DIPARTIMENTO DI FISICA ED ASTRONOMIA Corso di Laurea Magistrale in Fisica
Valutazione statistica della ripetibilità di
esperimenti di sequenziamento del DNA in specie
batteriche
Relatore:
Presentata da:
Prof. Gastone Castellani
Federico Mondaini
Correlatore:
Prof. Daniel Remondini
Italo Faria Do Valle
Abstract:
I progressi della biologia molecolare assieme alle nuove tecnologie di sequenziamento applicate su scala genomica alla genetica molecolare, hanno notevolmente elevato la conoscenza sulle componenti di base della biologia e delle patologie umane. All’interno di questo contesto, prende piede lo studio delle sequenze genetiche dei batteri, consentendo dunque, una migliore comprensione di ciò che si nasconde dietro le malattie legate all’uomo.
Il seguente lavoro di tesi si propone come obiettivo l’analisi del DNA del batterio Listeria monocytogenes, un microrganismo presente nel suolo e in grado di contaminare l’acqua e gli alimenti. Lo scopo principale è quello di confrontare la variabilità tecnica e biologica, al fine di capire quali siano gli SNPs reali (Single Nucleotide Polymorphism) e quali artefatti tecnici.
La prima parte, quindi, comprende una descrizione del processo di individuazione degli SNPs presenti nel DNA dei campioni in esame, in particolare di tre isolati diversi e tre copie.
Nella seconda parte, invece, sono effettuate delle indagini statistiche sui parametri relativi agli SNPs individuati, ad esempio il coverage o il punteggio di qualità assegnato alle basi. Il fine ultimo è quello di andare a verificare se sussistano particolari differenze tra gli SNPs dei vari isolati batterici.
Keywords: DNA| SNP | Next Generation Sequencing | Coverage | Quality by Depth| Strand Odds Ratio | ANOVA | Multiple Comparisons |
Indice
Introduzione
... 1Capitolo 1
... 21.1 Organismi e cellule ... 2
1.2 Struttura del DNA ... 3
1.3 SNP - Single Nucleotide Polymorphism ... 6
1.4 Cromosomi, geni e RNA ... 7
1.5 La sintesi proteica ... 8
Capitolo 2
... 122.1 Listeria monocytogenes ... 12
2.2 Analisi computazionale su sequenze di DNA... 13
2.3 Sequenziamento Next-Generation ... 14
2.4 Base calling & FastQC ... 14
2.5 Controllo di qualità - Trimmomatic ... 17
2.6 Allineamento al genoma di riferimento – BWA ... 20
2.6.1 Trasformata di Burrows – Wheeler (BWT) ... 21
2.7 Alignment Processing - PICARD ... 23
2.8 Variant Calling – GATK ... 24
2.8.1 File VCF ... 25
Capitolo 3
... 313.1 Analisi della Varianza (ANOVA) ... 31
3.1.1 One-way ANOVA ... 31
3.2 Test di Bartlett ... 33
3.3 Test di Kruskal - Wallis ... 34
3.4 Confronti multipli (Multiple Comparisons) ... 34
3.5 Box Plots ... 35
Capitolo 4
... 37 4.1 Depth of Coverage (DP) ... 38 4.1.1 DP: BedTools Coverage ... 42 4.1.2 DP: Test di Bartlett ... 44 4.1.3 DP: ANOVA ... 46 4.1.4 DP: Kruskal - Wallis ... 474.1.5 DP: Multiple Comparisons ... 48 4.1.6 DP: Box Plots ... 50 4.2 Quality by Depth (QD)... 53 4.2.1 QD: Test di Bartlett ... 57 4.2.2 QD: ANOVA & K-W ... 58 4.2.3 QD: Multiple Comparisons ... 59 4.2.4 QD: Box Plots ... 60
4.3 Strand Odds Ratio (SOR) ... 61
4.3.1 SOR: Test di Bartlett ... 65
4.3.2 SOR: ANOVA & K-W ... 66
4.3.3 SOR: Multiple Comparisons ... 67
4.3.4 SOR: Box Plots ... 68
Conclusioni
... 70Appendice
... 721
Introduzione
Il genoma, cioè il complesso dei geni che definiscono un individuo, è un documento immenso che viene trasmesso dalla cellula madre alla cellula figlia. Studiare il genoma di un organismo implica lo studio e la codifica del suo stesso DNA.
