1
Università degli Studi di Pisa
Facoltà di Scienze Matematiche, Fisiche e Naturali
Corso di Laurea Magistrale in Biologia Molecolare e
Cellulare
Tesi di Laurea Sperimentale
“Nuovi approcci di terapia genica per la
cura della discinesia ciliare primitiva”
Relatore: Candidata:
Prof. Mauro Pistello Ambra Del Grosso
2
Indice
1.Riassunto………...5
2.Abstract……….6
3.Introduzione………..7
3.1 La discinesia ciliare primitiva………..………7
3.1.1 Definizione e storia………...7
3.1.2 Epidemiologia………...8
3.1.3 Ultrastruttura e funzione ciliare normale………..8
3.1.4 Patologia e manifestazioni cliniche……….14
3.1.5 Patogenesi………...………18
3.1.5.1 Il gene DNAH11………..………21
3.1.7 Esami effettuati per confermare il sospetto clinico……….25
3.1.7.1 Test di screening………...25 3.1.7.2 Test diagnostici……….26 3.1.8 Terapie………33 3.2 Terapia genica………..33 3.3 Vettori virali……….39 3.3.1 Vettori retrovirali………41 3.3.2 Vettori lentivirali…..………...……43
3.4 I Transcriptor Activator-like Effectors………..………...……45
3.4.1 Introduzione………45
3.4.2 Funzione nativa………...……45
3.4.3 Struttura e funzionamento………...……46
3.4.3.1 Riparazione del DNA tramite ricombinazione………….50
3.4.4 Comparazione con altri metodi di manipolazione del genoma…...52
4. Materiali e metodi………..54
4.1 Progettazione della sequenza target……….………...54
4.1.1 Analisi sequenza e mutazioni nel gene DNAH11……...…………54
3
4.2 Costruzione delle TALEN……….………..56
4.2.1 I costrutti TALEN………...…56
4.2.2 Costruzione di una libreria di monomeri TALE...………..57
4.2.3 Assemblaggio degli esameri………...…59
4.2.4 Conferma e amplificazione degli esameri………...…60
4.2.5 Assemblaggio degli esameri all’interno del plasmide TALEN..…61
4.2.6 Screening delle colonie mediante PCR………...62
4.2.7 Estrazione del plasmide TALEN………63
4.2.8 Sequenziamento………..63
4.3 Verifica dell’efficienza di taglio delle TALEN………..65
4.3.1 Clonaggio della sequenza target in pcDNA3-GFP……….65
4.3.2 Trasfezione………..68
4.3.2.1 Linea cellulare………..70
4.3.3 Lettura al citofluorimetro (FACS)………..71
4.3.4 Western Blot……….………..71
4.4 Vettori lentivirali………..……73
4.4.1 Vettori con L-TALEN e R-TALEN………73
4.4.2 Vettore utilizzato per la prova di ricombinazione……...…………75
4.4.3 Vettore col frammento sano di ricombinazione (REC)…..………76
4.4.4 Preparazione dei vettori virali……….78
4.5 Verifica dell’efficienza di ricombinazione……….79
4.5.1 Trasfezione/Trasduzione……….80
4.5.2 Western Blot………...………81
4.6 Trasduzione di cellule ciliate ex-vivo………..81
4.6.1 Prelievo e coltura delle cellule ciliate……….81
4.6.2 Trasduzione delle cellule ciliate………..………81
4.6.3 Monitoraggio del battito ciliare………...………82
4.6.4 Estrazione del DNA dalle cellule ciliate trasdotte………..………83
4.6.5 Digital PCR……….83
5. Risultati………...………87
4
5.2 Sintesi dei domini TALE……….88
5.2.1 Amplificazione dei monomeri e degli esameri…………...………88
5.2.2 Verifica dell’assemblaggio degli esameri nel TALE backbone…..89
5.2.3 Sequenziamento………..……90
5.3 Verifica dell’efficienza di taglio delle TALEN………..……91
5.3.1 Clonaggio della sequenza target in pCDNA3-GFP………91
5.3.2 Analisi al FACS………..……92
5.3.3 Western Blot………...……93
5.4 Verifica dell’efficienza di ricombinazione……….94
5.4.1 Clonaggi dei vettori……….94
5.4.2 Analisi al FACS e al microscopio a fluorescenza…………...……96
5.4.3 Western blot………97
5.5 Trasduzione delle cellule ciliate………..……98
5.5.1 Clonaggio del vettore REC………..…...98
5.5.2 Verifica della trasducibilità………..……...…98
5.5.3 Valutazione della frequenza e della morfologia del battito ciliare in seguito a trasduzione con PLVX-EF1α-LTALE, PLVX-EF1α-RTALE e REC………100 5.5.4 Digital PCR………...103 5.5.5 Analisi statistica………106 6. Discussione………...107 7. Prospettive future………112 8. Bibliografia………...113
5
1. Riassunto
La discinesia ciliare primitiva (DCP) è una malattia genetica rara, autosomica e recessiva, che affligge globalmente circa 400.000 persone. La DCP è caratterizzata da disfunzione del movimento ciliare degli epiteli. Difetti nella motilità ciliare portano ad una scarsa clearance muco ciliare che danneggia la funzionalità polmonare, ad asma cronica, ed a serie infezioni respiratorie. La cura è solo sintomatica ed estrapolata da altre patologie del cavo orale. Molte speranze sono quindi riposte nella terapia genica. Questo lavoro di tesi è finalizzato allo sviluppo di un nuovo approccio di terapia genica per la DCP. Vi è una lunga lista di geni implicati nella struttura e movimento ciliare e che sono stati quindi associati a DCP. In questo studio ci siamo focalizzati su DNAH11, studiato dalla sezione di Pneumologia ed Allergologia Pediatrica dell’Azienda Ospedaliero-Universitaria di Pisa con cui abbiamo collaborato per questo studio. Tra i pazienti afferenti a questo centro sono stati scelti quindi soggetti con mutazioni che inattivano il gene DNAH11 che codifica per la catena pesante della dineina 11, componente essenziale della struttura ciliare.
Sono state allestite colture cellulari di cellule ciliate prelevate tramite brushing da due pazienti con mutazioni non-senso in DNAH11. Si è quindi cercato di correggerne il difetto genetico tramite l’utilizzo delle proteine TALEN (transcription
activator-like effector nucleases): il sistema è stato disegnato per tagliare il DNA a
monte del gene DNAH11, così da indurre ricombinazione sito specifica con una sequenza wild-type. Le TALEN e il frammento di DNA per la ricombinazione (Rec), sono stati forniti alle cellule tramite vettori lentivirali.
Le successive trasduzioni con i vettori lentivirali non hanno alterato la vitalità cellulare e la motilità ciliare. In un sistema surrogato su 293T si è dimostrato che le TALEN tagliano l’80% della sequenza mutata di DNAH11. Le cellule ciliate naïve di entrambi i pazienti mostravano battito ipercinetico (18-22 Hertz) che si è normalizzato (13 Hertz) in circa il 20% delle cellule dopo il trattamento con TALEN+Rec.
La presenza della proteina wild-type nel sistema surrogato è stata ricercata tramite Western blot, mentre l’avvenuta ricombinazione nelle cellule ciliate è stata monitorata tramite Digital PCR.
Avendo ripristinato tramite gene editing il normale battito ciliare, quindi, questo studio potrebbe aprire una nuova via al trattamento della DCP.
6
2. Abstract
Background Primary ciliary dyskinesia (PCD) is a rare autosomal recessive genetic disorder that manifests early in life and is characterized by dysfunction of the cilia lining the respiratory tract and other epithelia. Defects in ciliary motility cause poor muco-ciliary clearance and leads to extensive impairment of pulmonary functions, chronic asthma, and severe respiratory infections. There is no specific cure and gene therapy is the only hope to treat the disease.
Objective To develop a strategy that permanently corrects the genetic defect and normalizes ciliary beating in pediatric PCD patients with mutated dynein heavy chain 11 (DNAH11), a large gene that encodes a microtubule-dependent motor ATPase and is an essential component of ciliary structure.
Methods Two patients bearing a nonsense mutation in DNAH11, provided ciliated cells that were collected by nasal brushing and placed in culture. Correction of the genetic defect was pursued by gene editing using a transcription activator-like effector nucleases (TALENs) system designed to cleave the patient’s DNAH11 sequence upstream inactivating mutation and trigger site-specific recombination with the wild-type DNAH11 sequence (Rec). TALENs and Rec were delivered in the cultured ciliated cells by lentiviral vectors.
Results Transduction with lentiviral vector did not alter viability nor ciliary motility of ciliated cells cultured ex-vivo. In a surrogate system, transduction of TALENs alone cleaved over 80% mutated DNAH11 sequence, and transduction of TALENs+Rec replaced mutated with wild-type sequence. Naïve ciliated cells of both patients showed hyperkinetic beat (18-22 Hertz) that was stably normalized (13 Hertz) in 25 and 10% cells upon TALENs+Rec transduction.
Conclusions Albeit validation in an animal model is warranted, this study shows that ciliary beating can be rescued by gene editing and paves the way to devise novel approaches to cure PCD.
7
3. Introduzione
3.1 La discinesia ciliare primitiva
3.1.1 Definizione e storia
La discinesia ciliare primaria o primitiva (DCP) è una patologia respiratoria rara, eterogenea dal punto di vista clinico, strutturale e genetico, dovuta ad alterazioni della struttura e/o della funzione delle ciglia della mucosa respiratoria che determinano, a seguito del conseguente deficit del trasporto mucociliare, la comparsa di diversi quadri di patologia a carico delle vie aeree e del parenchima polmonare.
