INTRODUZIONE
GENERALE
ISCHEMIA MIOCARDICA
L’ischemia miocardica è determinata dall’alterazione del flusso sanguigno coronarico. Un’irrorazione cardiaca insufficiente, dovuta alla riduzione del flusso di sangue arterioso, comporta un apporto di ossigeno e nutrienti che non è in grado di soddisfare il fabbisogno cellulare. Spesso la diminuzione del flusso è dovuta ad un danneggiamento delle arterie coronarie, causato o da eventi trombotici o dalla formazione di placche alterosclerotiche nelle pareti vasali.
Fig. 1 Immagine dell’occlusione coronarica nel cuore infartuato.
L’ipossia e gli sconvolgimenti metabolici, dovuti all’ischemia, causano cambiamenti biochimici, morfologici e funzionali nella cellula del miocardio.1
La mancanza di ossigeno induce un rapido arresto della fosforilazione ossidativa mitocondriale e l’inibizione della sintesi di ATP, la principale fonte energetica cellulare. Per compensare la diminuzione di ATP, viene attivata una glicolisi anaerobica che porta ad un accumulo di ioni idrogeno e di lattato con conseguente acidosi intracellulare ed ulteriore inibizione della glicolisi.2
In presenza di ischemia possono verificarsi aritmie, soprattutto a carico del ventricolo, a seguito di un’alterazione dell’attività elettrica.3
Lo stato strutturale dei miociti ischemici è compromesso e il progressivo danneggiamento cellulare è evidenziato dalla rottura della membrana plasmatica mediante vari meccanismi.1-3Infatti, a causa dell’aumento della concentrazione
intracellulare di ioni Ca2+ e dell’accumulo di metaboliti derivanti dal metabolismo anaerobico, si ha l’attivazione di proteasi e fosfolipasi che degradano la struttura della membrana plasmatica. Inoltre il danno a carico della membrana fosfolipidica può essere causato dalla formazione di specie attive dell’ossigeno e di radicali liberi che si generano in miociti vitali in seguito alla riperfusione. Questi radicali causano la formazione di perossidi e superossidi che aumentano la superficie della zona lesa ed inducono necrosi tissutale.
Tutte queste degenerazioni alterano la permeabilità della membrana, comportano uno sconvolgimento dell’ equilibrio elettrolitico intracellulare ed un completo esaurimento dell’ATP. L’evento finale è quello di una rottura del sarcolemma dei miociti e conseguente morte cellulare.2
La gravità dell’evento ischemico e la grandezza della zona di necrosi dipende dall’entità dell’occlusione dei vasi coronarici e dalla durata dell’ipossia.4
PRECONDIZIONAMENTO ISCHEMICO DEL MIOCARDIO ( IPC)
Un breve periodo di ischemia innesca un processo cardioprotettivo in grado di incrementare la resistenza miocardica in previsione di un successivo e più acuto attacco ischemico.5 Questo meccanismo di auto-difesa è detto precondizionamento ischemico del miocardio (IPC) ed è caratterizzato da: una riduzione dell’ aritmia6, una diminuzione del metabolismo nella prima fase dell’ischemia7, un incremento del recupero post-ischemico8 ed una maggior protezione dell’endotelio coronarico9. Questo processo, osservato in molti mammiferi e anche nell’uomo, consta essenzialmente di due fasi: la prima fase detta ‘classica’ che dura da 1 a 3 ore dopo il precondizionamento ed una seconda che dura dalle 24 alle 96 ore.5
Molti agenti endogeni ed esogeni sono interessati nel processo di IPC, questi si possono distinguere in: recettore-dipendenti e recettore-indipendenti.10
Mediatori endogeni come l’adenosina, la bradichinina, la norepinefrina e gli oppiodi sono classificati come agenti recettore-dipendenti e sono capaci di innescare l’effetto cardioprotettivo attraverso l’interazione con specifici recettori a proteina G. Il legame di questi agenti con i loro recettori provoca l’attivazione della fosfolipasi C (PLC)11 che a sua volta catalizza l’idrolisi del fosfatidilinositolo 4,5-bifosfato (PIP2), generando i
secondi messaggeri inositolo 1,3,4-trifosfato (IP3) e diacilglicerolo (DAG) che vanno a
stimolare la proteina chinasi C (PKC). La PKC è un mediatore intracellulare che svolge un ruolo chiave nell’IPC.12 Attraverso un processo di fosforilazione, la PKC provoca l’attivazione di varie proteine e, tra queste, anche i canali al potassio ATP sensibili (KATP). Il coinvolgimento di tali canali KATP nell’ischemia è stato dimostrato attraverso
l’uso di farmaci bloccanti di questi canali, come la glibenclamide e il 5 –idrossi decanoato (5-HD).13
L’adenosina, che rappresenta uno dei mediatori recettore-dipendenti responsabile dell’IPC,14 in seguito all’interazione con specifici recettori A1 e A3 15, induce la
dilatazione dei vasi e riduce l’inotropismo cardiaco, aumentando l’apporto di ossigeno. Durante l’attacco ischemico, è stato osservato un incremento del livello di adenosina. Gli agenti recettori-indipendenti intervengono nel processo di IPC senza interagire con particolari recettori, tra questi un esempio è l’ NO, un fattore che aumenta durante il periodo ischemico. A testimonianza di questo se si induce una inibizione dell’enzima
NOS (NO-sintetasi) si ha una riduzione del precondizionamento ischemico nei ratti16, così al contrario con la somministrazione di farmaci donatori di NO si ha una significativa riduzione del danno miocardico post-ischemico in differenti specie animali. Il precondizionamento ischemico del miocardio comporta sostanzialmente una riduzione dell’accumulo di cataboliti come il lattato17 e quindi una diminuzione dell’ acidosi intracellulare e della glicolisi anaerobica, inoltre si ha un cambiamento della concentrazione di H+ e Na+, una down-regulation del TNFα e si ha l’attivazione dei canali al potassio ATP sensibili mitocondriali (mito-KATP).18
CANALI AL POTASSIO ATP SENSIBILI (KATP)
Struttura e caratteristiche generali
I canali al potassio ATP sensibili (KATP) sono stati scoperti per la prima volta nei
miociti cardiaci19 (1983), successivamente nelle cellule ß pancreatiche(1984) e in altri tessuti (cellule muscolari lisce vascolari, neuroni, cellule epiteliali).20-23
Il canale KATP è un complesso etero-ottamerico costituito da due subunità: una subunità
Kir6.x (Kir6.1 o Kir6.2 ) che costituisce il poro del canale e una subunità SUR che costituisce il sito recettoriale delle sulfaniluree.24 La subunità Kir 6.x è costituita da due
segmenti transmembranali, M1 e M2, connessi da una regione intracellulare selettiva per il K+, dove è presente un residuo amminoacidico comune a tutte le subunità Kir 6.x (Gly-Phe-Gly). La subunità SUR, appartiene alla superfamiglia ABC (ATP-binding cassette), ed è formata da tre domini transmembranali: TMD0, costituito da cinque segmenti, e TMD1 e TMD2, costituiti da 6.25 Tra le porzioni carbossiterminali dei domini TMD1 e TMD2 sono presenti due grandi loop intracellulari, detti NBF-1 e NBF-2.26 La subunità SUR esiste in differenti isoforme: SUR1 maggiormente localizzata a livello pancreatico, SUR2A presente soprattutto nelle cellule cardiache e SUR2B nei vasi.27
Fig. 2 a Rappresentazione schematica della struttura del canale KATP, b struttura
ottamerica formata da 4 subunità Kir 6.x e 4 subunità SUR, c immagine del modello molecolare del canale KATP nella membrana plasmatica.
Differenti combinazioni delle subunità Kir6.x e SUR portano all’espressione di varie isoforme di canali KATP localizzati in tessuti differenti e dotati di diverse caratteristiche
fisiologiche e farmacologiche28.(Tab. 1)
I canali KATP sensibili sono inibiti dall’elevata concentrazione intracellulare di ATP e
attivati dall’ MgADP.20
Subunità Kir Subunità SUR
Pancreas Neuroni Muscolo cardiaco Muscolo scheletrico Muscolo liscio Kir 6.2 Kir6.2 Kir6.2 Kir 6.2 Kir 6.2 SUR1 SUR1 SUR2A SUR2A SUR2B
Tab. 1 Isoforme dei canali KATP
Localizzazione dei canali KATP
I canali KATP sono localizzati in differenti tessuti nell’organismo dove svolgono
importanti funzioni fisiologiche. Sono maggiormente espressi in : Cuore
Cellule ß delle isole di Langerhans pancreatiche Cellule muscolari
Neuroni
Ruolo dei canali KATP nel pancreas e nel diabete.
Nella regione pancreatica i canali KATP sensibili sono ampiamente espressi a livello
delle cellule ß delle isole di Langerhans e sono formati dalla subunità Kir 6.2 e dalla SUR129.
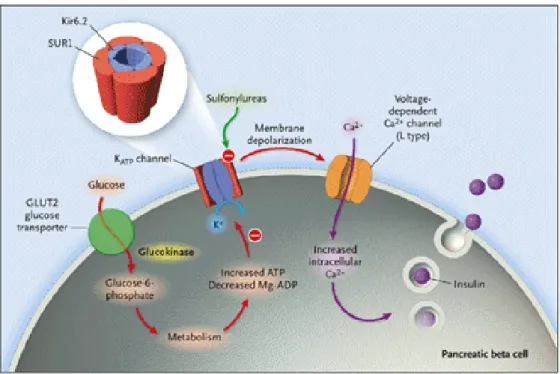
I canali KATP svolgono un ruolo chiave nella regolazione della secrezione di insulina
Nelle cellule ß pancreatiche un aumento della concentrazione di ATP, causata dall’incremento del metabolismo glucidico, comporta la chiusura dei canali KATP
causando depolarizzazione della membrana. Questa depolarizzazione provoca l’apertura dei canali al Ca2+ voltaggio-dipendenti (VDCCs) e conseguente aumento della
concentrazione di Ca2+ intracellulare che induce la produzione e l’esocitosi dell’insulina da parte delle cellule ß pancreatiche. (Fig. 3)
Fig. 3 Meccanismo di liberazione dell’insulina nella cellula ß pancreatica, mediato dalla
chiusura dei canali KATP.