Tale codificazione è, dunque, importante in quanto le informazioni riguardanti un essere vivente vengono immagazzinate lungo la sequenza genetica. In questi ultimi anni il rapido sequenziamento del DNA ha radicalmente cambiato la biologia, tant’è che si è scoperto che solo l’1.5 % dei geni che costituiscono il patrimonio genetico umano è codificante, cioè attivo, mentre il 98.5 % è non codificante e viene denominato “junk-DNA”.
Al contrario, nei batteri, quasi tutto il DNA risulta codificante, in quanto si è visto essere ricco di sequenze, e quindi informazioni, non ridondanti, cosa che invece è caratteristica del DNA non codificante. Quindi in termini di stringhe di nucleotidi, c’è proprio l’idea che dentro queste sequenze siano contenute delle informazioni importanti.
L’idea del seguente lavoro di tesi è stata sviluppata proprio all’interno di questo contesto. Lo scopo è stato quello di prendere l’intera sequenza genomica di un batterio, Listeria monocytogenes, e farne uno studio dal punto di vista informativo. In particolare, sono state effettuate delle analisi statistiche sulle variazioni della sequenza genetica del batterio considerato, ovvero sono stati studiati i cosiddetti SNP (single nucleotide polymorphism), al fine di confrontare ciò che differenzia la variabilità biologica da quella tecnica.
Su tali variazioni, o mutazioni, si sono accumulate sufficienti evidenze sperimentali, così da considerare il loro ruolo importante nell’indurre la suscettibilità delle risposte dell’organismo nei confronti degli stimoli interni (endogeni) ed esterni (esogeni). Ad esempio, alcuni di questi SNP possono rendere ragione della resistenza del batterio stesso a un determinato agente decontaminante.
Nel primo capitolo si descrive la struttura del DNA, le sue caratteristiche e il concetto che sta alla base degli SNPs. Lo scopo è quello di fornire al lettore gli strumenti necessari per comprendere i dati su cui verranno effettuate le analisi.
Nel secondo capitolo si presenta il metodo di lavoro che ha portato all’identificazione degli SNPs. Vengono, quindi, mostrati nel dettaglio tutti i passaggi di una determinata pipeline, alla fine della quale si giunge all’individuazione di tutte le varianti presenti nei campioni dei batteri studiati.
Nel terzo capitolo vengono presentati i metodi statistici utilizzati per esaminare gli SNPs ottenuti, in particolare si descrive in che cosa consiste il Test di Bartlett, l’Analisi di Varianza (ANOVA), il Test dei confronti Multipli e infine che cosa sono i Box plots.
Nel quarto capitolo vengono mostrati i risultati derivanti dall’ applicazione dei metodi statistici descritti, traendone infine le dovute conclusioni.
2
Capitolo 1
Il DNA
In questo capitolo si vuole dare una breve descrizione della struttura del DNA e del processo di sintesi delle proteine. Necessario a questo scopo è una esemplificata introduzione alla struttura della cellula e all’ambiente in cui sono situati e si svolgono i processi sui quali si vuole porre attenzione [1][2].
1.1 Organismi e cellule
La cellula è l’unità di struttura e di funzione di cui sono costituiti tutti gli organismi viventi: ogni loro attività dipende dal funzionamento delle cellule. Queste possono essere assai diverse tra loro, ma tutte hanno in comune le seguenti caratteristiche:
• ogni cellula è circondata da una membrana plasmatica, una sorta di sottilissima pellicola che separa la cellula dalle altre o dall’ambiente circostante, regolando anche l’ingresso e l’uscita dei materiali.
