o ha interagito con il virus in modo tale da essere indotta all’apoptosi (Li et al., 2005; Mattapallil et al., 2005). Questa massiva replicazione virale determina il picco di viremia dell’infezione primaria e si traduce in una drammatica riduzione dei linfociti T CD4+ nei siti extra-linfonodali tra il decimo ed il ventunesimo giorno dall’infezione (Picker et al., 2004; Li et al., 2005; Mattapallil et al., 2005). Contemporaneamente le cellule T CD4+ sopravvissute manifestano un incremento delle loro capacità proliferative (Picker et al., 2004). Ciò determina due conseguenze: la proliferazione linfocitaria contrasta la riduzione delle cellule T CD4+ nei siti extra-linfonodali, ma, allo stesso tempo, vengono prodotti nuovi bersagli capaci di sostenere la replicazione virale (Picker, 2006).
Con la deplezione dei linfociti T CD4+ CCR5+ si assiste alla comparsa di varianti virali capaci di colpire nuovi bersagli cellulari (Picker et al., 2004; Li et al., 2005), utilizzato come principale co-recettore non più il CCR5, ma il CXCR4 (Moore et al., 2004).
La massiva proliferazione delle cellule T CD4+ è solo una componente dell’attivazione generalizzata del sistema immunitario, che coinvolge, oltre ai linfociti T CD4+, Linfociti T CD8+, cellule NK, linfociti B ed altre cellule del sistema immunitario (Picker et al., 2004). L’intensità e la persistenza di tale attivazione potrebbero essere dovute alla distruzione da parte di HIV di cellule regolatrici del sistema immunitario (Eggena et al., 2005), al sovvertimento dell’architettura del sistema linfatico indotto da HIV (Shaker et al., 2002) o ad una compromissione dell’integrità delle mucose, tale da esporre cronicamente il sistema immunitario all’invasione di agenti esterni (Brenchley et al., 2006).
Questa attivazione generalizzata del sistema immunitario determina, inoltre, aumento dell’apoptosi e promozione di meccanismi di distruzione cellulare, riduzione delle capacità rigenerative ed alterazione delle funzioni cellulari (Brenchley et al., 2006; Grossman et al., 2006). E’ stato stimato che la metà delle cellule T CD4+ dell’organismo vengano irrimediabilmente perdute durante l’infezione acuta da HIV (Peter et al., 2006).
I meccanismi attraverso i quali si realizza la deplezione delle cellule T CD4+ sono numerosi. HIV è in grado di infettare direttamente le cellule T CD4+ e di indurne la morte per apoptosi, mediante la traduzione di segnali intracellulari aberranti (Ascher et al., 1990) o per effetto citotossico diretto. Inoltre, numerosi studi dimostrano che l’infezione da HIV determina un aumentato turnover delle cellule T CD4+ (Hoetal., 1995; Lempicki et al., 2000).
Anche fenomeni autoimmuni si pensa siano in grado di favorire la perdita di linfociti T CD4+ nei soggetti infetti (Golding et al., 1988; Golding et al., 1989).
Nel corso di un’infezione da HIV si assiste anche ad una ridotta produzione di cellule T CD4+, conseguente alla distruzione del microambiente timico (Stanley et al., 1993) ed alla deplezione virus-indotta dei timociti (Valentin et al., 1994).
Nonostante i linfociti T CD4+ siano sicuramente le cellule maggiormente colpite, HIV è in grado di danneggiare il funzionamento di tutto il sistema immunitario (Picker , 2006).
Durante la progressione della patologia è possibile osservare un’alterazione nel numero e nel funzionamento dei linfociti T CD8+ (Douek et al., 1998). Infatti, nei soggetti con un’infezione da HIV si riscontra la presenza di anormalità nel fenotipo delle cellule T CD8+. Tali anomalie sembrano avere un importante significato prognostico: dopo la sieroconversione, la mancata espressione da parte dei linfociti T CD8+ della molecola CD38 si associa ad una ridotta deplezione dei linfociti T CD4+ e ad una più lenta progressione della patologia (Giorni et al., 1994; Mocroft et al., 1997).
Nel corso dell’infezione primaria da HIV si assiste ad una riduzione nel numero dei linfociti B circolanti, anche se questo è generalmente un fenomeno transitorio, indicativo di una ridistribuzione cellulare nei tessuti linfatici (Amadori et al., 1990).