Nel diabete mellito (non insulino-dipendente) si ha una diminuzione della sensibilità dei canali all’aumento della concentrazione di ATP con il conseguente arresto del processo di liberazione dell’insulina.
Le sulfaniluree, farmaci di prima scelta nel trattamento del diabete mellito, come ad esempio la tolbutamide, inducono la liberazione di insulina andando proprio a bloccare i canali KATP dipendenti. Al contrario farmaci che portano all’apertura dei canali KATP
N H N S Cl O O S N H NH O O O
Fig. 4 Rappresentazione schematica dell’attività di farmaci bloccanti ed agonisti sul
canale KATP delle cellule ß pancreatiche.
Ruolo dei canali KATP nel cervello
Nel cervello i canali KATP si possono ritrovare in varie regioni: nella sostanza nigra31,
nella neocorteccia32, nell’ippocampo33 e nell’ipotalamo34.
L’attività dei canali KATP del cervelloè del tutto simile a quella descritta per i canali
delle cellule ß pancreatiche: la depolarizzazione della membrana plasmatica, dovuta alla chiusura dei canali KATP e all’ entrata di ioni Ca2+ attraverso i canali
voltaggio-dipendenti, porta ad attivazione neuronale. Ci sono inoltre analogie strutturali tra i canali neuronali e pancreatici, infatti in alcune zone del cervello questi sono costituiti dalla subunità Kir6.2 e SUR1.
I canali KATP delle cellule ß pancreatiche e i canali presenti a livello dell’ ipotalamo,
soprattutto quelli localizzati nei neuroni GRNs (glucouse-responsive neurons), hanno un’attività correlata. Infatti sia le cellule ß pancreatiche che l’ipotalamo svolgono un ruolo fondamentale nel mantenimento dell’omeostasi del glucosio attraverso la regolazione del rilascio ormonale, mediata dall’attivazione dei canali KATP.
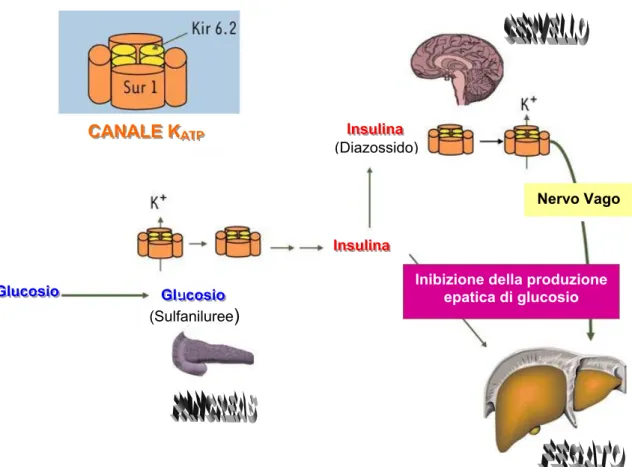
Quando il livello di glucosio nel sangue aumenta si ha l’inibizione dei canali KATP delle
cellule ß pancreatiche con conseguente liberazione di insulina e abbassamento della glicemia.
Insulina (Diazossido) IInnssuulliinnaa Insulina IInnssuulliinnaa CAAANNNALE KAALLEEKKAAATTTPPP CC Nervo Vago
Inibizione della produzione epatica di glucosio G GGlllucosio uuccoossiioo GGGuucosio (Sulfaniluree) l lluccoossiioo
Fig. 5 Rappresentazione schematica della regolazione metabolica mediata dai canali
KATP delle cellule pancreatiche e neuronali in caso di iperglicemia.
Quando invece la concentrazione di glucosio si abbassa, si ha l’attivazione dei canali KATP dei neuroni GRNs che mediante la stimolazione del sistema nervoso autonomo,
inducono la liberazione di glucagone e catecolamine da parte delle cellule α pancreatiche e si risolve così la situazione di ipoglicemia.
I canali KATP ipotalamici, analogamente a quelli localizzati in altre zone del cervello,
sono stimolati in caso di stress metabolico, ipossia o ipoglicemia. Il ruolo fondamentale svolto da questi canali è quello di prevenire i possibili danni neuronali e la neurodegenerazione conseguente alla mancanza di glucosio.35
Ruolo dei canali KATP nel sistema cardiovascolare
I canali KATP nel tessuto cardiaco svolgono varie ed importanti funzioni specialmente
nello stress metabolico causato dall’ischemia e dall’ipossia,36infatti la loro attivazione comporta un aumento della resistenza cardiaca al danno ischemico.
Durante l’ischemia si ha un decremento nella concentrazione di ATP che causa l’attivazione dei canali KATP miocardici e sarcoplasmatici. L’incremento dell’ afflusso
di ioni K+ causa l’accorciamento della durata del potenziale d’azione e l’inibizione dell’ingresso di ioni Ca2+ all’interno della cellula. Si previene così il sovraccarico di
Ca2+ e si riequilibria il bilancio tra la richiesta e l’apporto di energia alla cellula.37 Nel sistema vascolare, i canali KATP regolano il tono muscolare dei vasi e quindi anche
la pressione sanguigna. L’attivazione di questi canali, fisiologica o indotta da farmaci, porta all’iperpolarizzazione della membrana plasmatica che causa rilasciamento muscolare, forse dovuto all’inibizione dei canali al Ca2+ voltaggio-dipendenti.
Nei vasi coronarici, durante l’ischemia, i canali KATP sembrano essere coinvolti nel
processo di vasodilatazione e rilasciamento dei muscoli vasali. Infatti la glibenclamide, farmaco bloccante non selettivo dei canali KATP, inibisce questo processo di
vasodilatazione38; al contrario, la somministrazione di farmaci agonisti, come cromakalim e pinacidil, induce l’abbassamento della pressione arteriosa e il rilascio della muscolatura liscia vascolare.39
N N H N N N H PINACIDIL O N O N CROMAKALIM t-Bu N H N S Cl O O DIAZOSSIDO N N H O ONO2 NICORANDIL N H N HN CN N P-1075 OH
OMe Cl N H OMe S H N HN O O O O HMR 1098 NaOOC Me OH 5-HD N H S N H NH OMe Cl O O O O GLIBENCLAMIDE N N H N HNCN PNU-94750
Fig. 7 Struttura dei farmaci bloccanti selettivi (HMR 1098, 5-HD) e non selettivi
(glibenclamide) dei canali KATP.
Ruolo dei canali KATP nel muscolo scheletrico
La struttura dei canali KATP nel tessuto muscolare scheletrico40 non è stata del tutto
precisata, ma anch’essi come i canali KATP cardiaci sono inibiti dall’ATP e attivati da
adenosina e dai KCOs.
L’attivazione dei canali KATP risente del metabolismo glucidico e nel muscolo questo
subisce notevoli variazioni a seconda dell’attività svolta. In condizioni di riposo, i canali KATP sono chiusi, invece durante la contrazione o dopo affaticamento, questi vengono
attivati.41 L’apertura del canale aumenta l’afflusso di ioni K+ con conseguente diminuzione della durata del potenziale d’azione42 e dell’entrata di ioni Ca2+.43 Attraverso studi condotti utilizzando agonisti ed antagonisti dei canali KATP, è stato
mostrato che l’attivazione di questi canali durante lo sforzo, riduce la forza contrattile e accelera il recupero dopo la fatica, mantenendo costante il livello intracellulare di ATP.
CANALI AL POTASSIO ATP SENSIBILI (KATP) NEL TESSUTO
CARDIOVASCOLARE
Struttura dei canali al potassio ATP sensibili del tessuto cardiovascolare
I canali KATP dei cardiomiociti si possono a loro volta suddividere in: sarc-KATP , canali
localizzati nel sarcolemma e mito-KATP, localizzati invece nelle membrane interne dei
mitocondri. I canali sarc-KATP sono costituiti da un complesso ottamerico costituito da
quattro subunità Kir6.2 e quattro subunità SUR2A44(Fig. 8). La struttura dei canali mito–KATP non è ancora del tutto chiara anche se recenti studi hanno dimostrato che
sono composti dalle subunità Kir6.2 e SUR2, mentre la SUR1 sembra essere esclusa.45
Fig. 9 Struttura molecolare del canale sarc-KATP
Attività dei canali sarc-KATP e mito-KATP
L’attivazione dei canali sarc-KATP, mediata da ipossia, ischemia, o agenti farmacologici,
d’azione, e inibisce lo scambio Na+-K+, diminuendo l’entrata di ioni Ca2+ nella cellula. L’osservazione dei meccanismi intracellulari innescati dall’apertura del canale sarc-KATP, permette di ipotizzare che l’aumento della resistenza al danno ischemico sia
correlabile all’attivazione di questo canale;46 anche se studi più recenti attribuiscono ai canali mito-KATP il ruolo prominente nella protezione cardiaca contro l’ischemia.