• la membrana plasmatica racchiude il citoplasma, che occupa la maggior parte dello spazio interno e nel quale avviene gran parte delle funzioni cellulari. Queste funzioni, nelle cellule meglio organizzate ed evolute, sono affidate a speciali categorie di organelli1.
• tutte le cellule presentano un nucleo (che in quelle meno evolute è sostituito dal cosiddetto nucleoide) che contiene il DNA.
1Gli organelli costituiscono compartimenti delimitati da un tipo di membrana caratteristica.
3
Esistono due tipologie fondamentali di cellule: cellule eucariote (quelle che formano il corpo delle piante, degli animali e dell’uomo) e cellule procariote (per esempio i batteri).
Le cellule procariote, oltre a essere normalmente assai più piccole di quelle eucariote, sono anche molto più semplici dal punto di vista strutturale: in esse vi è il cosiddetto nucleoide sede del DNA e mancano del tutto organelli cellulari distinti.
Le cellule eucariote, al contrario, si caratterizzano proprio per la presenza di organelli distinti e provvisti di una loro membrana di separazione dal citoplasma; contengono un nucleo, che è separato dal resto con una membrana. Il nucleo contiene cromosomi che sono i portatori del materiale genetico.
Racchiusi nella membrana esterna ci sono organelli come centrioli, lisosomi, cloroplasti (che producono zucchero) e mitocondri che producono energia sotto forma di ATP, ecc.. .
Una singola cellula per produrre tessuti e organi deve: • crescere
• dividersi • differenziarsi
Queste tre fasi vanno a costituire il CICLO CELLULARE.
1.2 Struttura del DNA
Il DNA è la principale molecola portatrice di informazione in una cellula. Il DNA è un polinucleotide cioè una catena di piccole molecole chiamate nucleotidi. I nucleotidi sono composti da uno zucchero (che nelle molecole di DNA è il desossiribosio, uno zucchero a cinque atomi di carbonio) al quale si attaccano le basi azotate (adenina, timina, citosina, guanina) e un gruppo fosfato.
Esistono 4 differenti tipi di nucleotidi raggruppati in due generi chimici: • purine ADENINA [A] GUANINA [G] • pirimidine CITOSINA [C] TIMINA [T]
4
Figura 2: Purine e Pirimidine
Differenti nucleotidi si legano assieme in un qualche ordine a formare un polinucleotide, ad esempio2:
A − G − T − C − C − A − A − G − C − T − T
Il polinucleotide può avere una qualsiasi lunghezza e qualsiasi sequenza; la sequenza ha una direzione, come questa:
A → G → T → C → C → A → A → G → C → T → T
L’orientazione del polinucleotide è data marcando le estremità con 5’ a sinistra e 3’ a destra (le due estremità della sequenza sono chimicamente differenti).
Due stringhe sono dette complementari se una può essere ottenuta dall’altra cambiando A con T e C con G, e cambiando la direzione della molecola in senso opposto. Ad esempio:
← T ← C ← A ← G ← G ← T ← T ← C ← G ← A ← A
questa è la complementare della sequenza superiore.
Specifiche coppie di nucleotidi possono formare legami deboli fra loro. A si lega con T, C si lega con G (per essere più precisi, due legami idrogeno possono venire a formarsi fra le coppie A-T, e tre legami idrogeno fra C-G).
Sebbene tali interazioni siano legami deboli, quando due lunghi polinucleotidi complementari si incontrano, essi tendono ad attaccarsi, come in figura 3.
Figura 3: unione di polinucleotidi complementari.
Le linee verticali fra i due fili di DNA rappresentano le forze fra essi. Le coppie A-T e G-C sono chiamate coppie-base (bp). La lunghezza del DNA è misurata in coppie base di nucleotidi (nt).
5
Due catene di polinucleotidi complementari formano una struttura stabile, che assomiglia a un’elica ed è conosciuta come il DNA a doppia elica (circa 10 bp in questa struttura formano un intero giro che è lungo circa 3.4nm).