Anche anomalie delle cellule NK sono presenti durante tutti il corso dell’infezione da HIV. E’ stato dimostrato che la carica virale è inversamente proporzionale alla capacità delle cellule NK di sopprimere la replicazione di HIV (Kottilil et al., 2003).
Nel corso delle diverse fasi dell’infezione da HIV si assiste ad un’iperattivazione dei neutrofili (Elbim et al., 1994), associata ad un ridotta capacità di opsonizzazione degli stessi, che diviene sempre più evidente con il progredire della malattia (Tachavanic et al., 1996).
Controversa è la capacità del virus di ridurre l’abilità con cui le cellule dendritiche sono in grado di attivare i linfociti T (Macatonia et al., 1990; Roberts et al., 1994; Cameron et al., 1992).
Infine, risulta essere fondamentale il ruolo svolto dalle cellule della linea monocito-macrofagica nell’immunopatogenesi dell’infezione da HIV. Queste cellule costituiscono una riserva dell’infezione virale e la loro disfunzione
contribuisce al ridotto funzionamento dei linfociti T CD4+ e alla diminuita capacità di difesa dell’ospite nei confronti dei patogeni intracellulari (Fauci,1993). I monociti esprimono sulla loro superficie il CD4 e numerosi co-recettori, in particolare il CCR5 (Cheng-Mayer et al., 1997; He et al., 1997). Diversamente da quanto avviene per i linfociti T CD4+, HIV tende a non avere un’azione citopatica diretta nei confronti delle cellule della linea monocito-macrofagica, ma preferenzialmente si replica intensamente all’interno di tali cellule (Weinberg et al., 1991). I macrofagi cronicamente infettati possono sopravvivere a lungo e proteggere le particelle virali dalla degradazione per numerosi mesi (Smith et al., 2001). Tali riserve si stabiliscono entro il primo mese dall’inizio dell’infezione (Finzi et al., 1997).
E’ difficile riscontrare in vivo la presenza di monociti circolanti infetti (Weinberg et al., 1991), mentre il virus può essere facilmente ritrovato nei macrofagi tissutali ed in particolare nelle cellule della microglia (Koening et al., 1986), nei macrofagi degli alveoli polmonari e nei precursori dei monociti del midollo osseo (Armstrong et al., 1984; Plata et al., 1987; Collman et al., 1989). Le cellule della linea monocito-macrofagica hanno un ruolo chiave nella patogenesi delle malattie a carico del sistema nervoso centrale indotte da HIV (Britton et al., 1984; Neilson et al., 1984; Faulstich et al., 1986) e nel determinare alcune delle anomalie ematiche che si riscontrano nei soggetto con un’infezione da HIV (Orenstein et al., 1997).
Inoltre, l’infezione da parte di HIV dei monociti e dei macrofagi o la loro esposizione a proteine virali induce anomalie nella presentazione dell’antigene, con conseguente ridotta attivazione dei linfociti T CD4+ (Meyaard et al.,1993).
Oltre alle cellule della linea monocito-macrofagica, anche alcuni linfociti T CD4+ della memoria possono presentare il genoma virale integrato nel loro DNA. Le cellule con un’infezione latente non producono spontaneamente particelle virali, a meno che non vengano attivate (Zhang et al.,1999; Furtado et al.,1999). Queste riserve dell’infezione, che hanno una sopravvivenza superiore
ai 44 mesi, costituiscono uno dei principali ostacoli all’eradicazione del virus da parte delle attuali terapie antiretrovirali (Finzi et al.,1997; Smith et al.,2001).
L’intensa replicazione virale e l’estesa distruzione dei linfociti T CD4+ che si realizzano nel corso dell’infezione acuta da HIV colpiscono duramente il sistema immunitario agli esordi dell’infezione e pongono le basi per la sua successiva insufficienza (Picker,2006).
Controllo della risposta immunitaria contro HIV
Nonostante la risposta immunitaria ad HIV sia stata ampiamente studiata, non è ancora stato completamente chiarito come il virus riesca ad eludere il controllo immunologico nella maggior parte dei soggetti infettati.