Questa ipotesi è stata avanzata dall’osservazione che gli agonisti dei canali KATP, come
bimakalim e cromakalim, erano in grado di indurre un effetto cardioprotettivo anche a dosi prive di efficacia sul potenziale di membrana del sarcolemma.47-49
I canali mito-KATP sono localizzati nei mitocondri che costituiscono il centro di
produzione dell’energia cellulare. Quando si verifica una crisi energetica, la funzionalità mitocondriale può essere danneggiata, ma attraverso l’apertura dei canali mito-KATP si
ha un parziale ristabilimento del potenziale di membrana, così da prevenire la deplezione dei fosfati ad alta energia, creando un gradiente elettrochimico più favorevole per la sintesi di ATP.50-54
I canali sarc-KATP nell’ IPC.
L’aprikalim, farmaco agonista dei canali sarc-KATP, mima gli effetti del
precondizionamento ischemico in cani anestetizzati con barbiturici, comportando una significativa riduzione dell’entità dell’infarto; farmaci bloccanti di canali KATP, come la
glinbenclamide, invece aboliscono questo effetto protettivo.55 Questo risultato è stato confermato anche da altri studi che hanno inoltre determinato che la glinbenclamide comporta un danneggiamento delle funzioni ventricolari durante il periodo di riperfusione.56 Il ruolo dei canali sarc-KATP nell’IPC era già stato provato mediante un
bloccante di questi canali, HMR 1883, che ritardava la caduta del potenziale d’azione e riduceva l’effetto protettivo del diazossido.57
Nei miociti ventricolari di coniglio, durante un transitorio episodio ischemico, è stato notato un aumento dei livelli di adenosina che, mediante l’interazione con recettori A1 e
A358, attiva il PKC intracellulare. Questo porta ad un aumento della flusso ionico nei
sarc-KATP che può essere abolito somministrando antagonisti selettivi dell’adenosina.59
Da questo studio è stato evidenziato che l’apertura dei canali sarc-KATP è dovuta ad un
L’ NO è un altro fattore in grado di attivare i canali sarc-KATP con la differenza che ciò
può avvenire sia in condizioni di normale ossigenazione sia in ipossia.61
E’ stato scoperto che esiste un’interazione tra l’attivazione dei canali sarc-KATP e quella
dei mito-KATP, infatti l’iperpolarizzazione dovuta all’attivazione del primo può portare
all’attivazione del secondo e viceversa. L’iperpolarizzazione di membrana comporta l’attivazione della fosfolipasi D che causa a sua volta l’attivazione e la traslocazione del PKC che è responsabile dell’apertura sia dell’uno che dell’altro tipo di canale.62
Fig. 9 Rappresentazione del meccanismo di attivazione correlato del canale sarc-KATP e
del mito-KATP
Questa teoria è stata provata anche attraverso l’utilizzo di 5-HD (bloccante selettivo dei canali mito-KATP) e HMR 1098 (bloccante selettivo dei canali sarc-KATP); questi
farmaci solo se somministrati in associazione abolivano completamente l’effetto protettivo innescato dalla ipossia cronica.63
I canali mito-KATP nell’IPC
Il coinvolgimento dei canali mito-KATP nell’IPC fu confermata mediante l’utilizzo di
farmaci agonisti non selettivi (bimakalim) che, a basse dosi, non avevano effetto sui canali KATP del sarcolemma e sulla concentrazione di APD.64
L’apertura dei canali mito-KATP può essere regolata da vari fattori endogeni ed esogeni.
L’attivazione e la traslocazione di specifiche isoforme di PKC è uno dei meccanismi centrali dell’apertura di questi canali. Infatti la protezione indotta dall’ apertura del canale mito-KATP può essere abolita mediante la somministrazione di farmaci
antagonisti del PKC e nello stesso tempo la protezione dovuta all’attivazione del PKC può essere inibita da farmaci bloccanti dei mito-KATP: perciò PKC e i canali mito-KATP
sono due fattori necessari nell’evento di protezione e sono uno dipendente dall’altro.65 Inoltre è stato messo in evidenza che l’NO favorisce anch’esso l’apertura dei canali mito-KATP.66 Infatti, il diazossido è capace di indurre l’IPC, attraverso una via
NO-dipendente. Studi condotti in vivo sul cuore di coniglio hanno mostrato che la protezione indotta dal diazossido può essere abolita sia attraverso la somministrazione di 5-HD (agonista selettivo dei mito KATP) sia con inibitori dell’enzima NOS
(NO-sintetasi), confermando così il ruolo svolto dall’NO in questo fenomeno.67
Ci sono invece dati e teorie contrastanti a proposito del ruolo svolto dalle specie reattive dell’ossigeno (ROS). Recenti studi hanno evidenziato che la formazione di ROS risulta essere necessaria per l’attivazione della cardioprotezione, in quanto queste specie attivano l’IPC, aumentando la probabilità di apertura dei canali mito-KATP. Inoltre i
ROS sono responsabili dell’apertura dei pori di transizione di permeabilità mitocondriale (MPTP) durante la riperfusione68 e questo fenomeno è considerato uno delle prime cause della morte cellulare.( Fig. 10) L’apertura dei MPTP nella membrana mitocondriale comporta danni irreversibili alle cellule miocardiche, a causa del rilascio di proteine pre-apoptotiche, come il citocromo C, dai mitocondri al citosol. Però una transitoria apertura di questi pori, durante la fase di attivazione dell’IPC, potrebbe innescare l’effetto cardioprotettivo. Inoltre l’attivazione dei MPTP, ad un basso stato di conduzione, impedisce un precarico di ioni Ca2+, evitando così un ulteriore attivazione calcio-indotta che comporta una più ampia e dannosa apertura di MPTP.69
Fig. 10 Rappresentazione del poro di permeabilità mitocondriale transitorio (MPTP).
Nelle condizioni normali il poro è chiuso, mentre in seguito a stress ossidativo questo si apre, causando irreversibili danni cellulari.
La questione ancora irrisolta riguarda il ruolo dei canali mito-KATP se sono iniziatori, o
mediatori o effettori finali nell’IPC; molti autori pensano che questi canali abbiano tutti e tre questi ruoli.10
Meccanismo di protezione dei canali mito-KATP
In condizioni fisiologiche, l’entrata di K+ attraverso i canali mito-K
ATP è talmente bassa
che influenza in maniera trascurabile il potenziale di membrana mitocondriale, però causa un significativo aumento del volume della matrice. Durante l’ischemia, invece, l’afflusso di ioni K+, conseguente all’attivazione dei canali mito-KATP, produce
importanti conseguenze.70L’apertura del canale garantisce il mantenimento dell’omeostasi del volume della matrice che comporta una corretta regolazione della permeabilità della membrana ai nucleotidi (ADP e ATP). In condizioni normali, la permeabilità della membrana nei confronti di ADP e ATP è modesta, e lo scambio energetico tra mitocondri e citosol è efficientemente controllata da creatina e fosfo-creatina. In condizioni di ipossia, invece, la conduttanza della membrana esterna
diminuisce con conseguente contrazione della matrice ed espansione dello spazio tra la membrana esterna ed interna. Tutto ciò comporta un aumento della permeabilità della membrana esterna all’ADP e all’ATP e una successiva idrolisi di ATP.71-72
In queste condizioni, l’apertura dei canali mito-KATP induce un abbondante afflusso di
ioni K+ che comporta l’ingresso di acqua e ioni nel mitocondrio, causando rigonfiamento della matrice cellulare. Questo rigonfiamento diminuisce la permeabilità della membrana esterna ai nucleotidi e si ricrea un giusto gradiente per la sintesi di ATP.73
L’ apertura dei canali mito-KATP causa inoltre una depolarizzazione della membrana
interna che contrasta l’entrata di ioni Ca2+ all’interno del mitocondrio.74-75 Quando nella cellula si compromette il metabolismo energetico, come nel caso dell’ischemia, si ha una massiccia entrata di Ca2+ dal compartimento extracellulare. L’ aumento della concentrazione di Ca2+ nel citosol è in parte compensata da un passaggio di questi ioni all’interno dei mitocondri, attraverso dei canali Ca2+- selettivi.76 Alti livelli di Ca2+ mitocondriali causano la formazione di MPTP (pori mitocondriali transitori), durante la riperfusione, e questa apertura è considerata una delle prime cause che porta a morte cellulare. L’attivazione dei canali mito-KATP previene l’entrata di Ca2+ nella matrice
mitocondriale e inibisce l’apertura dei MPTP durante la riperfusione. L’inibizione dei MPTP è una degli effetti cardioprotettivi più importanti conseguenti all’attivazione dei canali mito-KATP.77
Infine recenti studi hanno mostrato che l’apertura dei canali mito-KATP comporta
un’alcalinizzazione della matrice della membrana e una conseguente diminuzione della produzione mitocondriale di specie reattive dell’ossigeno (ROS).78
AGONISTI DEI CANALI K
ATPGli agonisti dei canali KATP , detti KCOs ( Kallium channel Openers) , presentano una
pronunciata diversità chimica e appartengono a diverse classi strutturali, come i benzopirani, le cianoguanidine, le tioformammidi, le tiodiazine e i piridil-nitrati. La seconda generazione dei KCOs include i ciclobutenoidi, le strutture diidropiridiniche ed
entità chimiche completamente nuove come i carbinoli terziari (Fig. 11)
BENZOPIRANI CIANOGUANIDINE TIOFORMAMMIDI
N O N CROMAKALIM O OH N N NH N N H PINACIDIL t-Bu N S NH O APRIKALIM S
BENZOTIAZIDI PIRIDIL-NITRATI PIRIMIDIN SOLFATI
N H N S Cl O O DIAZOSSIDO N N H O N O O O NICORANDIL N N N NH2 O SO3 MINOXIDIL SOLFATO NH2
CICLOBUTENOIDI DIIDROPIRIDINICHE STRUTTURA CARBINOLI TERZIARI
H N H N O O Cl Cl WAY-15616 N H O O N ZM-244085 N H OH O O ZD-6169 CF3
RELAZIONE STRUTTURA ATTIVITA’ (SAR) A)BENZOPIRANI
I benzopirani presentano una vasta gamma di variazioni chimiche strutturali. Il cromakalim79, il prototipo di questa classe, contiene due carboni chirali; il gruppo 3-OH e l’anello 4-pirrolidinone sono in posizione trans tra loro. L’attività, come KCO, risiede nell’enantiomero 3S,4R- levcromakalim. O O N CROMAKALIM N OH
1. Variazioni strutturali in posizione 4
Maggiori modifiche sono state applicate in posizione 4 dell’anello benzopiranico.