Figura 4: Struttura del DNA
Notiamo che, dal momento che le stringhe sono complementari, ciascuna di esse determina completamente l’altra e quindi per avere l’informazione su tutto il DNA basterebbe prendere in considerazione una sola stringa della molecola del genoma. La massima quantità di informazione che può essere codificata in una tale molecola è quindi 2 bits volte la lunghezza della sequenza.
Durante il processo di sintesi del DNA (la replicazione del DNA) i due filamenti di DNA di ciascun cromosoma vengono srotolati e ciascun filamento dirige la sintesi del filamento di DNA ad esso complementare, generando così due doppie eliche di DNA, ciascuna delle quali è identica alla molecola parentale. Questo avviene durante la divisione cellulare e, dal momento che ciascuna doppia elica di DNA contiene un filamento che apparteneva alla molecola parentale e un filamento di DNA neosintetizzato, si dice che il processo replicativo è
semi-conservativo [3].
6
1.3
SNP -
Single Nucleotide Polymorphism
Le recenti scoperte inerenti al genoma umano hanno permesso di comprendere le basi che determinano le variazioni individuali nel funzionamento dell’organismo. E’ infatti emerso chiaramente che tutti gli individui sono uguali per il 99,9% del loro patrimonio genetico, mentre lo 0,1% di variabilità determina le differenze fra gli individui. La variabilità genetica fa sì che ogni individuo risponda in maniera diversa agli stimoli ambientali e, quindi, sia in grado di adattarsi o meno a particolari condizioni. Le differenze genetiche tra individui sono spesso rappresentate da varianti genetiche puntiformi dette SNP.
Un polimorfismo a singolo nucleotide (in inglese Single Nucleotide Polymorphism o SNP, pronunciato snip) è un polimorfismo, cioè una variazione, del materiale genico a carico di un unico nucleotide. La figura 6 mostra in dettaglio che cos’è uno SNP.
Ad esempio, se le sequenze individuate in due pazienti sono AAGCCTA e AAGCTTA, è presente uno SNP che differenzia i due alleli C e T.
Figura 6: Rappresentazione di uno SNP.
All'interno di una popolazione, è possibile determinare una minor frequenza allelica, il rapporto tra la frequenza della variante più rara e quella più comune di un determinato SNP. Solitamente ci si guarda con maggiore attenzione da SNPs aventi un valore di minor frequenza allelica minore all'1%, trascurando nelle analisi la maggior parte degli SNPs che, anche per il loro elevatissimo numero, risultano poco maneggevoli. È importante notare che possono esistere variazioni notevoli tra popolazioni umane. Uno SNP molto comune in un determinato gruppo etnico può, dunque, essere molto raro in un'altra popolazione.
Lo studio degli SNPs è molto utile poiché variazioni anche di singoli nucleotidi possono influenzare lo sviluppo delle patologie o la risposta ai patogeni, agli agenti chimici, ai farmaci. Per tale motivo gli SNPs possono avere una grande importanza nello sviluppo di nuovi farmaci
7
e nella diagnostica, in quanto consentono di conoscere l'effetto che può avere un farmaco su un individuo ancor prima della somministrazione, attraverso uno screening degli SNPs presenti nel gene responsabile della metabolizzazione del farmaco stesso; queste sono le basi della farmacogenomica. Dal momento che gli SNPs sono perlopiù ereditati di generazione in generazione, essi vengono utilizzati in alcuni studi genetici.
1.4 Cromosomi, geni e RNA
In questo paragrafo si vuole fare chiarezza su termini che si useranno d’ora in poi per descrivere il processo di sintesi delle proteine.