La relazione tra movimento ciliare e fisiopatologia respiratoria è stata evidenziata per la prima volta da Afzelius in una sindrome ereditaria a trasmissione autosomica recessiva descritta nel 1933 da Kartagener e caratterizzata clinicamente dalla triade: Situs Viscerum Inversus (SVI), bronchiectasie, sinusite cronica (Afzelius et al., 1979; Afzelius et al., 1986; Palmblad et al., 1984)
Inizialmente venne studiata l’ultrastruttura degli spermatozoi, dato che una parte dei pazienti con sindrome di Kartagener era affetta anche da sterilità; successivamente vennero studiate anche le ciglia dell’apparato respiratorio, vista l’analogia ultrastrutturale tra ciglia e flagelli. Nel 1975 la sterilità maschile dovuta ad immobilità degli spermatozoi venne associata alla sintomatologia bronchitica cronica di cui soffrivano i pazienti esaminati (Pedersen et al., 1974). Poiché l’alterazione prevalente, sia nelle code degli spermatozoi che nelle ciglia dell’apparato respiratorio, era la mancanza dei bracci esterni ed interni di dineina, nel 1976 Afzelius ipotizzò la presenza di un disturbo generalizzato dell’attività ciliare (Pedersen et al., 1976; Afzelius et al., 1976), definito poi nel 1977 da Eliasson come “sindrome delle ciglia immobili” e caratterizzata da: sterilità, bronchite cronica dalla prima infanzia, bronchiectasie, rinosinusite cronica e nel 50% dei casi da SVI (Eliasson et al., 1977).
In seguito a queste osservazioni l’esame ultrastrutturale dell’epitelio respiratorio divenne essenziale per studiare le caratteristiche morfologiche delle alterazioni ciliari in modo da distinguere le lesioni ciliari congenite (primitive) da quelle acquisite (secondarie) dovute a processi infiammatori, acuti o cronici, a carico delle vie aeree.
8
Inoltre la clearance muco-ciliare può essere compromessa in seguito all’esposizione ad agenti fisici o chimici (Burman et al., 1986; Kantar et al., 1994; Pedersen et al., 1990) e biologici (Wilson et al., 1986; Wilson et al., 1988).
E’ stato dimostrato, in seguito, che le ciglia non sono necessariamente immobili, ma possono presentare gradi qualitativamente e quantitativamente diversi di alterata motilità, rimanendo invariato l’effetto complessivo negativo sulle capacità di clearance ciliare della mucosa respiratoria. Il termine di “sindrome delle ciglia immobili” è stato allora sostituito con quello di DCP, che include tutte le forme di anomalie congenite delle ciglia che si traducono in alterato trasporto muco-ciliare; per cui la sindrome delle ciglia immobili e la sindrome di Kartagener sono dei sottogruppi (Cowan et al., 2001).
3.1.2 Epidemiologia
La DCP, pur essendo una malattia rara, è comunque la seconda malattia congenita dell’apparato respiratorio per frequenza dopo la fibrosi cistica. L’incidenza della DCP nella popolazione caucasica è stimata intorno a 1/15000-16000 nati vivi, ma questo dato sottostima la reale diffusione della malattia, soprattutto nelle comunità in cui la consanguineità è molto comune. A questo si somma la difficoltà nella diagnosi della DCP, pur in presenza della sintomatologia tipica, dovuta sia all’eterogeneità genetica e clinica, sia all’elevata frequenza delle comuni infezioni respiratorie tra i piccoli pazienti. Diventa comune, di conseguenza a quanto appena detto, giungere alla diagnosi di DCP, soprattutto nei paesi con una bassa spesa sanitaria, solo dopo un lungo periodo di malattia, sottostimando la reale incidenza della patologia nella popolazione generale. In Europa l’età media di diagnosi di DCP è di circa 3-5 anni. (Noone et al., 2004; Coren M.E. et al., 2004; Kuehni C.E. et al., 2010; Bush A. et al., 2007). Si può stimare che in Italia vi siano 70 nuovi nati con tale patologia ogni anno e 4000 soggetti affetti; si ritiene, inoltre, che larga parte di essi non sia ancora stata individuata (Bush et al., 2002).
3.1.3 Ultrastruttura e funzione ciliare normale
Le ciglia sono organuli altamente specializzati e complessi, strutturalmente correlati ai flagelli degli spermatozoi. Sono formate da più di 360 proteine e le ritroviamo poste sulla superficie di molte cellule eucariotiche. In particolare, nell’uomo, sono poste sulla superficie delle cellule dell’apparato respiratorio (sulla superficie della
9
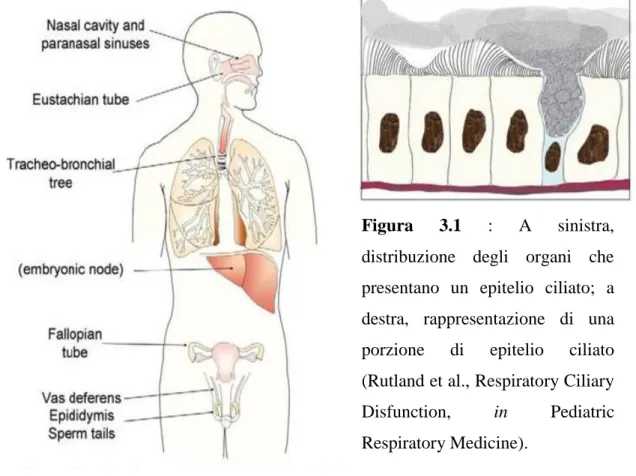
mucosa che riveste le cavità nasali, il rinofaringe, le vie aeree inferiori sino ai bronchioli terminali, l’orecchio medio e le trombe di Eustachio), e sulla superficie delle cellule endometriali delle tube di Falloppio (Fig 3.1). L’epitelio di rivestimento, dal naso ai bronchioli terminali, è di tipo pseudostratificato ed è composto da: cellule cilindriche ciliate (30% della popolazione cellulare), cellule caliciformi mucipare, cellule basali, cellule sierose e cellule a spazzola (Montgomery et al., 1990)
La precisa composizione proteica di ciglia e dei flagelli non è ancora completamente nota; molto di ciò che ad oggi si sa riguardo la struttura, la regolazione e la composizione proteica del complesso molecolare ciliare, deriva da studi effettuati su organismi modello (Pazour et al., 2005). Fondamentali sono stati gli studi effettuati su
Chlamydomonas reinhardtii, un’alga unicellulare di 10µm di diametro che si muove
grazie a due flagelli.
Le ciglia sono estroflessioni della membrana delle cellule epiteliali di forma tubulare, all’interno delle quali si trova l’assonema: un nucleo citoscheletrico costituito da un insieme di microtubuli. Si possono distinguere due tipi di ciglia in base alla
Figura 3.1 : A sinistra, distribuzione degli organi che presentano un epitelio ciliato; a destra, rappresentazione di una porzione di epitelio ciliato (Rutland et al., Respiratory Ciliary Disfunction, in Pediatric Respiratory Medicine).
10
diversa organizzazione dell’assonema: ciglia primarie e ciglia nodali a struttura “9+0” e ciglia mobili a struttura “9+2”. Le ciglia primarie sono immobili ed hanno un assonema formato da 9 coppie di microtubuli; nei mammiferi sono presenti su molti tipi cellulari dove svolgono un’importante funzione sensoriale, intervenendo nei processi fisiologici della visione e dell’olfatto (Pazour et al; 2005).
Un ciglio mobile, invece, presenta un assonema formato da 9 coppie di microtubuli periferici e due microtubuli singoli centrali. Le nove coppie periferiche di microtubuli sono costituite da un tubulo A, completo e formato da 13 protofilamenti di tubulina, e da un tubulo B, incompleto e formato da 10 protofilamenti, questi si compenetrano fra loro tre grazie ai 3 protofilamenti in comune che appartengono al tubulo A. I protofilamenti sono formati da dimeri di tubulina, polipeptide presente in forma di subunità globulari α e β che si allineano in successione, per poi affiancarsi in modo sfalsato e disporsi intorno ad una cavità centrale.
Ogni coppia di microtubuli periferici è stabilizzata da proteine associate ai microtubuli (MAP): la nexina, che interconnette il tubulo A al tubulo B della coppia di microtubuli successiva; i ponti radiali, che connettono ogni coppia periferica con quella centrale; la guaina interna, costituita da sottili bracci proteici, le proiezioni, che sporgono dalla coppia centrale e sono incurvati intorno ad essi a formare un anello e i bracci di dineina, responsabili della regolazione del battito ciliare (Avila et al., 1994). La guaina interna, i raggi e la nexina, intervengono nella determinazione della forma del battito ciliare (Fig 3.3).
11
Le MAP intervengono nella regolazione della stabilità dell' assonema, la più studiata è la dineina. Le molecole di dineina sono organizzate a formare coppie di bracci, uno interno, rivolto verso la guaina centrale, ed uno esterno, rivolto verso il margine, che se osservati dalla base del ciglio sporgono in senso antiorario dal tubulo A al tubulo B della coppia successiva. La dineina fa parte di un complesso macromolecolare composto da circa 12 subunità di varie dimensioni. Il braccio esterno, responsabile della frequenza del battito, è formato da: tre catene pesanti (HC) α, β e γ di 400-500 kDa, che possiedono attività ATPasica e costituiscono la ''testa'' del complesso; da due o più catene intermedie di 45-110 kDa che forniscono l'ancoraggio stabile sul microtubulo A; e da 4-8 subunità più piccole, dette catene leggere (LC), di 8-55 kDa. I bracci interni di dineina (che influenzano la conformazione del battito) hanno una struttura simile a quelli esterni ma presentano una maggiore variabilità. Sono state individuate sette isoforme, di cui una sola a ''due teste'' (composta da due HC, 1α e 1β, tre IC e tre LC) e le restanti sei a ''singola testa'' (Fig 3.3). Questi complessi multiproteici di dineina possiedono un dominio catalitico con attività ATP-asica, necessario per generare l’energia necessaria per lo scorrimento delle coppie di microtubuli e permettere la motilità ciliare.