Il gruppo carbonilico, in posizione 4 della pirrolidina del cromakalim, è stato considerato essenziale per l’attività biologica poiché i lattami sono vasodilatatori più potenti rispetto alle ammine cicliche originarie.
Le prime variazioni in posizione 4 comprendono lattami e sostituenti contenenti gruppi carbonilici in posizione α dal punto di attaccamento sul nucleo benzopiranico, mentre recenti variazioni includono sostituenti contenenti carbonili, ma non in posizione originale.
Le modifiche strutturali principali effettuate in posizione 4 comprendono: a) sostituzioni di natura ciclica;
b) sostituzioni con eteroatomi a loro volta sostituiti con raggruppamenti ciclici (sostituenti ‘a ponte’);
c) sostituzioni di tipo aciclico.
Le dimensioni del sostituente ciclico nel carbonio in posizione 4 del nucleo benzopiranico influenzano molto la potenza del farmaco: gli anelli a 6 termini mostrano una potenza superiore rispetto a quelli a 5 termini, che sono a loro volta più potenti degli anelli a 4, 7 e 8 termini.80 Ulteriori eteroatomi, sostituenti ciclici a 4 termini o
sostituenti biciclici, riducono la potenza ad eccezione del celikalim. Alcune importanti informazioni, riguardanti le conformazioni biologicamente attive dei benzopirani, sono state ottenute in seguito alla sintesi di composti spirociclici, come U96501.
O N O O CELIKALIM O N HN N O H N O O U96501 OH F F F
La forzata ortogonalità all’interno dei composti spirociclici, sostiene fortemente il presupposto che la conformazione bioattiva di sostituenti in posizione 4 con libera rotazione presenta una corrispondente ortogonalità.
La presenza di un gruppo etereo o di un ponte NH tra il sostituente in posizione 4 e l’anello benzopiranico conferisce a questi nuovi composti una maggiore potenza. I rapporti struttura-attività per i KCOs, appartenenti a questa serie di derivati, differiscono
dai composti che presentano un sostituente ciclico direttamente legato al C481. In questi
composti, la posizione occupata dal gruppo carbonilico nei composti A e B è di secondaria importanza. O N OH O N NH O A O N N H B N N N NH2 OH
La presenza di eteroatomi supplementari nell’anello, o di anelli sostituiti, non diminuisce la potenza. L’introduzione di un metile supplementare in posizione 3 (composto A) è conveniente nel caso di derivati “a ponte”, mentre è inefficace per quelli che presentano una struttura non a “ponte”.82
La sostituzione sul C4 del nucleo benzopiranico con raggruppamenti aciclici,
rappresenta il metodo più efficace per realizzare nuove strutture molto potenti (C-D). La classe delle tioammidi è particolarmente interessante perché include parecchi composti estremamente attivi, come il KC-399(C), che presenta un gruppo N-β-cianoetilico all’interno della funzione tioammidica e un sostituente -CH2F in posizione 2. KC-399 è
circa 100 volte più potente del levcromakalim e presenta un effetto antipertensivo più lungo e duraturo.83 Un profilo farmacologico simile è descritto per KC-515, che possiede anche una struttura chimica simile a C. Questo composto mostra un’analoga carbossiammide in posizione 4 e un gruppo -C2F5 in posizione 6.
O F2 C CH2F CH2F O HN N F3C KC-515 O N CH2F CH2F O -O S HN N KC-399 (C) O N O O S HN D
2. Variazioni strutturali in posizione 3
La maggior parte dei benzopirani risultano non sostituiti in posizione 3 oppure presentano un gruppo idrossilico. In una serie di derivati benzopiranici, diversamente sostituiti in posizione 3, è stato osservato che in presenza di gruppi come –CHO o –CH2OH si mantiene una moderata potenza ipotensiva mentre con gruppi come –Br o
–CH3 si ha una totale perdita di efficacia.84 Sono stati eseguiti studi sugli effetti della
trasposizione dei sostituenti dalla posizione 3 alla 4; questi hanno condotto ad una serie di composti, tipo BRL 49381, che risultano avere una potenza simile al cromakalim. L’importanza del gruppo –OH è ambigua. Mentre la presenza del 3–OH nel levcromakalim aumenta la potenza di 15 volte rispetto all’analogo con 3–H, i cromeni come KC-399, che non hanno sostituenti in questa posizione, sono di gran lunga più potenti del levcromakalim. Di conseguenza, è improbabile che il gruppo 3-OH interagisca con il sito di legame. E’ ancora più improbabile che questo gruppo stabilizzi la conformazione bioattiva. Solo in presenza del gruppo –OH in 3 è stato osservato un elevato valore del rapporto eudismico tra i due enantiomeri del cromakalim, mentre nei corrispondenti analoghi con –H in 3 il rapporto eudismico è minimo.
O N O N OH LEVCROMAKALIM O N H N HN O OH BRL49381
3. Variazioni strutturali in posizione 2
L’influenza sulla potenza dei derivati benzopiranici del sostituente nella posizione 2, dipende dalla natura del sostituente in posizione 4.
Per il cromakalim il sostituente ottimale in posizione 2 è il gruppo dimetilico, che risulta essere più attivo di quello monometilico e di quello etilico. Il composto di-idro è relativamente privo di attività.85
4. Sostituzioni aromatiche
La posizione e la natura del sostituente aromatico (E-F), sono due fattori capaci di influenzare notevolmente la potenza dei derivati benzopiranici.
L’attività è abolita da sostituzioni aromatiche in posizione 5 e 8, mentre aumenta passando alle posizioni 7 e 6. Nelle prime ricerche, la sostituzione ottimale è stata attribuita ai sostituenti elettronegativi con il seguente ordine: -NO2, -CF3, -CN, -OCF3,
-C2F5, -MeCO, -CHO, -H.
Nel NIP-121(E), la parte cianofenilica è sostituita da un anello benzossidiazolico.86 Successivamente, è stato scoperto che i sostituenti più ingombranti, come il fenilsulfonile in posizione 6 del rilmakalim, sono capaci di conferire un’elevata potenza.87 Il derivato 6-N-fenil-N-metil-sulfonamidico (F) rappresenta il più potente KCO tra i 4-piridoni-cromeni. Da studi di relazione struttura-attività e dall’analisi conformazionale, è possibile ritenere che i sostituenti in 6 accrescano l’interazione al sito di legame
. O N O OH N O N O N O OH S O O NIP-121(E) RILMAKALIM O S N O O N O F
5. Trasformazioni del nucleo benzopiranico
Il sito di legame per i benzopirani accoglie cambiamenti sia dell’anello aromatico che dell’anello pirano. La piridina può sostituire il cianofenile, come mostrato nel composto
G. La posizione 6 dell’azoto piridinico è migliore di quello in posizione 7, le posizioni 5
e 8 danno composti inattivi. I tio[3,2-b]pirani, come RWJ 29009, contengono sostituenti come il gruppo nitro sull’anello tiofenico che aumentano l’attività. La sostituzione dell’ossigeno piranico con uno zolfo, rappresenta uno dei pochi esempi di composti che mantengono una certa potenza. Rispetto al cromakalim, l’introduzione di NH o CH2
riduce la potenza di circa 10 volte, mentre il carbonile di circa 30 volte. L’ossidazione a solfossido o solfone è dannosa. Le sostituzioni più vantaggiose dell’anello piranico sono state effettuate con sistemi eterociclici, come le 1,4-benzossazine, tra cui il YM 934 che è un potente vasodilatatore con particolari effetti sulle coronarie88, e le 1,4-benzotiazine come H che mostra una buona attività sia in vitro che in vivo.