Anticorpi anti-HIV iniziano ad essere rilevati nel plasma dopo 2-4 settimane dall’inizio dell’infezione (Henrad et al., 1995; Fiebig et al., 2003; Rybarczyk et al., 2004). Questi anticorpi non sono comunque in grado di ridurre la carica virale nel sangue, nelle secrezioni genitali o in altri compartimenti fino all quarta settimana dall’inizio dell’infezione (Pilcher et al., 2001; Pullium et al., 2001; Pilcher et al., 2004). Oltre alla scarsa immunogenicità delle glicoproteine dell’envelope, anche la loro struttura e diversità contribuiscono a determinare l’incapacità da parte della risposta immunitaria anticorpale nel neutralizzare il virus.
Tra gli anticorpi diretti contro le proteine di HIV si pensa che solo quelli rivolti contro le glicoproteine dell’envelope virale, che compaiono tra l’ottava e la ventiquattresima settimana dall’inizio dell’infezione (Stafford et al., 2000), conferiscano una protezione immunitaria, impedendo a gp120 di legarsi al CD4 ed ai co-recettori (Trkola et al., 1996). I siti di legame di gp120 rimangono inaccessibili nella conformazione nativa della glicoproteina e, affinché gli anticorpi siano in grado di legarvisi, occorrono dei cambiamenti conformazionali indotti dal legame con il CD4 (Sattentau et al., 1993; Saphire et al., 2001; Kwong et al., 2002). Secondo alcuni studi anticorpi in grado di legare
le particelle virali ma non i siti di legame del CD4 e dei co-recettori sarebbero ugualmente capaci di bloccare le interazioni tra virus e cellula in modo tale da neutralizzare HIV (Stott, 1994; Arthur et al., 1995; Parrei et al., 1998).
Linfociti T citotossici CD8+ HIV-specifici si ritrovano nel sangue periferico nei primi mesi dell’infezione (Walker et al., 1987; Koup et al., 1994) e svolgono un ruolo importante nel controllo della replicazione virale. Esiste infatti un’associazione temporale tra il picco della risposta citotossica specifica nei confronti del virus e la riduzione della viremia nel corso dell’infezione acuta (Walker et al., 1986; Walker et al., 1987). In scimmie infettate da SIV è stato dimostrato come la deplezione delle cellule T CD8+ corrisponda ad una ripresa della replicazione virale (Matano et al., 1998; Schmitz et al., 1999): è stato ipotizzato che un numero particolarmente basso di linfociti T CD8+ HIV-specifici consentirebbe ad HIV di sfuggire alla risposta immunitaria (Kalams et al., 1999). Il controllo della replicazione virale da parte dei linfociti T CD8+ sarebbe invece favorito dalla capacità delle cellule T CD8+ di reagire nei confronti di un elevato numero di peptidi virali. Durante l’infezione acuta l’espansione delle cellule T CD8+ HIV specifiche è limitata ad un numero ridotto di cloni. Un’espansione monoclonale nel corso dell’infezione acuta è associata ad una prognosi peggiore (Pantaleo et al., 1994). Inoltre, numerosi studi sostengono che una precoce risposta immunitaria da parte dei linfociti T CD8+ specifica nei confronti di prodotti dei geni virali tat o rev sia associata ad una migliore evoluzione della patologia (Van Baalen et al., 1997; Addo et al., 2001).
I linfociti T CD8+ sono anche in grado di secernere fattori solubili capaci di inibire la replicazione virale. Le chemochine MIP-1α, MIP-1β e RANTES sono capaci di bloccare l’ingresso del virus nelle cellule bersaglio (Cocchi et al., 1995). Oltre alla chemochine esistono altri fattori secreti dalle cellule T CD8+ capaci di controllare la replicazione virale sopprimendo la trascrizione di HIV nelle cellule infettate (Zhang et al., 2002; Chang et al., 2003).
Numerosi studi sugli animali hanno dimostrato che le cellule T CD4+ HIV-specifiche hanno un ruolo fondamentale nell’indurre e nel mantenere una risposta immunitaria citotossica (Matloubian et al., 1994; Cardin et al., 1996). Nella maggior parte dei pazienti non trattati l’infezione da HIV è caratterizzata dall’assenza di una risposta proliferativa da parte dei linfociti T CD4+ HIV-specifici (Warhen et al., 1987). Poiché il principale bersaglio di HIV sono proprio le cellule T CD4+, si pensava che la mancata proliferazione dei linfociti T CD4+ virus-specifici fosse dovuta alla deplezione virus-indotta dei linfociti T CD4+ nei tessuti linfatici; alcuni studi hanno invece dimostrato che l’assaenza della proliferazione delle cellule T CD4+ HIV-specifiche sia in parte da attribuire agli elevati livelli di antigene virale (Carney et al., 1981; Boni et al., 1998).