Sono stati individuati numerosi ceppi mutanti di C. reinhardtii che hanno permesso di identificare numerosi geni codificanti per dineine assonemali (Silflow e Lefebvre 2001; Holzbaur e Vallee, 1994; King, 2000). Gran parte di tali geni presentano gli omologhi nei mammiferi, in cui mantengono invariata la loro funzione all'interno del complesso assonemale: i geni umani DNAH5 e DNAI1, ad esempio, (responsabili di circa un terzo dei casi di DCP) sembrano essere gli omologhi, in C. reinhardtii, rispettivamente, dei bracci esterni HCγ e IC78 (Omran et al., 2000; Pennarun et al., 1999). In tali geni sono state identificate mutazioni ultrastrutturali ritrovate poi anche nei geni omologhi umani riscontrati in alcuni pazienti con difetti ciliari.
12
Figura 3.3: Struttura assonemale e suoi componenti in C. reinhardtii. (A) Rappresentazione schematica dell'assonema ciliare in lunghezza e sua sezione trasversale, con indicate le varie componenti. (B) Il braccio esterno (a sinistra) è composto da: tre catene pesanti (HC) di dineina α, β e γ; due catene intermedie (IC) e da otto catene leggere (LC). Il braccio interno (a destra) è più variabile, esistono almeno otto diverse HC organizzate in sette isoforme: una a ''due-teste'' (in arancione chiaro) e sei a ''singola-testa'' (in arancione scuro) (Ibanez -Tallon, 2003).
In vivo esistono centri di organizzazione, detti corpuscoli basali, che agiscono come stampo per disporre i microtubuli a coppie con il pattern 9+2. Il corpuscolo basale è una struttura cilindrica posta nella porzione apicale del citoplasma della cellula ciliata che contiene un anello di 9 triplette di microtubuli (A, B, C) ed ogni tripletta contiene un microtubulo completo A fuso con altri due incompleti: B e C. Come nell’assonema, anche nel corpuscolo basale esistono legami proteici di stabilizzazione, in particolare tra la subfibra A di una tripletta e la subfibra C di quella vicina. L’assetto 9+2 dei microtubuli dell’assonema si modifica nella porzione apicale del ciglio: le coppie periferiche perdono progressivamente la subunità B del microtubulo, la coppia centrale ed i bracci di dineina (Alberts B, Bray D, Lewis J, et al. Biologia molecolare della cellula. II ed. Zanichelli. 1991; 767-777).
13
Le ciglia dell’epitelio respiratorio sono dotate di movimento, con il quale provvedono al drenaggio verso l’esterno del muco presente nelle vie aeree, impedendone il ristagno. Il battito ciliare, che nei soggetti sani ha frequenza di 12 Hz, deriva dallo scorrimento coordinato delle coppie di microtubuli rispetto all'asse centrale. E’ stato dimostrato che i responsabili del movimento ciliare sono i bracci di dineina. Infatti l’assonema isolato è in grado di compiere movimenti di flessione se immerso in una soluzione salina contenente ATP e ioni magnesio, per cui il corpuscolo basale e la membrana plasmatici non sono necessari. Inoltre, esponendo assonemi isolati all’azione di enzimi proteolitici che distruggono nexina e ponti radiali, ma lasciano intatti microtubuli e dineina, l’assonema è in grado di allungarsi fino a 9 volte rispetto alla sua lunghezza iniziale in presenza di ATP. Al contrario, negli assonemi isolati e senza bracci di dineina il movimento non è più presente. Questo ricompare con l’aggiunta di dineina e ATP (Alberts B, Bray D, Lewis J, et al. Biologia molecolare della cellula. II ed. Zanichelli. 1991; 767-777).
Tramite idrolisi dell’ATP ogni braccio di dineina genera una forza che agisce in modo da spostare verso l’apice del ciglio la coppia di microtubuli adiacenti con cui prende contatto. Se tutti i bracci si attivassero nello stesso momento, l’assonema si avvolgerebbe a formare un’elica stretta, per questo è proprio la loro attivazione asincrona a permettere il tipico movimento a frusta del ciglio (Fig. 3.4). Grazie alla presenza di nexina e di ponti radiali, che offrono punti fermi di ancoraggio, il movimento di scorrimento si trasforma in movimento di flessione.
Sulla superficie apicale delle cellule sono presenti centinaia di ciglia disposte in file che battono simultaneamente, mentre quelle della fila limitrofa si muovono con un Fig 3.4: Rappresentazione del battito ciliare normale. Movimento a frusta: consiste in una prima fase di flessione ciliare molto rapida e in una fase finale di recupero più lenta; il muco si muove nella stessa direzione del battito (www.dcp-pisa.it).
14
leggero ritardo, assumendo un movimento ad onda. Questo particolare movimento è presente anche sull'apice delle cellule ependimali cerebrali, lungo le tube di Falloppio e nel flagello dello spermatozoo.
Un altro tipo di movimento ciliare esistente è quello di tipo rotazionale, presente solo in una determinata fase dello sviluppo embrionale. Durante l'embriogenesi dei mammiferi alcune cellule presenti sulla superficie ventrale del nodo embrionale possiedono un monociglio con una struttura assonemale di tipo "9+0". La mancanza della coppia centrale di microtubuli crea un movimento rotazionale che determina il flusso verso sinistra del fluido extracellulare, fondamentale per il corretto patterning di segnali essenziali alla determinazione dell'asse destro-sinistro dell'individuo. Un’alterazione delle ciglia nodali correla, infatti, con difetti di lateralizzazione.
Complessi di dineina sono presenti anche nel citosol (dineina citoplasmatica), dove generano e controllano il movimento intracitoplasmatico di diversi organelli ed intervengono nel processo di divisione cellulare (Karki e Holzbaur, 1999).
3.1.4 Patologia e manifestazioni cliniche
La DCP è una malattia respiratoria rara con trasmissione autosomica recessiva. Alla base della malattia ci sono alterazioni della struttura e/o funzione delle ciglia della mucosa respiratoria. L’epitelio ciliato riveste la mucosa in tutte le varie porzioni delle vie respiratorie compresi i seni paranasali, la tuba uditiva e l’orecchio medio.
Attività ciliare e muco bronchiale costituiscono un’unità funzionale detta
clearance muco-ciliare che contribuisce alle difese dell’apparato respiratorio. Essa,
infatti, è responsabile del normale trasporto del muco e della conseguente eliminazione dalle vie respiratorie di particelle di polvere, cellule morte, batteri e virus. Per un’efficace clearance muco-ciliare sono condizioni essenziali: la composizione ottimale del muco (composto da: glicoproteine, proteoglicani e lipidi), ed una valida attività ciliare. In condizioni normali il muco si organizza sulla superficie delle vie aeree in 2
strati a diversa viscosità, che vengono spostati verso l’orofaringe dal movimento delle ciglia presenti all’apice delle cellule dell’epitelio respiratorio.
Circa l’80% delle cellule presenti nell’epitelio respiratorio sono ciliate, mentre il 20% è rappresentato da cellule secernenti muco. Ne consegue che alterazioni della funzionalità
15
ciliare danno luogo a sintomi clinici a carico di diversi apparati, predisponendo a frequenti infezioni polmonari e delle alte vie respiratorie, con conseguenti bronchiectasie e sinusiti croniche. Se il paziente viene trascurato, può degenerare in broncopneumopatia cronica ostruttiva e insufficienza respiratoria, che necessita trapianto polmonare.
Figura 3.5: Malfunzionamento ciliare nelle patologie umane. Sono rappresentati gli organi in cui è presente l'azione del movimento ciliare e le patologie associate; sono indicate anche le differenti strutture dell'assonema di ciascun tipo di ciglia. Nel cervello sono presenti le cellule ependimali che rivestono i ventricoli cerebrali e possiedono sulla superficie apicale ciglia mobili con ultrastruttura "9+2". Nella retina i fotorecettori sono costituiti da segmenti interni ed esterni in connessione a un cilio con ultrastruttura "9+0". Nei tratti respiratori superiori ed inferiori le cellule epiteliali sono ricoperte da ciglia mobili con ultrastruttura "9+2". Nei reni, ciglia con struttura "9+0" sono presenti nei glomeruli e nelle cellule tubulari. Il flagello degli spermatozoi e le ciglia nei tubi efferenti hanno una struttura "9+2", lo stesso tipo di ciglia mobili si trova sull'epitelio dell'utero e delle tube di Falloppio (da Ibanez -Tallon et al., 2003).
16
Epiteli ciliati, oltre che a livello delle vie respiratorie, si trovano anche a livello del sacco lacrimale, del pavimento ependimale, del canale centrale del midollo spinale, dell'endometrio, della cervice, delle tube di Falloppio e dei dotti efferenti tra testicolo ed epididimo (Fig. 3.5); pertanto la DCP è considerata una malattia multisistemica che può compromettere il funzionamento dei diversi organi.
La presentazione clinica della DCP varia a seconda dell’età del paziente al momento della diagnosi (Hogg et al., 2009).
In età prenatale è il riscontro di situs inversus nel nascituro durante l’esame ecografico che pone il sospetto di DCP.