N O N O OH G N O UR-8225 O N O S N O O RWJ 29009 N O S N O + O H N O O N N O + O N YM 934 N S N O KOCN7
TRASFORMAZIONI DELL'ANELLO AROMATICO
TRASFORMAZIONE DELL'ANELLO PIRANICO
O
B)BENZO- E PIRIDO-TIADIAZINE
Il diazossido, è il prototipo della classe delle benzotiadiazine, è in grado di indurre l’apertura dei canali mito-KATP con una buona selettività, senza andare ad attivare i
canali sarc-KATP. Se usato a piccole dosi, questo causa l’apertura dei canali mito-KATP;
tuttavia se si aumentano le dosi è attivo su tutto il cuore, anche sui canali KATP dei
muscoli dell’endotelio vasale e debolmente anche nei canali sarc-KATP.89 Recenti studi
hanno mostrato che questo farmaco può causare l’attivazione dei canali sarc-KATP
durante momenti di inibizione metabolica, ma questo effetto non è dovuto ad un’ azione diretta sul canale, ma è probabilmente associato all’attivazione mitocondriale o all’inibizione della succinil deidrogenasi.90 Questo farmaco è l’unico tra gli agonisti dei canali KATP che presenta la stessa affinità di legame sia per la subunità SUR1 che per la
diazossido si ha anche l’induzione di vasorilasciamento e l’inibizione del rilascio di insulina. Infatti per lungo tempo l’uso di questa benzotiadiazina era collegato ai suoi effetti iperglicemici.91 DIAZOSSIDO S N H N Cl O O
Partendo dal diazossido, i ricercatori hanno sviluppato alcuni derivati selettivi per il pancreas e per la muscolatura liscia.92 La più ampia serie di diazossidi piridotiadiazinici, che possiede una varietà di catene laterali 3- o 4-alchil e 3-aminoalchil, è stata sintetizzata e testata su cellule ß del pancreas e in preparazioni di aorta di ratto. Da questi studi è stato ipotizzato un modello farmacoforico per l’attività agonista sui canali KATP delle cellule ß del pancreas. In particolare, la sostituzione della porzione
7-clorobenzenica con il nucleo bioisosterico piridinico ha migliorato significativamente la selettività; un esempio è il prototipo BPDZ-44. Cambiando la posizione dell’azoto sull’anello piridinico si conferisce ai derivati una selettività opposta nei confronti dei tessuti: sia BPDZ-79 e sia BPDZ-83 sono più selettivi per il rilascio dell’aorta che per l’inibizione del rilascio d’insulina dalle cellule ß del pancreas. BDPZ-79 e BPDZ-44 differiscono strutturalmente per la posizione dell’azoto piridinico, dimostrando come piccole variazioni possano modificare la selettività del tessuto.
N N H N S N H O O BPDZ-44 N N H N S N H O O N N H N S N H O O Cl BPDZ-79 BPDZ-83
Successivamente sono stati sviluppati dei composti ancora più potenti e selettivi sostituendo la piridina con il tiofene. Il composto L attiva i canali KATP pancreatici ed è
almeno 1000 volte più potente del diazossido per quanto riguarda il rilascio d’insulina.
N N NH S O O S N H N H O O O O O N H N S N H O O S Cl L I
AGONISTI SELETTIVI DEI CANALI MITO KATP (BENZOPIRANIL
CIANOGUANIDINE)
Per aumentare le capacità farmacologiche e la selettività degli agonisti verso i canali mito-KATP, sono stati sintetizzati farmaci con struttura ibrida, composti
dall’anello benzopiranico e la catena delle cianoguanidine.93 Il primo composto interessante tra questi è il BMS 180448, che presenta scarse attività vasorilascianti, ma mantiene un soddisfacente potere cardioprotettivo.94 Questo farmaco ha un effetto anti-ischemico comparabile a quello del cromakalim: accelera il recupero post-anti-ischemico delle funzioni inotropiche e diminuisce il rilascio della lattato deidrogenasi (LDH). La somministrazione di cromakalim comporta un apprezzabile aumento del flusso sanguigno coronarico, invece BMS 180448 non ha nessun effetto vascolare apprezzabile. Inoltre il cromakalim causa, a differenza dell’BMS 18448, una riduzione del APD e dell’intervallo QT. 95
O NC OH N HN CN H N Cl BMS-180448
A partire da questo farmaco, i ricercatori hanno effettuato dettagliati studi di SAR96 per ottimizzare le proprietà cardioselettive. Per quanto riguarda le modifiche sull’anello benzopiranico, la sostituzione dell’ossigeno in posizione 1 con un metilene è tollerata, invece la sostituzione con NH porta a composti inattivi. I gruppi metilici geminali sono essenziali, infatti gli analoghi demetilati sono privi di attività. La presenza del gruppo –OH in posizione 3 non è necessaria ai fini dell’efficacia del farmaco, ma se è presente, il trans-OH risulta essere più attivo del cis-OH. L’introduzione di un doppio legame 3-4 abolisce l’attività antischemica suggerendo che un carbonio sp3 è preferibile ad un C
O
R OH
HN
HN NCN
piccolo gruppo elettron-attrattore gruppo solfonammido ingombrante
essenziale gruppo lipofilo
cianoguanide simile all'urea
anello arilico importante per la cardioselettività
Al fine di aumentare l’attività vasodilatante di BMS 180448, sono stati sintetizzati una serie di composti con l’anello benzopiranico 4-(N-aril)sostituito e fra questi quello che ha rivelato la maggior attività anti-ischemica e la maggior selettività nei confronti del cuore è il BMS 191095. Questa molecola ha una selettività 30 volte maggiore rispetto al BMS 180448.97 L’attività cardioprotettiva di questo farmaco è simile a quella del cromakalim e del BMS 180448, ed è antagonizzata dall’azione di farmaci come la glibenclamide e 5-HD. Le caratteristiche farmacologiche del composto BMS 191095, rivelate mediante test in vitro, sono: miglioramento della funzione cardiaca post-ischemica, diminuzione del rilascio di LDH e l’aumento del tempo di contrazione. Inoltre, questo farmaco ha mostrato un modesto effetto su APD e sull’intervallo QT e un leggero potere vasorilasciante. Sfortunatamente il composto BMS 191095 ha mostrato tossicità neuronale che non ne permette un impiego clinico, ma resta comunque un interessante strumento per lo studio e lo sviluppo di nuove entità dotate di attività cardioprotettiva.98 O NC N N NH OH Cl BMS-191095
INTRODUZIONE
ALLA
I canali al potassio ATP dipendenti sono proteine di membrana che vengono attivate dalla presenza intracellulare di ATP e che permettono il passaggio selettivo di ioni K+ attraverso la membrana plasmatica.
Sono localizzati nella membrana sarcoplasmatica (sarc-KATP) e nella membrana
mitocondriale interna (mito-KATP) delle cellule di molti tessuti, come il tessuto
vascolare, le cellule ß delle isole di Langerhans pancreatiche, il cervello e il cuore, e la loro attivazione è quindi coinvolta nella regolazione di molte attività cellulari:
l’eccitabilità neuronale e muscolare; la regolazione del potenziale d’azione; le funzioni secretive;
la modulazione dell’omeostasi degli ioni Ca2+;
In considerazione di queste molteplici funzionalità, i canali KATP sono considerati dei
bersagli farmacologici interessanti per la cura di molte patologie. In particolare, l’attivazione dei canali KATP dei cardiomiociti comporta un effetto cardioprotettivo
durante l’ischemia e per questo vennero studiati farmaci agonisti di tali canali con attività antiischemica e cardioprotettiva.
I canali KATP dei cardiomiociti svolgono un importante ruolo nel processo di
precondizionamento ischemico del miocardio (IPC).L’IPC è un meccanismo endogeno in cui brevi periodi d’ischemia determinano un aumento della resistenza delle cellule cardiache al danno indotto da un successivo e più prolungato attacco ischemico, riducendo così l’estensione della necrosi cellulare. Questo fenomeno risulta essere attivato sia da processi recettore-dipendenti, in cui sono coinvolti mediatori endogeni come l’adenosina e la bradichinina, sia da processi recettore-indipendenti, mediati da NO e radicali liberi, che portano all’attivazione dei canali KATP.
Fig. 12 Meccanismo di attivazione dei canali KATP mediata da agenti
recettore-dipendenti.
Studi sperimentali, condotti mediante l’utilizzo di farmaci agonisti (diazossido) ed antagonisti dei canali sarc-KATP (HMR 1098) e mito-KATP (5-HD), hanno indicato che
sono i canali mito-KATP a svolgere un ruolo chiave nell’induzione dell’IPC.47-49La
possibilità di innescare questo evento protettivo per il cuore, grazie all’uso di attivatori esogeni dei canali mito-KATP, rappresenta quindi un buon punto di partenza per lo
sviluppo di farmaci antischemici innovativi.
Gli attivatori dei canali al potassio, KCOs (Kallium channel Openers), comprendono un
gruppo eterogeneo di composti appartenenti a varie classi strutturali. I KCOs di prima
generazione, come per esempio il cromakalim, presentavano effetti cardioprotettivi dovuti all’attivazione dei canali mito-KATP, però la loro somministrazione comportava
anche la comparsa di gravi effetti collaterali, tra cui un potente effetto ipotensivo dovuto essenzialmente all’azione vasorilasciante legata all’attivazione dei canali KATP
vascolari.
Successivamente la ricerca si è indirizzata verso la sintesi di derivati dotati di maggiore selettività tissutale verso i canali mito-KATP per ottenere un buon effetto anti ischemico.
Osservando le caratteristiche strutturali peculiari dei principali KCOs, si può evidenziare
che per ottenere una selettiva attività cardiaca sono necessarie le seguenti caratteristiche strutturali:
1) nucleo eterociclico benzopiranico
2) sostituenti lipofili in posizione 2 ( R', R'') 3) sostituente elettron-attrattore in posizione 6 (R) 4) sostituente lipofilo ricco di elettroni in posizione 4 (X)
X
O
R''
R'
X= catena ricca di elettroni
R'=R''=CH3 R'=CH3, R''=C2H5
R
R= CN, NO2,Br..
Fig. 13 Requisiti strutturali importanti per l’attività ischemica.
Negli ultimi anni, in letteratura sono apparsi esempi di derivati a struttura benzopiranica che presentano una buona selettività. Tra questi, i due derivati benzopiranil-cianoguanidinici, BMS 180448 e BMS 191095, presentano un’ elevata attività cardioprotettiva, una buona selettività per i canali mito-KATP e un ridotto effetto
vasorilasciante. O NC N N NH OH Cl O NC OH N HN CN H N Cl BMS-191095 BMS-180448
Fig. 14 Struttura di BMS 191095 e di BMS 180448, agonisti selettivi dei canali
Il sostituente in posizione C4 dell’anello benzilico sembrerebbe rivestire un ruolo
importante nella selettività di questi nuovi derivati.