Meccanismi virali di elusione della risposta immunitaria
Lo straordinario polimorfismo di HIV contribuisce a rendere difficile la neutralizzazione del virus. La variabilità genetica virale è molto elevata sia nelle popolazioni geograficamente definite, sia nei singoli pazienti. Le mutazioni che compaiono durante la retrotrascrizione, l’alto tasso di replicazione virale e la prolungata durata dell’infezione sono responsabili della genesi di varianti virali che coesistono nel plasma del medesimo soggetto e non vengono riconosciute dal sistema immunitario (Coffin, 1995). Infatti, i livelli di anticorpi capaci di neutralizzare il virus aumentano precocemente durante l’infezione, ma HIV è ugualmente in grado di eludere la risposta immunitaria umorale mutando le sequenze delle proteine dell’envelope delle particelle virali circolanti (Richman et al., 2003). Inoltre, è proprio la risposta immunitaria HIV-specifica responsabile della selezione di mutanti virali capaci di sfuggire alla neutralizzazione (Richman et al., 2003; Frost et al., 2005).
HIV non soltanto è in grado di eludere la risposta immunitaria umorale, ma anche di sfuggire alla risposta immunitaria cellulo-mediata. Studi delle
sequenze virali hanno dimostrato la comparsa di mutazioni che impediscono il legame tra le molecole dell’MHC di classe I e gli epitopi virali (Borrow et al., 1997; Goulder et al., 1997). Inoltre, è stato osservato che prodotti dei geni virali
nef, tat e vpu sono in grado di ridurre l’espressione sulla superficie cellulare delle molecole dell’MHC di classe I, necessarie per il riconoscimento delle cellule infettate (Howcroft et al., 1993; Schwartz et al., 1996; Kerkau et al., 1997). Questo consentirebbe alle cellule infettate di evitare la lisi cellulo-mediata (Kalams et al., 1999).
Valore predittivo dei marker immunologici e virologici
I marker di laboratorio considerati predittivi della progressione dell’infezione sono la carica virale, la conta delle cellule T CD4+ ed i marker di attivazione del sistema immunitario, come l’espressione del CD38 da parte dei linfociti T CD8+ (Giorni et al., 2002).
Alcuni studi hanno dimostrato che la viremia e l’espressione del CD38 abbiano un valore predittivo maggiore rispetto alla conta dei CD4 nelle fasi precoci dell’infezione (Giorni et al., 2002). E’ stato ipotizzato che questo sia dovuto al fatto che la carica virale all’inizio dell’infezione sia in grado di condizionare l’entità dell riduzione delle cellule T CD4+ nelle fasi successive della malattia (Mellors et al., 1997).
La conta dei linfociti T CD4+ ha invece un valore predittivo superiore rispetto a quello della viremia in una fase più tardiva dell’infezione ed è in grado di prevedere con maggior precisione l’evoluzione verso l’AIDS e la progressione della patologia verso la morte (Bruiste net al., 1997; Spijkman et al., 1997).
L’espressione del CD38 da parte delle cellule T CD8+ sembra avere un elevato valore predittivo sia nelle fasi precoci che in quelle più avanzate dell’infezione (Giorni et al., 2002).
Nei soggetti con una bassa carica virale è stato osservato che la conta delle cellule T CD4+ ha un valore predittivo di evoluzione della malattia maggiore rispetto alla viremia e all’espressione del CD38 (Giorni et al., 2002).
L’attivazione del sistema immunitario svolge un ruolo importante nel condizionare la progressione della patologia in una fase tardiva della malattia (Liu et al., 1998; Giorni et al., 1999). Uno studio condotto su individui affetti da AIDS, che presentavano una conta dei CD4<50 cell/mmc, ha messo in evidenza come l’attivazione dei linfociti T, misurata come espressione del CD38 da parte dei CD4 e dei CD8, sia in grado di predire l’evoluzione della patologia verso la morte (Giorni et al., 1999).