In età neonatale, invece, si sospetta DCP in seguito alla manifestazione nel neonato di: distress respiratorio, atalettasia, alterazioni congenite cardiache e/o spleniche e difetti di lateralizzazione (Bush A. et. al.,., 2007)
In età pediatrica circa l’84-100% dei pazienti manifesta tosse conica produttiva con espettorato muco-purulento, rinite cronica, ostruzione nasale, otite media, bronchiectasie e un possibile ritardo della crescita durante i primi anni di vita. Ci sono, inoltre, una serie di patogeni più frequentemente identificati all’esame colturale dell’escreato, che possono colonizzare in maniera cronica le vie aeree del paziente: Haemophilus influenzae, Staphylococcus aueus, Strptococcus
pneumoniae e pseudomonas aeruginosa (Jain K. et al., 2007; Brown et al.,
2008).
Il paziente con DCP diagnosticata in età adulta, a causa della lunga storia della malattia, presenta: bonchiectasie, emottisi, sinusite cornica e ippocratismo digitale. La broncopneumatia può evolvere verso insufficienza respiratoria. Il 50% circa dei pazienti maschi, inoltre, può presentare infertilità secondaria, associata alla mancata motilità degli spermatozoi. Alcuni pazienti con DCP, quindi, sono fertili, suggerendo l’esistenza di un diverso controllo genetico tra ciglia e flagelli (Munro et. al.,.,1994). Le donne adulte, invece possono presentare subfertilità causata dal ritardato trasporto dell’ovocita attraverso le tube uterine. (halberts S.A. et al., 1997).
Oltretutto, nel paziente con DCP in cui sono presenti contemporaneamente sia alterazioni strutturali delle ciglia mobili che delle ciglia primarie, la sovrapposizione tra
17
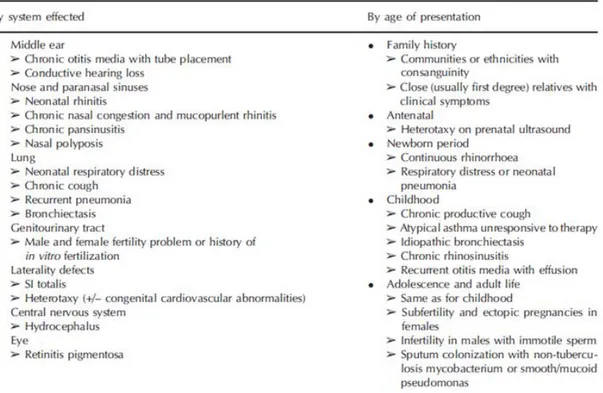
i diversi fenotipi comporta che la malattia si possa presentare associata ad altre condizioni patologiche: cardiopatie congenite, atresia delle vie biliari, retinite pigmentosa, idrocefalo, rene policistico, malattia cistica del fegato, grave malattie da reflusso gastro-esofageo e atresia esofagea. Tutti i possibili sintomi clinici di DCP sono riassunti nella tabella sottostante (Tab. 3.1).
Tabella 3.1: Sintomi e segnali clinici della DCP (Lobo et al., 2014)
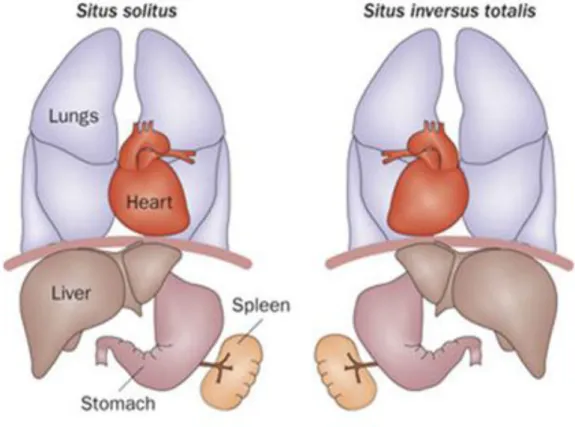
Circa il 38% dei pazienti affetti da DCP presenta disposizione invertita degli organi interni sull’asse sinistro/destro (Fig. 3.6). I meccanismi molecolari alla base della determinazione dell’asimmetria sinistra/destra non sono ancora del tutto chiariti, ma l’ipotesi più accreditata sostiene che il flusso nodale sia necessario per stabilire un gradiente di uno o più morfogeni (come l’acido retinoico), i quali avvierebbero una cascata di segnali per la laterizzazione dell’embrione (Hirokawa et al., 2006). La superficie ventrale del nodo embrionale dei mammiferi, infatti, è ricoperta da ciglia che ruotano in senso orario, generando un flusso antiorario nel fluido extracellulare. Se le ciglia nodali sono immobili o assenti non si crea il flusso nodale e la determinazione del
18
letteratura sono riportati casi di gemelli omozigoti in cui solo uno dei due presentava
situs inversus (Lee e Anderson, 2008; Noone et al., 1999).
Figura 3.6: Confronto della disposizione degli organi interni in un soggetto normale (a sinistra) e in soggetto con situs inversus (a destra) (Patel e Honorè, 2010).
3.1.5 Patogenesi
Il Progetto Genoma Umano e gli studi sull’alga unicellulare Chlamydomonas hanno incoraggiato la ricerca e l’identificazione dei geni potenzialmente responsabili della DCP. L’analisi genetica e la consulenza genetica sono importanti per confermare il sospetto diagnostico ma anche per stabilire l’eventuale stato di portatore e informare quindi sui rischi riproduttivi per effettuare eventualmente la diagnosi prenatale.
Le analisi di linkage genetico e gli studi delle mutazioni nei soggetti affetti dalla malattia, tuttavia, hanno dimostrato che tale condizione è estremamente eterogenea dal punto di vista genetico, anche all’interno di specifici fenotipi ultrastrutturali.
19
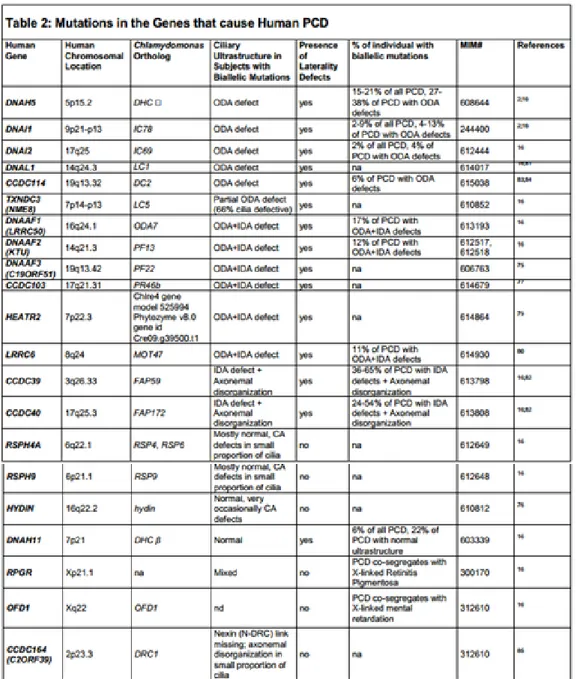
Dal 1999 al 2010 erano state identificate mutazioni causanti DCP in 11 geni; dal tardo 2011 in poi, grazie alle nuove tecnologie che permettono il sequenziamento dell’esoma in maniera molto più rapida, sono stati scoperti altri 10 geni in cui sono state rinvenute mutazione causative di DCP (Vedi tabella 3.2). La grande maggioranza di queste mutazioni (85%) determina loss-of-function della proteina codificata dal gene
wild-type (sono mutazioni non-senso, causanti frameshift o difetti di splicing); il 15% ,
invece, sono mutazioni missenso conservative. Alcune di queste mutazioni sono comuni in vari pazienti, ma la maggior parte di esse viene ritrovata solo in un paziente o in una famiglia (Knowles et al., 2013). Knowles e collaboratori, in una studio del 2013 hanno identificato le mutazioni causative di DCP in più di 200 pazienti, stimando che circa il 65% dei pazienti affetti da DCP possa essere identificato in quanto possessore di una mutazione biallelica, appunto, in uno di questi 21 geni. Nuove high-throughput genetic
technologies, unite alla scoperta recente di ulteriori nuovi geni, potrebbero innalzare
questa percentuale all’80%.
Osservando la tabella 3.2, emerge una forte correlazione tra mutazioni in specifici geni e effetto sull’ultrastruttura ciliare. Molti di questi geni codificano per proteine dell’assonema ciliare (ODA: Outer dynein arms, IDA: Inner dynein arms o raggio radiale), ma 6 di essi sono espressi nel citoplasma e giocano ruolo nel preassemblamento delle ciglia, determinando perdita sia di ODA che di IDA (sono associati ad immobilità ciliare o a severa dismotilità). Si nota, inoltre, che mutazioni nei geni che codificano per i bracci di dineina, sono associati con anormalità del situs, mentre mutazioni in geni che codificano per componenti dell’apparato centrale non lo sono. Oltretutto, si nota la presenza di mutazioni in pazienti affetti ma che mantengono una normale ultrastruttura ciliare (DNAH11, RSPH4A, CCDC40). In questi pazienti la forma dell’onda di battito ciliare appare normale e la frequenza di battito può essere normale oppure maggiore del normale (battito ipercinetico).
20
Tabella 3.2: Mutazioni nei geni che causano DCP; (ODA: Outer dynein arms, IDA:
Inner dynein arms, CA: Central apparatus (Knowles et al., 2013).