Su questa base, nel laboratorio presso il quale ho svolto questa tesi di laurea erano stati progettati derivati che presentano in posizione 4 una struttura spiromorfolonica (A) e spiromorfolinica (B) variamente funzionalizzati. I composti sintetizzati presentavano differenti sostituenti a livello dell’anello benzilico legato all’atomo d’azoto dello spirociclo come ad esempio: il gruppo nitro (R= NO2), gruppi elettronattrattori forti
(R=-CF3, -Br), gruppi elettronattrattori deboli (R= -NHCOCH3, -NHSO2CH3) e gruppi
elettron-donatori (R = -NH2,-CH3). O X N O O O X N O A B H,p-NH2,p-CF3... X= H, Br R R R= R= 4 R= 4 R R
Alcuni dei derivati, sintetizzati in precedenza, avevano mostrato una buona attività farmacologica di protezione nei confronti dell’insulto ischemico. Tra questi, i derivati morfolonici di tipo A che presentano come sostituenti in posizione para dell’anello benzilico un gruppo amminico (-NH2) 1c, solfonamidico (-NHSO2CH3) 1b e
acetamminico (-NHCOCH3) 1a hanno mostrato una buona attività cardioprotettiva ed
un’efficacia paragonabile a farmaci come il cromakalim e diazossido.
O X N O O A 1a: -NHAc 1b: -NHSO2CH3 1c: =4-NH2
Questi composti sono stati testati come miscele racemiche alla dose di 40 mg K-1 su cuori di ratto isolati alla Langendorff sottoposti a cicli di ischemia/riperfusione.99
In particolare il derivato 1a aveva mostrato una buona attività cardioprotettiva e modesta riduzione del danno tissutale in seguito all’ischemia ( cromakalim: RPP 120' (%) heart: 31± 4; 1a: RPP 120' (%) heart: 62± 20).99 Questo derivato aveva inoltre mostrato ridotti effetti vasodilatatori tipici dei KCOs non selettivi come il cromakalim.
Sulla base del promettente profilo farmacologico di questo derivato, in questa tesi di laurea, è stato nuovamente sintetizzato il composto 1a per approfondire gli studi farmacologici anche su modello d’infarto in vivo.
In questa tesi è stato inoltre sintetizzato l’analogo di 1a (2a) in cui il carbonile dello spiromorfolone è stato sostituito con l’isostero tiocarbonile, al fine di valutare l’effetto sull’attività cardioprotettiva di questo tipo di modificazione strutturale.
O O N NHCOCH3 1a O O N NHCOCH3 2a O S
SCHEMA 1
O O O O NC OTMS O HO NH2 O HO N H O Cl O O NH O O O N O NO2 OH 3 4 5 a b 6 7 8 9 c d e fa: acetone, pirrolidina, toluene, 24h, riflusso; b: TMSCN, ZnI2, CH2Cl2, 4h, t.a.;
c: LiAlH4, THF, 2h, t.a.;
d:ClCOCH2Cl, CH2Cl2, 2h, t.a.; e: t-BuOK, toluene, 2h, t.a.;
Il p-nitrobenzil derivato 9, intermedio necessario alla sintesi di 1a e 2a, è stato sintetizzato come indicato nello SCHEMA 1.
L’ acetofenone commerciale 3 per reazione con acetone e pirrolidina a riflusso ha fornito il 2,2-dimetilcromano 4 che è stato successivamente sottoposto a reazione con il trimetilsilicianuro per ottenere il cianoderivato 5. La reazione di riduzione con LiAlH4
ha portato alla formazione dell’amminoalcool 6; tale derivato è stato fatto reagire con il cloroacetilcloruro ottenendo il derivato 7. La successiva reazione di ciclizzazione in presenza di t-BuOK ha fornito il morfolone 8 che è stato sottoposto a reazione di benzilazione con il p-nitrobenzilbromuro che ha portato alla formazione dell’intermedio
SCHEMA 2
O O N O NH2 O O N S NHCOCH3 O O N S NH2 O O N O NHCOCH3 O O N O NO2 O O N S NO2 9 1a 2a III IV I II II 11 10 12I: NH2-NH2 · H2O, FeCl3, MeOH, 24h, riflusso; II: Ac2O, K2CO3, acetone, 2h, N2;
III:Reattivo di Lawessons, clorobenzene, 24h, riflusso; IV: H2, Pd/C, EtOH ass., 3h, t.a.;
La sintesi dei composti 1a, 2a è stata effettuata seguendo la procedura riportata nello
SCHEMA 2.
Il p-nitrobenzil derivato 9 è stato sottoposto a riduzione con idrazina in presenza di FeCl3 per dare il derivato amminico 10. La successiva reazione di acetilazione con
anidride acetica e carbonato di potassio ha fornito il prodotto desiderato 1a.
La reazione del p-nitrobenzil derivato 9 con il reattivo di Lawessons ha fornito il derivato 4'- nitrotiomorfolonico 11 che in seguito a reazione con H2 in presenza di Pd/C
10% ha fornito il derivato amminico 12. Analogamente a quanto descritto per il derivato
1a, la successiva reazione di acetilazione di 12 con anidride acetica ha fornito il
tiomorfolone desiderato 2a.
Il composto 1a, che ha mostrato un buon profilo farmacologico in vitro, sarà valutato per le sue proprietà farmacologiche su modelli sperimentali di infarto del miocardio su ratti anestetizzati.
Il composto 2a verrà testato come miscela racemica alla dose di 40 mg Kg-1 su cuori di ratto isolato alla Langendorff, sottoposti a cicli di ischemia /riperfusione (30 minuti e 120 minuti rispettivamente).
MATERIALI E METODI
La struttura di tutti i composti è stata controllata per mezzo della spettrometria di massa e ¹H-NMR. Degli spettri ¹H-NMR e MS sono stati riportati i particolari più significativi. Tutti i composti sintetizzati presentano dati spettroscopici in accordo con le strutture assegnate.
Gli spettri di risonanza magnetica nucleare sono stati eseguiti con uno spettrofotometro Varian Gemini 200 operante a 200 MHz in CDCl3, i chemical shift δ sono espressi in
ppm (scala δ).
Gli spettri di massa sono stati registrati con uno spettrofotometro Hewelett Packard 5988° per introduzione diretta di un’energia nominale di 70 eV ad una temperatura di 350ºC.
I punti di fusione sono stati determinati al microscopio di Kofler e non sono stati corretti.
Le analisi elementari sono state eseguite nel nostro laboratorio di analitica: la differenza tra i valori teorici e quelli calcolati è risultata essere compresa nell’intervallo di ± 0,4%. Le evaporazioni sono state eseguite sottovuoto in evaporatore rotante e le disidratazioni delle fasi organiche sono state eseguite usando Na2SO4.
Le TLC analitiche sono state effettuate usando lastre Merck di gel di silice G60 contenente un indicatore fluorescente 20×20.2 mm; le varie macchie sono state evidenziate da una lampada UV (256 nm).
Per le cromatografie su colonna è stato usato un gel di silice Merck 70- 230 Mesh. Per la filtrazione su celite è stata usata celite ® 521.
SINTESI DEL DERIVATO
2-2 DIMETILCROMANICO 4 ( SCHEMA 1)
Ad una soluzione del 2-idrossiacetofenone commerciale 3 (5.00 g, 36.72 mmoli), sciolto in CH3CN(8 ml), è stato aggiunto acetone (4.05 ml, 54.95 mmoli) e pirrolidina (0.78 g,
10.99 mmoli). La soluzione risultante è stata lasciata in agitazione a temperatura ambiente per 1 h e successivamente posta a riflusso per 24 h. Trascorso tale periodo il solvente è stato evaporato e il residuo è stato ripreso con AcOEt e lavato con una soluzione acquosa di HCl 6N, NaOH 2N e H2O. La fase organica è stata essiccata,
filtrata ed evaporata ottenendo un olio grezzo costituito essenzialmente dal prodotto desiderato.
Resa: 87%
1H NMR (CDCl
3): δ 1.46 (s, 6H, CH3); 2.72 (s, 2H, CH2); 6.90-7.01 (m, 2H, Ar); 7.43-7.51 (m, 1H, Ar); 7.83-7.88 ( m, 1H, Ar) ppm.MS
(m/s): 177 (M+, 100%)Analisi elementare:
C11H12O2 C H Calc. % 74.98 6.86 Trov. % 74.84 7.07SINTESI DEL
TRIMETIL SILILDERIVATO 5 ( SCHEMA 1)
Ad una soluzione del dimetilcromanone 4 (4.77 g, 27.10 mmoli) in CH2Cl2 (15 ml) è
stato addizionato TMSCN (4.03 g, 40.65 mmoli) e ZnI2 (1.30 g, 4.07 mmoli). La
miscela di reazione è stata lasciata sotto agitazione a temperatura ambiente per 4 h. Trascorso tale periodo, la soluzione è stata lavata con H2O, essiccata ed evaporata a p.r.
ottenendo un solido grezzo costituito essenzialmente dal prodotto desiderato.
Resa: 78%
1
H-NMR
(CDCl3): δ 0.25 (m, 9H, CH3); 1.43 (s, 3H, CH3); 1.46 (s, 3H, CH3); 2.33 (d,
1H, J = 14.2 Hz, CH2); 2.45 (d, 1H, J = 14.2 Hz, CH2); 6.81 (dd, 1H, J = 8.2, 1.5 Hz,
Ar); 6.94-7.02 (m, 1H, Ar); 7.22-7.32 (m, 1H, Ar); 7.52-7.57 (m, 1H, Ar) ppm.