MANIFESTAZIONI CLINICHE DELL’INFEZIONE PRIMARIA
I segni ed i sintomi che compaiono durante l’infezione primaria da HIV possono essere provocati dall’elevata carica virale e dall’intensa risposta immunitaria che si osservano nelle prime settimane dal contagio oppure possono essere il risultato della severa ma transitoria immunosoppressione che si verifica nelle fasi precoci dell’infezione.
Manifestazioni cliniche associate all’infezione primaria vennero per la prima volta descritte nel 1984 (anonimo, 1984). Nel 1985 uno studio condotto da Cooper et al. metteva in evidenza la comparsa di una sindrome simil mononucleosica durante la fase acuta dell’infezione da HIV.
Sindrome retrovirale acuta
Nelle settimane successive al contagio, circa i due terzi dei pazienti sviluppano segni e sintomi attribuibili ad una sindrome retrovirale acuta (Daar et al., 2001; Hecht et al., 2002; Celum et al., 2001). La sintomatologia compare dopo un periodo d’incubazione di 1-6 settimane dall’esposizione al virus, con un picco intorno alla terza settimana.
Le manifestazioni cliniche della sindrome retrovirale acuta sono aspecifiche ed estremamente variabili (Kahn et al., 1998; Quinn, 1997). I sintomi più frequenti sono: febbre, sudorazione notturna, mialgia, anoressia, nausea, diarrea e faringite non essudativa. Numerosi pazienti presentano anche segni e sintomi d’irritazione meningea, cefalea e fotofobia. In più della metà dei casi si verifica la comparsa di un esantema maculopapuloso, simile a quello della rosolia, che coinvolge prevalentemente il tronco e gli arti superiori. Una minoranza dei pazienti può presentare meningite asettica ed altre patologie neurologiche, tra cui encefalite, neuropatia periferica e sindrome di Guillain-Barrè.
Generalmente l’esame obiettivo mette in evidenza la presenza di linfoadenopatia generalizzata, rash cutaneo e, meno frequentemente, epatosplenomegalia. In numerosi casi è stata segnalata anche la comparsa di ulcere orali e genitali e di candidosi orale ed esofagea. La linfoadenopatia generalizzata compare precocemente nel corso dell’infezione da HIV e tende a persistere sino a fasi avanzate della malattia. I linfonodi principalmente interessati sono i cervicali anteriori e posteriori, i sottomandibolari, gli occipitali e gli ascellari; meno frequentemente sono coinvolti anche i linfonodi femorali ed epitrocleari.
In alcuni casi la sindrome retrovirale acuta può associarsi allo sviluppo di infezioni opportunistiche, come polmoniti batteriche, meningiti criptococciche ed esofagiti da Candida, probabilmente dovute alla riduzione dei CD4 che generalmente accompagna l’infezione acuta da HIV.
Solitamente la sintomatologia della sindrome retrovirale acuta regredisce in 10-15 giorni.
Nelle fasi iniziali della sindrome i dati di laboratorio mettono in evidenza un calo di tutta la popolazione linfocitaria. Dopo alcune settimane dalla comparsa della sintomatologia sia i linfociti CD4 che i CD8 iniziano ad aumentare; la crescita dei CD8 è però più rapida di quella dei CD4, per cui si assiste ad un’inversione del rapporto CD4/CD8, che persisterà anche dopo la risoluzione della sindrome acuta.
L’antigene p24 di HIV può essere rilevato nel siero e nel liquor del 75% dei pazienti con un’infezione primaria da HIV dopo due settimane dall’esposizione al virus, spesso in concomitanza con la comparsa dei sintomi (Goudsmit et al., 1986; Kessler et al., 1987). Comunque, il marker di laboratorio più sensibile per la diagnosi di sindrome retrovirale acuta è la carica virale plasmatica. Infatti, durante la fase acuta dell’infezione da HIV i livelli di RNA virale sono fortemente aumentati nella maggior parte dei pazienti e possono raggiungere valori superiori a 106
La presenza o assenza di sintomi durante l’infezione primaria da HIV può avere un importante significato prognostico. E’ stato dimostrato che sia l’intensità che la durata della sindrome retrovirale acuta sono in grado di influenzare la progressione della patologia (Koup et al., 1994; Borrow et al., 1994). Infatti, la durata e l’intensità della sintomatologia durante l’infezione acuta correlano con l’entità del picco di viremia raggiunto nelle prime 2-3 settimane dall’inizio dell’infezione. Un picco di viremia elevato nella fase acuta dell’infezione si associa ad un elevato livello di replicazione virale (viral set
point), che si traduce, a sua volta, in una drastica riduzione dei linfociti T CD4+: l’intensa replicazione virale e l’estesa distruzione dei linfociti T CD4+ che si realizzano nel corso di un’infezione acuta da HIV colpiscono duramente il sistema immunitario ponendo le basi per la sua successiva insufficienza (Picker, 2006).