Alcuni dei geni causativi di DCP:
DNAI1 (catena intermedia della dineina assonemale 1), omologo del gene IC78 di C. reinhardtii, è localizzato nel cromosoma 9 in posizione p21-p13. Mutazioni di tale gene si ritrovano all’incirca nel 9% dei pazienti; tra queste l'inserzione di una timina in posizione +3 del primo introne rappresenta il 55% delle mutazioni del gene, determinando il deficit del braccio esterno di dineina (Zariwala et al., 2006).
21
DNAH5 (catena pesante della dineina assonemale 5), omologo del gene HCγ di C. reinhardtii, è localizzato nel cromosoma 5; si ritrova mutato nel 21% dei pazienti (Olbrich et al., 2002).
TXNDC3 (tioredoxin-nucleoside difosfato chinasi) codifica per una proteina, appartenente alla famiglia delle tioredoxine, espressa nel braccio corto dei flagelli degli spermatozoi e delle ciglia dell'epitelio respiratorio. Una particolare mutazione non-senso è stata ritrovata associata a eterotassia e deficit parziale del braccio esterno della dineina (Duriez et al., 2007). DNAI2 (catena intermedia dineina assonemale 2), omologo del gene IC69 di
C. reinhardtii, è localizzato sul cromosoma 17. Le mutazioni su questo gene
rappresentano il 2% degli affetti totali, in cui si manifesta con situs inversus associato a deficit del braccio esterno della dineina (Pennarun et al., 2000). KTU (kintoun) codifica per una proteina citoplasmatica necessaria per
l'assemblaggio del complesso di dineina ed è coinvolto nel 12% dei pazienti con deficit di entrambi i bracci di dineina (Omran et al., 2008).
RSPH9 (proteina 9 della testa del ponte radiale) è localizzato sul cromosoma 6; le sue mutazioni causano significative alterazioni dei raggi di connessione con deficit della coppia centrale di microtubuli (Leigh et al.,2009).
RSPH4A (proteina 4A della testa del ponte radiale) è stata associata alla presenza di alterazioni dei raggi di connessione dell'assonema (Leigh et al.,2009).
RPGR (regolatore GTPasico della retinite pigmentosa) codifica per una proteina che, oltre a garantire la funzione dei fotorecettori, è espressa anche nell'epitelio respiratorio. In pazienti di sesso maschile è stata riscontrata una una rara forma di DCP associata alla retinite pigmentosa, dimostrando che la trasmissione della DCP può essere anche X-linked (Krawczyoski et al., 2004).
3.1.5.1 Il gene DNAH11
Il gene DNAH11 (dynein axonemal heavy chain 11) codifica per la catena pesante del braccio esterno di dineina, è composto da 83 esoni codificanti e copre una regione genomica di oltre 353 kb sul cromosoma 7p15.3-21.
22
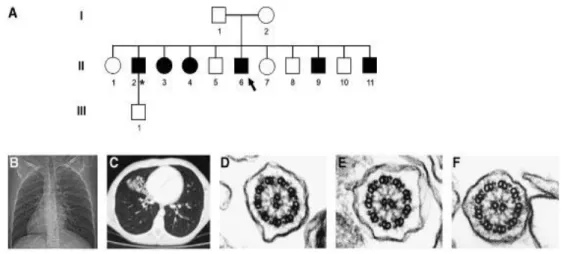
La prima mutazione nel gene DNAH11 è stata identificata mediante sequenziamento diretto. Tale mutazione omozigote non-senso R2852X nell'esone 52 è stata evidenziata in un paziente con disomia uniparentale paterna (UPD7) e diagnosi genetica di fibrosi cistica (mutazione deltaF508 in omozigosi nel gene CFTR). Il paziente presentava anche caratteristiche cliniche della DCP, quali situs inversus totalis ed una grave compromissione respiratoria, ma la microscopia elettronica dimostrava un'ultrastruttura ciliare normale (Fig.3.6) (Bartoloni et al., 2002).
A B
C
Figura 3.6: (A) Albero genealogico del paziente con UPD7 e rappresentazione schematica di parte del cromosoma 7 con mutazioni nei geni CFTR eDNAH11 (alleli mutanti in rosso, alleli normali in bianco). (B) Cromatogramma della sequenza di DNA dell’esone 52 di DNAH11 con mutazione non-senso R2852X. (C) Situs
23
Le catene pesanti della dineina presentano diversi domini strutturali. La coda N-terminale interagisce con gli altri domini della dineina e con il dominio che costituisce il motore. Il dominio motore contiene 6 domini ATPasi della famiglia AAA (ATPases associated with cellular activities) nella testa che formano un anello. In ogni dominio AAA è presente un sito ATPasico, ma solo quello in AAA1 idrolizza ATP. Tra i domini AAA4 e AAA5 protrude un gambo che termina con il sito di legame al microtubulo (Fig. 3.7) (Stephen et al., 2000).
Figura 3.7: (A) Rappresentazione schematica della proteina DNAH11; (B) Rappresentazione schematica del dominio globulare di DNAH11 (Schawabe et al., 2008).
La proteina mutata dovuta alla mutazione omozigote non-senso R2852X è predetta contenere un normale dominio N-terminale e tre dei 6 domini AAA (Fig. 3.8). Se la proteina mutata è stabile dovrebbe essere incorporata correttamente nel complesso del braccio della dineina, ma essendo assente il sito di legame al microtubulo non si legherà a questo. L’ipotesi è compatibile con la microscopia elettronica che evidenzia un assonema e bracci di dineina normali.
24
Figura 3.8: Rappresentazione schematica dei domini di DNAH11 e posizione della mutazione R2852X (Schawabe et al., 2008).
Il possibile coinvolgimento del gene DNAH11 rimaneva comunque dubbio, per la concomitante presenza della fibrosi cistica in questo paziente con DCP atipica. Tuttavia, uno studio successivo condotto su un'ampia famiglia tedesca con diversi individui affetti da DCP (Fig. 3.9) ha dimostrato che anche in questo caso era coinvolto il gene DNAH11 e l'ultrastruttura delle ciglia era normale, mentre il movimento ciliare era ipercinetico (Schwabe et al., 2008). In questi pazienti sono state evidenziate due mutazioni in eterozigosi composta ed è stato dimostrato che l’mRNA di entrambi gli alleli mutati è trascritto e quindi espresso. Se viene anche tradotto, l’effetto predetto delle mutazioni sarebbe: la mutazione p.Y4128X dovrebbe troncare l’intero dominio C-terminale della proteina DNAH11, la mutazione p.A4518_A4523delinsQ dovrebbe far mancare parte del dominio C-terminale e quindi il contatto tra questo e il dominio AAA1 (Fig. 3.10).
Figura 3.9: Albero genealogico della famiglia con DCP e immagini relative al situs inversus, bronchiectasie, ultrastruttura ciliare normale (Schawabe et al., 2008).
25
Figura 3.10: (A) Rappresentazione schematica della proteina DNAH11 e posizione delle mutazioni p.Y4128X e p.A4518_A4523delinsQ; (B) Rappresentazione schematica del dominio globulare di DNAH11 nei casi delle due mutazioni sopracitate (Schawabe et al., 2008).
3.1.7 Esami effettuati per confermare il sospetto clinico
Nei casi in cui sia presente un fondato sospetto e siano state escluse altre cause compatibili con il quadro clinico (fibrosi cistica, deficit immunitari) è necessario effettuare inizialmente dei semplici test di screening (valutazione del trasporto muco-ciliare e determinazione dell’ossido nitrico nasale) seguiti eventualmente da test diagnostici più sofisticati (analisi dell’attività ciliare, esame ultrastrutturale delle ciglia dell’epitelio respiratorio, colture cellulari).
3.1.7.1 Test di screening
Valutazione del trasporto muco-ciliare
Il test alla saccarina è un test semplice che si basa sulla valutazione del tempo che intercorre tra il posizionamento di una piccola quantità di saccarina a livello del turbinato nasale inferiore e la rilevazione della stessa nel cavo orofaringeo, attraverso la
26
percezione del suo sapore da parte del soggetto. Nei bambini può risultare difficile ottenere risposte attendibili (Bush et al., 2002).
La scintigrafia con tecnezio-99 è sicuramente un test più obiettivo poiché non richiede la collaborazione del paziente. Una goccia di sospensione di particelle colloidali marcate con tecnezio-99 viene posta circa 1 cm dietro la giunzione mucocutanea della cavità nasale, sul turbinato inferiore o lungo il pavimento laterale. Viene poi registrato il movimento della radioattività.
Determinazione dell’ossido nitrico nasale
La determinazione dell’ossido nitrico (NO) nasale può essere effettuata fin dai primi mesi di vita. Fisiologicamente l’NO viene prodotto dalla NO-sintetasi a livello del corpuscolo basale delle ciglia dell’epitelio respiratorio ed è coinvolto nella modulazione del battito ciliare. Nella DCP i livelli di NO nasale sono tipicamente ridotti (inferiori a 250 ppb) (Ludberg et al., 1994; Jain et al., 1993).
E’ comunque opportuno ricordare che l’anormalità sia del test alla saccarina sia della determinazione del NO nasale non è esclusiva della DCP. Infatti possono riscontrarsi valori alterati anche in altre condizioni patologiche quali fibrosi cistica, rinosinusiti, mentre valori normali permettono comunque di escludere la DCP.
3.1.7.2 Test diagnostici
Analisi dell’attività ciliare
Questo test deve essere effettuato nei bambini con problemi respiratori cronici perché è l’unico modo per distinguere le alterazioni primitive da quelle secondarie. Dal punto di vista diagnostico sono importanti sia la frequenza del battito che il tipo di movimento ciliare.