Analisi elementare:
C15H21NO2Si C H N
Calc. % 65.41 7.69 5.09
SINTESI
DELL’ AMMINOALCOOL 6 (SCHEMA 1)
Ad una soluzione di LiAlH4 1M in THF (21.82 ml, 21.82 mmoli), raffreddata a 0°C, è
stata aggiunta goccia a goccia il derivato 5 (3.00 g, 10.91 mmoli) sciolta nella minima quantità di THF. La miscela di reazione è stata lasciata in agitazione a temperatura ambiente per 2 h. Trascorso tale periodo la soluzione è stata posta a 0°C e addizionata di H2O e di una soluzione acquosa di NaOH 1N per precipitare i sali di litio e di alluminio.
I sali formatisi sono stati filtrati e il solvente è stato evaporato ottenendo un olio grezzo costituito essenzialmente dal prodotto desiderato 6.
Resa:
99% 1H-NMR (CDCl
3): δ 1.37 (s, 3H, CH3);1.43 (s, 3H, CH3); 2.01 (s, 2H, CH2NH2); 2.80 (d, 1H, J = 12.8 Hz, CH2N); 3.01 (d, 1H, J = 12.8 Hz, CH2N); 6.78- 6.97 (m, 2H, Ar), 7.14- 7.26 (m, 1H, Ar); 7.40 (d, 1 H, J = 7.9 Hz, Ar) ppm.MS
(m/z): 207 (M+, 25%); 177(M+- CH 2NH2, 100%)Analisi elementare:
C12H17NO2 C H N Calc. % 69.54 7.96 5.09 Trov. % 69.29 7.71 4.82SINTESI DEL
CLOROACETAMMIDO 7 (SCHEMA 1)
Ad una soluzione dell’amminoalcol 6 (2.60 g, 12.56 mmoli) in CH2Cl2 è stata
addizionata H2O (27 ml) e NaOH (0.60 g, 15.10 mmoli). Alla soluzione risultante,
raffreddata a 0°C, è stato aggiunto, goccia a goccia, il 2-cloroacetilcloruro commerciale (1.99 g, 17.58 mmoli) e la miscela di reazione è stata lasciata a temperatura ambiente per tutta la notte. Trascorso tale periodo le due fasi sono state separate e la fase organica è stata lavata con una soluzione acquosa di HCl 1N e H2O. La fase organica è stata
essiccata, filtrata ed evaporata a p.r. ottenendo un solido grezzo che corrisponde al prodotto desiderato.
Resa: 73%
1H-NMR (CDCl
3) : δ 1.36 (s, 3H, CH3); 1.43 (s, 3H, CH3); 2.03 (s, 2H, CH2); 3.57 (dd, 1H, J = 13.7, 7.3 Hz, CH2NH); 3.75 (dd, 1H, J = 13.7, 5.0 Hz, CH2NH); 4.09 (s, 2H, CH2Cl); 6.83-7.01 (m, 2H, Ar); 7.19-7.27 (m, 1H, Ar); 7.44 (dd, 1H, J = 7.7, 1.6 Hz, Ar) ppm.Analisi elementare:
C14H18ClNO3 C H N Calc. % 59.26 6.39 4.94 Trov.% 59.50 6.15 5.12SINTESI DELLO
SPIROMORFOLONE 8 (SCHEMA 1)
Ad una soluzione del derivato 7 (2.55 g, 9.46 mmoli) nella minima quantità di toluene è stato aggiunto t- BuOK (5.52 g, 49.19 mmoli) in piccole porzioni. La miscela di reazione risultante è stata lasciata sotto agitazione a temperatura ambiente per 2 h. Trascorso tale periodo è stato evaporato il toluene e il residuo è stato ripreso con AcOEt e lavato con H2O. La fase organica è stata essiccata, filtrata ed evaporata ottenendo un
solido vetroso grezzo.
Resa: 76 %
1
H-NMR (CDCl
3): δ 1.40 (s, 3H, CH3); 1.43 (s, 3H, CH3); 2.04 (d, 1 H, J = 14.7 Hz,
CH2); 2.43 (d,1H, J = 14.7 Hz, CH2); 3.25 (dd, 1H, J = 12.4, 4.3 Hz, CH2NH); 3.93 (d,
1H, J = 12.4 Hz, CH2NH); 4.24 (d, 1 H, J = 17.6 Hz, CH2O); 4.35 (d, 1H, J = 17.6 Hz,
CH2O); 6.85-7.00 (m, 2H, Ar); 7.21-7.30 (m, 1H, Ar); 7.46 (dd, 1H, J = 7.9, 1.6 Hz, Ar)
ppm.
MS
(m/z): 248 (M+, 20%)Analisi elementare:
C14H17NO3 C H N Calc. % 68.00 6.99 5.66 Trov. % 67.89 7.21 5.30SINTESI DEL
p-NITROBENZILDERIVATO 9 (SCHEMA 1)
Ad una sospensione di NaH (dispersione in olio minerale al 60%, 92.69 mg, 3.87 mmoli) in DMF (3 ml), posta sotto atmosfera di N2, è stato aggiunto il morfolone 8 (320
mg, 1.29 mmoli), solubilizzato nella minima quantità di DMF. La miscela è stata mantenuta sotto agitazione a temperatura ambiente per 30 minuti. Quindi è stata raffreddata a 0°C ed addizionata del p-nitrobenzilbromuro commerciale (334 mg, 1.55 mmoli ). La reazione è stata mantenuta a temperatura ambiente sotto agitazione per 2 h. Trascorso tale periodo è stata aggiunta H2O e la fase acquosa è stata estratta con AcOEt
e lavata con H2O. La fase organica è stata essiccata, filtrata ed evaporata ottenendo un
olio giallo.
Il grezzo è stato purificato mediante cromatografia su colonna usando come eluente Esano/ AcOEt (6: 4)
Resa : 46%
1H-NMR (CDCl
3): δ 1.26 (s, 3H, CH3); 1.35 (s, 3H, CH3); 1.76 (d, 1H, J = 14.5 Hz, CH2); 2.32 (d, 1H, J = 14.5 Hz, CH2); 3.07 (d, 1H, J = 12.3 Hz, CH2N); 3.84 (d, 1H, J = 12.3 Hz, CH2N); 4.31 (d, 1H, J = 17.6 Hz, CH2O); 4.43 (d, 1H, J = 17.6 Hz, CH2O); 4.55 (d, 1H, J = 14.8 Hz, CH2N); 4.92 (d, 1H, J = 14.8 Hz, CH2N); 6.80-6.95 (m, 2H,Ar); 7.18-7.27 (m, 1H, Ar); 7.35 (dd, 1H, J = 7.8, 1.5 Hz, Ar); 7.49 (d, 2H, J = 8.6 Hz, AA'XX'); 8.22 (d, 2H, J = 8.6 Hz, AA'XX') ppm.
MS (m/z): 382 (M
+, 60%); 177(100%)Analisi elementare:
C21H22N2O5 C H N Calc. % 65.96 5.80 7.33 Trov. % 66.01 5.73 6.88SINTESI DEL
p- AMMINO BENZILDERIVATO 10 (SCHEMA 2)
Ad una soluzione del composto 9 (0.15 g, 0.39 mmoli) solubilizzata nella minima quantità di MeOH (8 ml) è stato aggiunto il carbone (21 mg) ed FeCl3 (7 mg). La
miscela di reazione è stata scaldata a riflusso per 15 minuti, quindi è stata addizionata di una soluzione di idrazina idrata (0.33 ml, 6.71 mmoli) e la reazione è stata lasciata a riflusso e sotto agitazione per tutta la notte. Trascorso tale periodo, la miscela è stata filtrata ed il filtrato è stato evaporato. Il residuo è stato ripreso con AcOEt e lavato con H2O; la fase organica è stata essiccata, filtrata ed evaporata a p.r. ottenendo un solido
bianco vetroso corrispondente al prodotto desiderato.
Resa : 99%
1H-NMR (CDCl
3): δ 1.14 (s, 3H, CH3); 1.32 (s, 3H, CH3); 1.77 (d, 1H, J = 14.6 Hz, CH2); 2.23 (d, 1H, J = 14.6 Hz, CH2); 3.07 (d, 1H, J = 12.6 Hz, CH2N); 3.75 (d, 1H, J = 12.6 Hz, CH2N); 4.12 (d, 1H, J = 14.1 Hz, CH2Ph); 4.26 (d, 1H, J = 17.4 Hz, CH2O); 4.38 (d, 1H, J = 17.4 Hz, CH2O); 4.91 (d, 1H, J = 14.1 Hz, CH2Ph); 6.63 (d, 2H, J = 8.3Hz, AA'XX'); 6.77- 6.95 (m, 2H, Ar); 7.07 (d, 2H, J = 8.3 Hz, AA'XX'); 7.16-7.26 (m, 1H, Ar); 7.37 (dd, 1H, J = 7.9, 1.5 Hz, Ar) ppm.
MS
(m/z): 352 (M+,29%);Analisi elementare:
C21H24N2O3 C H N Calc. % 71.57 6.80 7.95 Trov. % 71.44 6.67 7.78SINTESI
DEL COMPOSTO 1a (SCHEMA 2)
Ad una soluzione dell’ammina 10 (66 mg, 0.19 mmoli) in acetone (8 ml) è stato aggiunto K2CO3 (40 mg, 0.29 mmoli) e l’anidride acetica (41 mg, 0.19 mmoli). La
miscela di reazione è stata lasciata sotto agitazione e in atmosfera di N2 per 2 h. Quindi
è stata ripresa con AcOEt e lavata con H2O. La fase organica è stata essiccata, filtrata ed
evaporata a p.r. ottenendo un solido bianco vetroso grezzo che viene purificato mediante triturazione in Et2O.