Manifestazioni cutanee e mucose
Oltre all’esantema maculopapuloso caratteristico della sindrome retrovirale acuta, un’altra manifestazione cutanea che può presentarsi nel corso dell’infezione primaria da HIV è la dermatite seborroica. Questa patologia della cute si sviluppa a seguito di un processo infiammatorio a carico delle ghiandole sebacee e si manifesta con la comparsa di lesioni eritematose ed aree di disepitelizzazione localizzate a livello della faccia e della regione inguinale.
Ulcere orali, genitali ed anali possono comparire in ogni fase dell’infezione da HIV. Le ulcere orali sono piccole, hanno un fondo eritematoso e si localizzano sulle labbra, sul palato duro e sulla mucosa geniena. Le ulcere genitali ed anali sono spesso localizzate nel sito d’ingresso di HIV (Gains et al., 1988).
Gengiviti severe e periodontiti sono state osservate in pazienti con infezione primaria da HIV. L’esame obiettivo può mettere in evidenza arrossamento ed erosione gengivale, necrosi ed ulcerazione delle papille
interdentali, perdita di denti. L’eziologia di queste manifestazioni non è stata ancora chiarita.
Infine, durante l’infezione primaria da HIV, così come in qualsiasi altra fase dell’infezione, è possibile osservare la comparsa di candidosi orale ed esofagea. La forma di candidosi orale che si manifesta più frequentemente nelle fasi precoci dell’infezione da HIV è il mughetto: caratteristiche placche biancastre, facilmente asportabili, si localizzano tipicamente sul palato molle, sulle tonsille e sulla mucosa geniena.
Manifestazioni muscoloscheletriche
La polimiosite è una complicanza dell’infezione da HIV e può comparire anche in fasi precoci dell’infezione stessa (Dalakas et al., 1986). Si manifesta clinicamente con la comparsa di mialgia, debolezza dei muscoli prossimali, astenia e calo ponderale (Kaye, 1989).
Piuttosto frequente è l’interessamento delle articolazioni nel corso dell’infezione primaria da HIV. Sebbene alcuni retrovirus siano stati associati allo sviluppo di artropatie negli animali, la capacità di HIV di provocare direttamente un’infezione articolare rimane dubbia.
Manifestazioni renali
In alcuni pazienti con infezione primaria da HIV è stato descritto lo sviluppo di una nefropatia HIV relata (Del Rio et al., 1990; Rao, 1998), la cui patogenesi non è stata ancora chiarita: HIV potrebbe essere in grado di danneggiare direttamente le cellule dei tubuli e dei glomeruli renali. Inoltre, è stato osservato come la nefropatia da HIV sia più comune nella razza nera, suggerendo l’esistenza di una predisposizione genetica capace di favorire lo sviluppo di questa patologia (Kimmel et al., 2003).
Manifestazioni neurologiche
In alcuni casi l’infezione primaria da HIV può manifestarsi con la comparsa di sintomi neurologici.
La meningite asettica è sicuramente la patologia neurologica che compare più frequentemente durante le fasi precoci dell’infezione da HIV. Si manifesta con cefalea, rigidità nucale e febbre, spesso in associazione con nausea e vomito. In alcuni casi possono comparire sintomi dovuti all’interessamento del quinto, sesto ed ottavo paio dei nervi cranici. Generalmente questa meningite tende a risolversi spontaneamente, anche se raramente può complicarsi con lo sviluppo di encefalopatia.
Nel periodo della sieroconversione alcuni pazienti possono sviluppare una polineuropatia infiammatoria demielinizzante (IDP). La patologia insorge con la comparsa di parestesie, seguita da progressiva debolezza muscolare ed ariflessia. Anche i muscoli respiratori possono essere coinvolti ed in alcuni casi bisogna ricorrere alla ventilazione assistita. La sintomatologia raggiunge il suo acme entro le prime quattro settimane, anche se a volte si può assistere ad una cronicizzazione della patologia.