La tecnica maggiormente utilizzata per il prelievo è quella del brushing nasale mediante uno spazzolino citologico. Nei bambini il campionamento viene effettuato a livello dei turbinati nasali inferiori dato che le alterazioni ultrastrutturali delle ciglia nasali correlano in maniera significativa con quelle bronchiali (Verra et al., 1993). E’ una tecnica semplice, poco invasiva e non necessita di anestesia, ma richiede comunque una serie di accorgimenti che permettano di ottenere un campione adeguato: la mucosa dovrebbe essere prelevata lontano da infezioni respiratorie acute o da riacutizzazioni di
27
una patologia cronica e i pazienti non dovrebbero assumere farmaci nelle 48 ore precedenti.
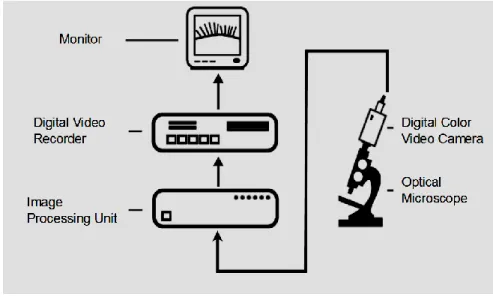
I campioni di ciglia prelevati vengono messi in un mezzo di coltura e analizzati al microscopio ottico dotato di contrasto interferenziale di fase con obiettivo ad immersione (x100). Il microscopio è connesso con una videocamera che consente la ripresa in tempo reale delle immagini ad elevato ingrandimento (Fig. 3.11).
Figura 3.11: Strumentazione per l’analisi dell’attività delle ciglia respiratorie in vitro (www.dcp-pisa.it).
Per valutare l’attività ciliare vengono valutati sia la frequenza del battito che il pattern motorio. La frequenza normale del battito ciliare a livello della mucosa respiratoria è di circa 12 Hz (12 battiti al secondo). La DCP invece è caratterizzata da una frequenza del battito molto bassa o assente, ma in alcuni casi possono essere presenti ciglia dotate di un movimento iperfrequente (Pifferi et al., 2001).
Nel normale pattern motorio le ciglia della stessa fila battono in modo sincrono, quelle della fila contigua battono nella stessa direzione e nella stessa fase, anche se con un piccolo ritardo che determina il tipico movimento a onda metacronale, paragonata ad un campo di grano mosso dal vento.
28
Figura 3.13: Battito ciliare normale: onda metacronale (www.dcp-pisa.it).
Nella DCP sono state riscontrate diverse anomalie del pattern motorio: “a frullino”, “a metronomo, “a cavaturaccioli”, “di prensione dell’apice”, “di sottile tremolio” (figura 3.14). Questi difetti del pattern motorio sono stati correlati, anche se non sempre, con specifiche alterazioni della ultrastruttura ciliare (Schildow et al., 1994).
Figura 3.14: Anomalie del pattern motorio ciliare (www.dcp-pisa.it).
Il tipo di movimento o la presenza di ciglia immobili viene giudicata significativa se è osservata in almeno il 40% dei campi microscopici.
L’analisi dell’attività ciliare in vitro con la valutazione della frequenza del battito ciliare e del pattern di movimento è essenziale nell’iter diagnostico della DCP. Infatti, rappresenta un test diagnostico in grado di individuare i soggetti da sottoporre ad esame ultrastrutturale delle ciglia dell’epitelio respiratorio, ma è anche essenziale per giungere ad una diagnosi di certezza di DCP. Infatti, in pazienti con significative alterazioni del movimento ciliare ma senza alcuna anomalia ultrastrutturale dimostrabile, viene effettuato nuovamente il test per l’analisi dell’attività ciliare in vitro
29
dopo un prolungato periodo di trattamento fisioterapico e farmacologico (Pifferi et al., 2001). Esistono comunque casi di DCP con ultrastruttura normale per cui la diagnosi definitiva è posta in seguito alla valutazione complessiva degli elementi clinici e delle indagini di laboratorio.
Esame ultrastrutturale delle ciglia dell’epitelio respiratorio
Una parte delle cellule poste in coltura dopo il prelievo viene utilizzata per l’esame ultrastrutturale al microscopio elettronico a trasmissione ad ingrandimenti iniziali variabili da 45000 a 91000. Il campionamento mediante brushing nasale è attendibile dal punto di vista diagnostico poiché le alterazioni ultrastrutturali delle ciglia nasali correlano in maniera significativa con quelle bronchiali (Verra et al., 1993).
La struttura dell’assonema è estremamente complessa per cui sono possibili molti tipi di lesioni. Questa eterogeneità strutturale implica, in ogni caso, la perdita del ruolo fisiologico protettivo e lo stesso difetto funzionale, cioè l’incapacità del ciglio di rimuovere il muco e di conseguenza il suo ristagno, che comporta l’insorgere di quadri patologici a carico delle alte e basse vie respiratorie.
Le anomalie ultrastrutturali caratteristiche di patologia ciliare congenita, riviste da Afzelius nel 1987 (Afzelius et al., 1985) sono rappresentate da:
I. Assenza, riduzione o accorciamento dei bracci interno e/o esterno di dineina.
II. Assenza dei ponti radiali e/o assenza della guaina centrale, con conseguente decentramento dei microtubuli centrali.
III. Assenza o accorciamento dei microtubuli centrali e spostamento della coppia n. 1 di microtubuli periferici al centro (trasposizione).
IV. Assenza dei legami di nexina, dei ponti radiali, dei bracci interni di dineina, con conseguente perdita della disposizione circolare delle coppie di microtubuli e “disorganizzazione” dell’assonema.
V. Assenza dei microtubuli centrali e dei bracci interni di dineina. VI. Abnorme lunghezza delle ciglia.
VII. Assenza delle ciglia e dei relativi corpuscoli basali. VIII. Alterazione del normale orientamento delle ciglia.
E’ stata descritta una certa correlazione tra specifiche alterazioni ultrastrutturali e tipo di anomalia del movimento (Tab. 3.3) (Rossman et al., 1984).
30
Ultrastruttura ciliare Difetto di motilità
Assenza dei bracci di dineina Movimento a frullino Movimento a metronomo Rigido movimento su un piano Rigido movimento vibratorio
Difetto dei ponti radiali Movimento di rotazione a cavaturaccioli Rigido movimento su un piano
Difetto di traslocazione Movimento di prensione dell’apice con base rigida
Movimento rotazionale Ultrastruttura normale Movimento di sottile tremolio
Tabella3.3: Correlazione tra ultrastruttura ciliare e difetto di motilità.
Correlato allo stato di flogosi esiste un pattern di lesioni sostanzialmente diverso da quello rilevato nelle forme congenite. Sono alterazioni secondarie, aspecifiche, presenti fino ad un 10% anche nelle ciglia di soggetti normali ed in percentuale maggiore nei vari tipi di patologia acquisita, sia acuta che cronica, dell’apparato respiratorio (discinesie ciliari secondarie; DCS).
Il pattern di queste lesioni è rappresentato da:
I. ciglia con aumento della matrice citoplasmatica (swollen cilia)
II. ciglia con più di un assonema all’interno di una singola membrana (assonemi multipli)
III. ciglia che si proiettano in larghe cavità intracellulari (ciglia intracitoplasmatiche)
IV. alterazioni a carico delle coppie periferiche o dei microtubuli periferici (assenti o aggiuntivi)
V. dislocazione di una o più coppie periferiche VI. microtubuli incompleti
VII. alterazioni della coppia o dei microtubuli centrali (assenti o aggiuntivi).
Inoltre, esiste un gruppo di soggetti definito “borderline” che presenta un pattern intermedio. Presentano cioè una percentuale di elementi patologici superiore a quella
31
riscontrata nelle forme acquisite di flogosi cronica ed un pattern ultrastrutturale caratterizzato dalla presenza di alterazioni “specifiche” dei bracci di dineina e/o della coppia centrale, associato ad un numero variabile di lesioni secondarie. Tale gruppo, eterogeneo dal punto di vista clinico, comprende sia soggetti con una maggiore predisposizione ad ammalarsi ed una minore tendenza alla risoluzione del quadro clinico, sia soggetti con deficit congenito parziale.
L’esame ultrastrutturale delle ciglia ha un ruolo essenziale nell’iter diagnostico delle affezioni respiratorie croniche in quanto permette di distinguere le forme congenite da quelle acquisite sia da un punto di vista qualitativo che quantitativo. Infatti, difetti comunemente considerati di natura congenita possono essere anche conseguenza di una flogosi cronica (Mierau et al., 1992). Esistono comunque delle forme congenite con ultrastruttura normale che rendono più difficoltosa la diagnosi differenziale (Jorissen et al., 2000). Inoltre esiste la possibilità che alterazioni ultrastrutturali considerate tipiche di DCP siano reversibili con il miglioramento clinico. Per questi motivi lo studio ultrastrutturale non può essere considerato un criterio assoluto per la diagnosi di DCP (Cangiotti et al., 2002).
Recentemente sono stati sviluppati anche anticorpi fluorescenti diretti contro i componenti principali dell’assonema, al fine di identificare anormalità strutturali delle ciglia: se il componente contro cui è diretto il determinato anticorpo è mutato, non si avrà il corretto legame dell’anticorpo. Sono in recente sviluppo panelli di anticorpi diretti contro le varie componenti proteiche ciliari (Lobo et al., 2014).
Colture cellulari delle ciglia dell’epitelio respiratorio
Nei casi in cui la diagnosi di DCP risulti particolarmente complessa, a causa di un esame ultrastrutturale non informativo e di un esame dell'attività ciliare influenzato da un’importante flogosi persistente a livello delle cavità nasali, possono essere impiegate le colture di cellule epiteliali ciliate in ambiente privo di agenti nocivi. In presenza di rigenerazione ciliare è infatti possibile escludere la natura congenita delle alterazioni precedentemente evidenziate.