Resa: 58%
P.f. : 85-88 °C
1H- NMR (CDCl
3) : δ 1.18 (s, 3H, CH3); 1.32 (s, 3H, CH3); 1.73 (d, 1H, J = 14.7 Hz, CH2); 2.18 (s, 3H, NHCOCH3); 2.26 (d, 1H, J = 14.7 Hz, CH2); 3.05 (d, 1H, J = 12.4 Hz, CH2N); 3.77 (d, 1H, J = 12.4 Hz, CH2N); 4.28 (d, 1H, J = 17.6 Hz, CH2O); 4.26-4.35 (m, 4H, CH2Ph); 4.39 (d, 1H, J = 17.6 Hz, CH2O); 4.90 (d, 1H, J =14.8 Hz, CH2Ph); 6.79-6.94 (m, 2H, Ar); 7.17-7.40 (m, 4H, Ar); 7.48 (d, 2H, J = 8.4 Hz, AA'XX') ppm.MS (m/z): 394 (M
+, 12%); 146 (100%)Analisi elementare:
C23H26N2O4 C H N Calc. % 70.03 6.64 7.10 Trov. % 69.85 6.36 7.02SINTESI DEL
COMPOSTO 11 ( SCHEMA 2)
Ad una soluzione del composto 9 (140 mg, 0.36 mmoli) in clorobenzene (8 ml) è stato aggiunto il Reattivo di Lawessons commerciale (148 mg, 0.36 mmoli) e la miscela risultante è stata posta a riflusso per tutta la notte. Trascorso tale periodo, il clorobenzene è stato evaporato ottenendo un solido giallo vetroso grezzo che è stato purificato mediante cromatografica su colonna usando come eluente la miscela Esano/ AcOEt (7:3)
Resa : 73%
1H-NMR (CDCl
3): δ 1.22 (s, 3H, CH3); 1.33 (s, 3H, CH3); 1.65 (d, 1H, J = 14.6 Hz, CH2); 2.25 (d, 1H, J = 14.6 Hz, CH2); 3.20 (d, 1H, J = 13.3 Hz, CH2N); 3.84 (d, 1H, J = 13.3 Hz, CH2N); 4.73 (d, 1H, J = 19.2 Hz, CH2O); 4.88 (d, 1H, J = 19.2 Hz, CH2O); 5.03 (d, 1H, J = 14.5 Hz, CH2Ph); 5.78 (d, 1H, J = 14.5 Hz, CH2Ph); 6.80-6.96 (m, 2H,Ar); 7.19-7.32 (m, 2H, Ar); 7.59 (d, 2H, J = 8.4 Hz, AA'XX'); 8.23 (d, 2H, J = 8.4 Hz, AA'XX') ppm.
SINTESI DEL
COMPOSTO 12 (SCHEMA 2)
La soluzione del composto 11 (100 mg, 0.27 mmoli ) in EtOH assoluto (10 ml) è stata sottoposta ad idrogenazione, utilizzando come catalizzatore Pd/ C 10% (16 mg), per 3 h a temperatura ambiente. Trascorso tale periodo, il catalizzatore è stato filtrato su un filtro compatto di celite ed il filtrato è stato evaporato ottenendo un solido bianco corrispondente al prodotto desiderato.
Resa : 71%
1H-NMR
(CDCl3): δ 1.09 (s, 3H, CH3); 1.30 (s, 3H, CH3); 1.59 (d, 1H, J = 14.5 Hz, CH2); 2.13 (d, 1H, J = 14.5 Hz, CH2); 3.25 (d, 1H, J = 13.5 Hz, CH2N); 3.73 (d, 1H, J = 13.5 Hz, CH2N); 4.52 (d, 1H, J = 13.9 Hz, CH2Ph); 4.68 (d, 1H, J = 18.9 Hz, CH2O); 4.84 (d, 1H, J =18.9 Hz, CH2O); 5.83 (d, 1H, J = 13.9 Hz, CH2Ph); 6.63 (d, 2H, J = 8.3Hz, AA'XX'); 6.80 (dd, 1H, J = 8.7, 1.1 Hz, Ar); 6.88-6.96 (m, 1H, Ar); 7.20 (d, 2H, J = 8.3 Hz, AA'XX'); 7.25-7.34 (m, 2H, Ar) ppm.
SINTESI
DEL COMPOSTO 2a (SCHEMA 2)
Ad una soluzione del composto 12 (30 mg, 0.08 mmoli) nella minima quantità di acetone (3 ml) è stato aggiunto K2CO3 (16.6 mg, 0.12 mmoli) e anidride acetica (0.02
ml, 0.08 mmoli). La reazione è stata lasciata sotto agitazione e in atmosfera di N2 per 2
h. Trascorso tale periodo l’ acetone è stato evaporato e il residuo, ripreso con AcOEt, è stato lavato con H2O. La fase organica è stata essiccata, filtrata ed evaporata ottenendo
un olio grezzo corrispondente al prodotto desiderato.
Resa : 38%
1H-NMR
(CDCl3): δ 1.13 (s, 3H, CH3); 1.31 (s, 3H, CH3); 1.60 (d, 1H, J = 14.5 Hz, CH2); 2.17 (d, 1H, J = 14.5 Hz, CH2); 2.18 (s, 3H, NHCOCH3); 3.22 (d, 1H, J = 13.4 Hz, CH2N); 3.76 (d, 1H, J = 13.4 Hz, CH2N); 4.65-4.89 (m, 3H, CH2Ph); 5.78 (d,1H, J = 13.9 Hz, CH2Ph); 6.78- 6.95 (m, 2H, Ar); 7.17-7.39 (m, 4H, Ar); 7.50 (d, 2H, J = 8.2 Hz, AA'XX') ppm.Analisi elementare:
C23H26N2O3S C H N S Calc. % 67.29 6.33 6.83 7.80 Trov. % 67.07 6.58 7.90 7.63BIBLIOGRAFIA
1. Buja LM. Modulation of the myocardial response to ischemia. Lab invest 1998;
78;1345-73
2. L.Maximilian Buja, Myocardial ischemia and reperfusion injury
3. Buja LM, Hagler HK, Willerson JT. Altered calcium homeostasis in the
pathogenesis of myocardial ischemic and hypoxic injury. Cell Calcium
1988;9:205– 17.
4. Reimer KA, Jennings RB. The bwavefront phenomenonQ of myocardial
ischemic cell death: II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and collateral flow. Lab Invest 1979;40:633–44.
5. Vincenzo Calderone, Lara Testai, Alma Martelli, Simona Raposelli and Maria
Cristina Breschi. Activators of cardiac mitochondrial ATP-sensitive potassium canne: promising drugs for anti-ischaemic therapy. Vol. 11,1,2007
6. Shiki, K.,and Hears, D. J. 1987, Am. J. Physiol., 253, H1470
7. Murry, C.E., Jennings, R.B., and Reimer, K. A. 1986, Circulation, 74, 1124 8. Cave, A. C., and Hearse, D. J. 1992, J. Mol. Cell Cardiol., 24, 1113
9. Richard, V.; Karffer,N.; Tron, C.; Thuillez, C. Circulation, 1994, 89,1254
10. L.Testai, S. Raposelli and V. Calderone. Cardiovascular and Hematological
Agent in Medical Chemistry, 2007,5,000-000.
11. Thorton, J. D. , Thorton, C.S., and Downey, J.M. 1993, Am J. Physiol., 265,
H504
12. McCully, J.D.; Toyoda, Y.; Uematsu, M.; Stewart, R.D.; Levitsky, S. Am. J.
Physiol. Heart Cic. Physiol., 2001,280,H591.
13. Ohnuma, Y.; Miura, T.; Miki, T.;Tanno, M.;Kuno,A.; Tsuchida, A.; Shimamoto,
K. Am. J.Physiol. Heart Circ. Physioli., 2002, 283, H440
14. Schulz, R.; Post, H.; Vahlhaus, C.; Heusch, G. Circulation, 1998, 98,1022
15. Liu, G.S.;Thornton,J.; Van Winkle, D.M.; Stanley, A.W.; Olsson, R.A.;
Downey, J.M. Circulation,1991,84,350
16. Lochner, A.; Marais, E.; Genade, S.;Moolman, J.A. Am. J.Physiol.Heart Circ.
Cardiol, 2000, 279, H2752
17. Murry, C.E.; Richard, V.J.; Reimer, K.A.;Jennings, R.B Circ. Res., 1990, 66,
1133
18. Belosjorow, S.; Schulz, R.; Dirge, H.; Shade, F.U.; Heusch, G. Am. J Physiol.
Ren. Phusiol., 1999,276, 33369
19. Noma, A., 1983. ATP-regulated K+ channel in cardiac muscle. Nature 305 85930), 147-148
20. Noma A., Nature, 1983, 305: 147.
21. Bajgar R., Seetharamen S., Kowaltowski A.J., Garlid K.D., Paucek P.J.,
Biol.Chem.; 2001, 276: 33369.
22. Brochiero E.; Wallendorf B., Gagnon D., Laprade R., Lapointe J.Y., Am. J.
Physiol.Ren.Physiol., 2002, 282, F289.
23. Mironova G.D., Grigoriev S.M., Skarga Y.Y., Negoda A.E., Kolomytkin O.V.,
Membr. Cell. Biol., 1997, 10: 583
24. Susumu Seino, Takashi Miki, Progress in Biophysics & Molecular Biology 81