Infine, anche la mononeurite multipla può essere una manifestazione dell’infezione primaria da HIV. I pazienti affetti da questa patologia sviluppano un deficit motorio o sensitivo a carico di uno o più nervi periferici, anche se sono stati descritti dei casi con interessamento del nervo faciale e laringeo. Nei pazienti con infezione primaria la patologia tende a risolversi spontaneamente (Simpson et al., 1992).
TERAPIA ANTIRETROVIRALE
La storia della terapia antiretrovirale, rivolta ai pazienti affetti da immunodeficit HIV relato, inizia nel 1987, con l’approvazione da parte della Food and Drug Administration (FDA) del primo farmaco a dimostrata efficacia terapeutica: l’azidovudina (AZT). Negli anni successivi sono stati approvati ed introdotti altri agenti antiretrovirali, capaci di bloccare il ciclo replicativo di HIV. I primi regimi terapeutici utilizzati comprendevano uno o due farmaci e spesso erano destinati al fallimento, ma, a partire dal 1995, con l’introduzione dell’HAART (highly active antiretroviral therapy) , che consiste nell’associazione terapeutica di almeno tre agenti antiretrovirali, il trattamento dell’infezione da HIV ha subito una svolta radicale, con un forte incremento nella sopravvivenza dei soggetti trattati.
FARMACI ANTIRETROVIRALI
Le classi di farmaci attualmente utilizzate nella terapia dell’infezione da HIV sono quattro:
• inibitori nucleosidici e nucleotidici della trascrittasi inversa (NRTI) • inibitori non nucleosidici della trascrittasi inversa (NNRTI)
• inibitori della proteasi (PI) • inibitori della fusione.
Inibitori nucleosidici e nucleotidici della trascrittasi inversa
Gli analoghi nucleosidici della trascrittasi inversa inibiscono la replicazione di HIV, interferendo con la funzione polimerasica della trascrittasi inversa (RT) virale. Questi farmaci, una volta entrati nella cellula bersaglio, devono essere attivati nella forma trifosfato da parte di chinasi cellulari (Yarchoan et al., 1989). Dal momento che l’attività della nucleoside chinasi
varia a seconda della cellula bersaglio e del suo stato di attivazione, ugualmente la capacità di bloccare la replicazione virale da parte degli analoghi nucleosidici varia nei diversi compartimenti cellulari (Gao et al., 1993). Le forme trifosfato dei farmaci hanno un’elevata affinità per la trascrittasi inversa virale e competono per il legame con i substrati naturali dell’enzima. Gli analoghi nucleosidici vengono incorporati nella catena di DNA nascente, provocando il precoce arresto della sintesi del DNA (Yarchoan et al., 1989; Palmer et al., 1998).
A differenza dei nucleosidici,gli analoghi nucleotidici necessitano di sole due fosforilazioni per essere attivati e, dal momento che gli enzimi responsabili della loro attivazione sono ubiquitari, gli inbitori nucleotidici della trascrittasi inversa possono svolgere la loro attività antiretrovirale nei confronti di un maggior numero di tessuti e bersagli cellulari.
Condividendo il meccanismo d’azione, gli NRTIs hanno anche effetti collaterali analoghi. Questi farmaci possono essere associati ad iperlactatemia asintomatica e, più raramente, ad acidosi lattica sintomatica e a severa epatomegalia con steatosi (Lonergan et al., 2000; Carr et al., 2003). Tali effetti avversi possono essere dovuti alla capacità degli NRTIs di inibire la DNA polimerasi γ, coinvolta nella replicazione mitocondriale (Cui et al., 1996; Birkus et al., 2002).
L’azidovudina (3’-azido-2’,3’-dideossitimidina; AZT) è un analogo del nucleoside timidina. La forma trifosfato del farmaco è un inibitore competitivo con affinità per la trascrittasi inversa 100 volte superiore rispetto al substrato naturale dell’enzima, la deossitimidina trifosfato (dTTP). L’azione del farmaco è dovuta alla sostituzione del gruppo ossidrile in posizione 3’ del ribosio con un gruppo azide. Ciò impedisce la formazione del legame 3’-5’ fosfodiesterico con il nucleoside successivo, provocando l’arresto della trascrizione della catena di DNA nascente (Furman et al., 1986; St Clair et al., 1987).