32
33
3.1.8 Terapie
Ad oggi non esistono trattamenti specifici in grado di correggere la disfunzione ciliare che è alla base delle manifestazioni presenti nella DCP. La terapia è, pertanto, sintomatica: consiste nel miglioramento della clearance muco-ciliare e nella prevenzione e trattamento delle infezioni delle vie aeree. E’ importante, infatti, intervenire con l’applicazione di misure fisioterapiche in modo da rimuovere le secrezioni che, accumulandosi nelle vie aeree, favoriscono il ripetersi di processi infettivi responsabili sia del danno strutturale che del decadimento della funzione respiratoria e quindi della progressione della malattia. Le vaccinazioni regolari e i trattamenti antibiotici precoci, associati alla fisioterapia respiratoria, in caso di infezioni polmonari, aiutano a limitare lo sviluppo della bronchiectasia. In casi estremi di insufficienza respiratoria può essere necessario il trapianto di polmone come terapia risolutiva (Date et. al., 2001).
Tutte queste terapie sono, però, solo cure palliative, che tengono sotto controllo i sintomi senza attuare una terapia risolutiva. L’unica strategia che può essere adottata per correggere definitivamente la DCP, come le altre malattia genetiche, sembra essere la correzione del difetto genetico.
3.2 Terapia genica
La terapia genica consiste nel trasferimento di uno o più geni sani in una cellula malata, al fine di curare una patologia causata dall'assenza o dal difetto di uno o più geni mutati.
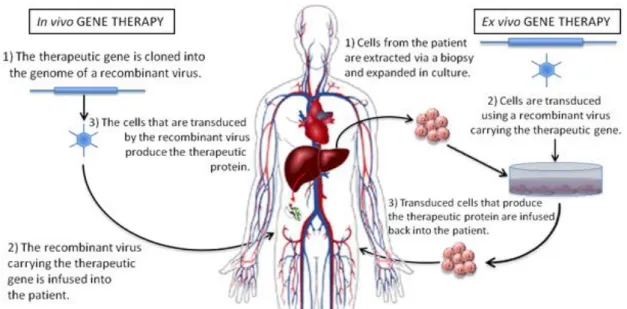
I metodi che possono essere utilizzati per il trasferimento di geni sono di due tipi: ex vivo e in vivo. Nel trasferimento ex vivo si trasferiscono geni clonati in cellule in coltura. Normalmente si usano cellule autologhe per evitare che vengano rigettate dal sistema immunitario del paziente trattato. Il sistema è quindi applicabile ai soli tessuti che possono essere prelevati dal corpo, modificati geneticamente, e reintrodotti nel paziente. E’ una procedura è lunga e costosa ma permette di selezionare ed amplificare le cellule d’interesse, inoltre gode di una elevata efficienza. La terapia genica in vivo, invece, viene attuata in quei casi in cui le cellule non possono essere né messe in coltura, né possono essere prelevate e reimpiantate (come quelle del cervello, del cuore e della maggior parte degli organi interni). In questo caso il gene d’interesse viene
34
inserito nell’organismo tramite un opportuno vettore, per via locale o sistemica. E’ un metodo più economico rispetto al metodo ex vivo, ma meno efficiente e con effetti indesiderati più gravi (Naldini, 2011).
Figura 3.9: Rappresentazione schematica della terapia genica in vivo ed ex vivo (http://global-biotherapeutics.com/ry.php).
L’inserimento del “nuovo” acido nucleico all’interno della cellula può avvenire tramite trasferimento non virale oppure tramite trasferimento virale. Le metodologie adottate per trasferire il DNA senza ricorrere a virus comprendono: l'iniezione di DNA nudo, l'inserimento tramite liposomi, l'inserimento attraverso l'uso di polimeri cationici e il bombardamento tramite particelle (gene gun). L'iniezione di DNA nudo consiste nell'iniettare il gene terapeutico, inserito all’interno di un plasmide, direttamente nella cellula, tramite l'utilizzo di una micropipetta. E’ una procedura semplice, lineare, e permette di trasferire costrutti genici di grandi dimensioni. Lo svantaggio di questa metodica consiste nel fatto che è necessario iniettare il DNA in ogni cellula singolarmente. Il rendimento, inoltre, è molto basso. I liposomi sono vescicole sferiche la cui parete è composta da un doppio strato fosfolipidico. Usando liposomi cationici è possibile far complessare ad essi il DNA, che a pH neutro presenta carica negativa. Il complesso DNA-liposoma può fondersi con la membrana cellulare oppure essere internalizzato tramite endocitosi. Successivamente il DNA viene liberato nel citoplasma, entra nel nucleo e viene espresso. Molto simile è la procedura che utilizza polimeri cationici: sono polimeri dotati di cariche positive che interagiscono con il
35
DNA provocandone la condensazione e proteggendolo da aggressivi chimici, enzimatici, e da radiazioni ionizzanti. Anche i complessi DNA-policatione vengono internalizzati dalla cellula per endocitosi, e possono essere attivamente indirizzati verso specifiche linee cellulari o tessuti utilizzando anticorpi o altre molecole direzionanti. Il quarto metodo consiste nell'utilizzo di particolari strumenti elettrici o ad alta pressione, dette pistole geniche (gene gun), che permettono di inviare nella cellula microscopiche particelle d'oro o di tungsteno ricoperte di DNA.
Il trasferimento di DNA virale, invece, si basa appunto sull’utilizzo di opportuni virus ricombinanti. Questi vengono ingegnerizzati in modo che mantengano inalterate le capacità di veicolazione (delivery) e perdano le proprietà patogene. Il genoma virale, quindi, deve essere riarrangiato completamente, così da eliminare i geni essenziali per la replicazione e la patogenicità del virus e fare spazio per i geni eterologhi. I virus hanno un'ottima tendenza naturale ad infettare le cellule e ad inserirvi il proprio DNA, sia integrandolo sia sotto forma d'episoma. Rispetto ai sistemi di trasferimento non virali hanno un'efficienza nettamente maggiore. Il trasporto di un acido nucleico eterologo da una cellula all’altra è chiamato trasduzione; essa è stata individuata sin da subito come un sistema con il quale correggere difetti genetici (terapia genica) o far esprimere proteine terapeutiche (es. ormoni, fattori di crescita ecc.) e immunogeni per vaccinare contro agenti infettivi o tumori (Pistello et al.,).
L’approccio classico di terapia genica effettuata mediante l’utilizzo di vettori virali consiste nel trasferimento di uno o più geni sani in una cellula malata. Queste metodiche si sono affermate nei primi anni ’90, e negli ultimi dieci anni sono state ampiamente sviluppate e studiate. Il primo caso di successo di terapia genica risale al 1990, quando una bambina affetta da deficit dell'enzima adenosina deaminasi (ADA-scid), una malattia genetica rara che compromette il sistema immunitario al punto che l'organismo è incapace di difendersi da qualsiasi agente infettivo, è stata sottoposta alla terapia genica con vettori virali che hanno ripristinato l’enzima wild-type e curato la paziente, permettendole di avere una vita normale (Aiuti et al., 2002). I metodi tradizionali sono stati utilizzati anche per sviluppare nuovi sistemi per combattere l’anemia falciforme: vettori virali contenenti il gene wild-type (codificante per la catena β dell’emoglobina) sono stati iniettati nel sangue in animali da laboratorio per la correzione del difetto genico. Da qualche anno, inoltre, è stato approvato dalla Food
36
la cura di una rara malattia genetica metabolica: il deficit della lipoproteina lipasi. Nella figura seguente sono indicate le “pietre miliari” che hanno contribuito all’attuazione della terapia genica per malattie monogeniche.
Figura 3.10: Storia della terapia genica e le “pietre miliari” che hanno contribuito all’attuazione della terapia genica per malattie monogeniche. In blu: le “pietre miliari” nel campo della HSCT (Hematopoietic stem cell transplantation); in viola: i maggiori contributi per quanto riguarda il trasferimento genico (Wirth et al., 2013).
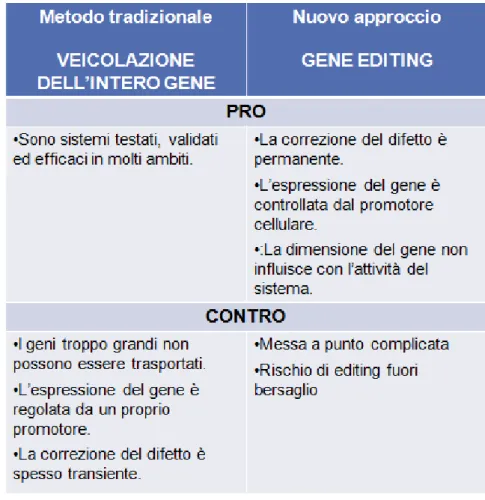
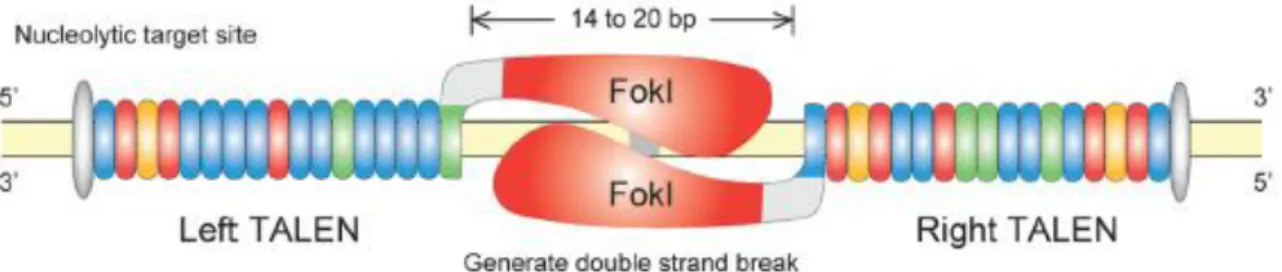
Questi metodi di terapia genica tradizionale, però, presentano alcuni limiti: i geni troppo grandi non possono essere veicolati dai vettori, la trascrizione del gene è regolata da un proprio promotore, e la correzione del difetto è spesso transiente. Questi limiti sono stati parzialmente superati da nuove tecnologie che, mediante ricombinazione sito-specifica, riescono ad indurre la sostituzione della regione genica mutata con un’analoga porzione di DNA contenente la sequenza corretta (gene editing). Queste nuove, eccitanti, tecnologie, prevedono l’utilizzo di proteine con attività endonucleasica (Zinc Finger, TALEN, CrispR) in grado di modificare il DNA genomico, inducendo tagli e/o la sostituzione di un frammento genico con un altro (se, oltre all’endonucleasi, viene fornito anche un frammento di DNA omologo a quello che subisce il taglio). Queste tecnologie in breve tempo si sono affermate nella comunità scientifica e sono diventate un potente strumento per la terapia genica. Possono essere utilizzate per indurre la distruzione di un gene, per creare organismi chimerici o per sostituire un allele mutato con uno wild-type.
37
Un’applicazione di questa metodica, che viene attualmente utilizzata in trial clinici su pazienti e sta dando ottimi risultati, è usata per il trattamento di infezioni dovute ad HIV (il virus della immunodeficienza umana). Quest’ultimo infetta i linfociti T CD4+ riconoscendo specifici recettori presenti sulle loro membrane, tra cui, uno dei più importanti, è CXCR4. La sua assenza sulla superficie cellulare interferisce, quindi, col processo infettivo del virus. E’ possibile indurre un blocco nella produzione di recettore CXCR4 utilizzando delle proteine Zinc Finger che distruggono l’mRNA di CXCR4, inducendo resistenza all’ingresso del virus.
Rispetto alla terapia genica tradizionale (veicolazione dell’intero gene) queste nuove metodiche presentano vantaggi rilevanti: la dimensione del gene non influisce con l’attività del sistema, le modificazioni indotte sono permanenti e il gene ripristinato verrà espresso secondo un controllo cellulare. Nonostante ciò rimangono tecniche più complesse e più costose, e con una certa possibilità di off-target
I principali vantaggi e svantaggi dei due diversi approcci di terapia genica sono riassunti nella seguente tabella (Tab. 3.4):
Tabella 3.4: in questa tabella sono riassunti i principali vantaggi e svantaggi dell’approccio tradizionale e del nuovo approccio di terapia genica.
38
Attualmente, sono più di 1800 i clinical trials di terapia genica approvati, condotti oppure ancora in corso. I vettori che vengono maggiormente adoperati sono: vettori adenovirali, retrovirali, e plasmidi “nudi” (Fig. 3.11). Per quanto riguarda le malattie che occupano il maggior numero di clinical trials di terapia genica, sebbene inizialmente il primo posto fosse occupato dalle malattia monogeniche, ad oggi, invece, il 60% dei clinical trials di terapia genica riguarda il cancro, seguito da malattie monogeniche e cardiovascolari (vedi figura 3.11).
Figura 3.11: Situazione attuale dei clinical trials di terapia genica. (A) Rappresentazione grafica delle malattie che occupano attualmente i clinical trials di terapia genica (Wirth et al., 2014); (B) I vettori usati in terapia genica (Wirth et. al.,., 2013).
39
3.3 Vettori virali
I vettori virali sono un efficiente strumento biotecnologico utilizzato per il rilascio di materiale genetico in una cellula. Questi, sfruttano la naturale capacità che hanno i virus di aderire alla superficie cellulare e rilasciare il proprio materiale genetico all’interno della cellula per la propria sopravvivenza.
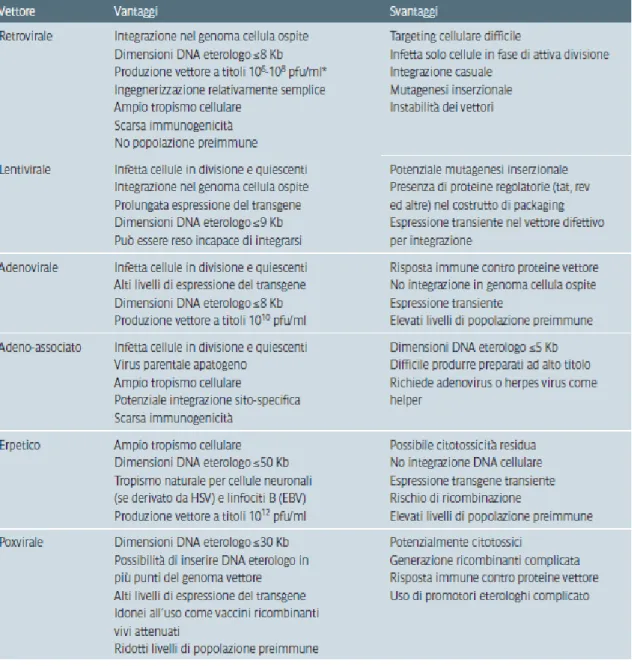
Ad oggi sono stati sviluppati vettori virali da diversi virus (retrovirus, lentivirus, adenovirus, virus adenoassociati, herpesvirus e poxvirus), ognuno con peculiari proprietà funzionali, di versatilità e applicabilità (Tab. 3.5).
I vettori adenovirali, derivati da virus a doppio filamento di DNA, sono tra i vettori più utilizzati attualmente per vaccini e terapia genica. A loro favore depone l'elevata capacità infettante, l’ampio tropismo cellulare e la bassa patogenicità, ma la popolazione è diffusamente immune verso i sierotipi più comuni, e ciò limita il suo utilizzo come vettore per terapia genica a causa della possibilità che si sviluppino risposte immuni dell'ospite.
I vettori erpetici sono derivati dal virus herpes simplex; quest’ultimo possiede un genoma a doppio filamento di DNA e grazie alle glicoproteine di membrana è in grado di infettare un gran numero di tipi cellulari diversi. Può trasportare fino a 50 Kbp di DNA esogeno ed il genoma permane in stato episomiale determinando un’espressione transiente del transgene. Ha un impiego limitato a causa di riscontrati effetti citotossici nelle cellule infettate e per le risposte immuni che sono state rinvenute nella quasi totalità della popolazione.
40
Tabella 3.5: Vantaggi e svantaggi dei vari tipi di vettori virali (Pistello M. in Principi di microbiologia medica. Antonelli G, Clementi M.,Pozzi G., Rossolini G.M., eds. Casa Editrice Ambrosiana, Milano.2012 Cap. 67, page B-370).
41
3.3.1 Vettori retrovirali
I retrovirus sono virus muniti di envelope lipidico e di un capside, al cui interno sono presenti due molecole a singola catena di RNA a polarità positiva di 7-11 Kb. In seguito all’ingresso nelle cellule target, il genoma a RNA è retrotrascritto in una molecola lineare di dsDNA ed è poi integrato nel genoma della cellula. Questa famiglia di virus include diverse specie utilizzate per la terapia genica: oncoretrovirus (virus della leucemia murina di Moloney: MLV), lentivirus (come HIV) e spumavirus.
Tutti i genomi retrovirali hanno alle estremità due promotori virali: le “long
terminal repeat” (LTR) che agiscono in cis durante l’espressione genica virale,
l’incapsidamento, la retrotrascrizione e l’integrazione del genoma. Le LTR sono collocate in tandem con le open reading frame (ORF) gag (group specific antigen), pol (polimerasi) ed env (envelope) che codificano rispettivamente per le proteine strutturali, enzimatiche e per le glicoproteine di superficie (Fig. 3.12). La maggior parte delle sequenze agenti in cis è collocata nelle regioni terminali; questo ha permesso una semplice ed efficace ingegnerizzazione di questi vettori, che consentono l’inserimento di oltre 8 Kb di DNA esogeno. Una proprietà importante dei vettori retrovirali è la loro capacità di integrarsi efficientemente all’interno della cromatina delle cellule target Sebbene l’integrazione non garantisca l’espressione stabile del gene trasdotto, questa strategia risulta efficace per mantenere l’informazione genetica in un tessuto che si auto-rinnova o in cellule tumorali. La disgregazione della membrana nucleare è necessaria affinché il complesso di preintegrazione (che comprende la retrotrascrittasi, il genoma virale e altre proteine virali) possa accedere alla cromatina, per questo motivo la trasduzione da parte dei vettori retrovirali è strettamente dipendente dall’entrata della cellula in mitosi subito dopo l’ingresso del virus. Poiché solo una piccola frazione delle cellule entra in mitosi in un dato momento, questo fattore limita le applicazioni dei vettori retrovirali a target selezionati ex vivo come linfociti e cellule progenitrici della linea ematopoietica.
I primi vettori retrovirali prodotti erano basati su MLV (virus della leucemia murina di Moloney). Per questi vettori è stato sviluppato per la prima volta il processo di pseudotipizzazione, ora largamente utilizzato in tutti gli altri sistemi vettore, che consiste nella produzione di vettori virali in combinazione con differenti proteine d'envelope. Solitamente questi vettori sono pseudotipizzati con la glicoproteina G dell’envelope del virus della stomatite vescicolare (VSV-G), che permette un tropismo