In una cellula tipica ci sono uno o parecchi DNA a doppia elica arrotolati e organizzati come cromosomi. Nelle cellule eucariote i cromosomi hanno una struttura complessa in cui il DNA è arrotolato intorno a proteine strutturali chiamate istoni. Nell’uomo, ad esempio, ci sono 23 coppie di cromosomi, grandi abbastanza da essere visti al microscopio. La lunghezza totale del DNA umano, se potessimo srotolarlo, dovrebbe essere più di un metro. Anche i mitocondri contengono DNA ma in quantità assai minore rispetto ai cromosomi. IL DNA dei cromosomi e dei mitocondri forma il GENOMA dell’organismo. Tutte le cellule in un organismo contengono identici genomi, con alcune eccezioni, come risultati della replicazione del DNA a ciascuna divisione cellulare.
Il gene può essere visto come una sorta di messaggio in codice dal quale si parte per arrivare alla sintesi delle proteine. Una definizione di gene appropriata non è ancora stata concordata; si propone la seguente, con la consapevolezza della sua imprecisione:
In pratica, il messaggio contenuto nel DNA del nucleo viene trascritto in un altro acido nucleico, il cosiddetto RNA o acido ribonucleico. Similmente al DNA, l’RNA è costituito anch’esso da nucleotidi ma al posto della timina (T) ha l’uracile (U), al posto del desossiribosio è presente il ribosio e, inoltre, non ha una struttura ad elica ma è un’unica stringa. Comunque l’RNA può avere strutture spaziali complesse dovute a legami complementari fra le parti della stessa stringa.
L’RNA può codificare l’informazione genetica, è replicabile, forma complesse strutture 3D, e può anche agire come catalizzatore per certe reazioni chimiche relative allo splicing (una fase della sintesi proteica che verrà esposta più avanti).
Un gene è un tratto continuo del DNA dal quale un complesso macchinario molecolare può leggere una informazione (codificata come una stringa di A, T, G e C) e produrre un particolare tipo di proteina o più differenti proteine.
8
In una rappresentazione lineare l’RNA si presenta come un insieme di geni uniti da sequenze dette regioni intergeniche la cui funzione è quella di essere riconosciute da molecole regolatrici, solitamente proteine. Sono chiamate anche regioni non codificanti in quanto non codificano per le proteine; qui la funzione del DNA è determinata direttamente dalla sua sequenza, non per mezzo di un qualche codice intermediario.
1.5 La sintesi proteica
Figura 7: La sintesi proteica.
Analizziamo ora il processo che porta alla creazione di una proteina. Possiamo schematizzare la sintesi delle proteine in tre fasi (trascrizione, splicing, traduzione) per le cellule eucariote; in due (trascrizione, traduzione) per le cellule procariote:
1. Trascrizione: Il messaggio genetico, che è codificato nella molecola di DNA, non viene utilizzato direttamente per la sintesi delle proteine che, tra l’altro, avviene nel citoplasma. Nelle cellule eucariote esso viene infatti trascritto in un complementare pre-mRNA dalla complessa proteina RNA polimerasi (per cellule procariote l’RNA è direttamente l’mRNA). Siccome il linguaggio del DNA e quello dell’RNA sono basati su sequenze di nucleotidi sostanzialmente simili, non vi è, nei due acidi nucleici, una sostanziale differenza di linguaggio molecolare e per questo il processo è chiamato trascrizione, cioè sostanzialmente “copiatura”. Questo processo è simile alla replicazione del DNA, con la sola differenza che, ad essere copiato, è uno solo dei due filamenti del DNA e che la copiatura, o trascrizione, produce una molecola di mRNA complementare al tratto copiato, dove le basi complementari sono G − C e A − U (invece di A − T). Dei due filamenti di DNA, dunque, solo uno viene copiato, quello cioè che codifica la proteina e che è riconosciuto dall’enzima della trascrizione, la RNA polimerasi, per la presenza di una sequenza, chiamata promotore, collocata all’inizio del gene. La trascrizione termina in corrispondenza di un’altra sequenza specifica che dà il segnale di terminazione.
9
Nei batteri, dove non vi è una membrana nucleare, l’mRNA così prodotto viene immesso direttamente nel citoplasma ed è pronto per avviare la sintesi proteica. Negli eucarioti, invece, prima di essere inviato nel citoplasma, il pre-mRNA viene processato nella cosiddetta fase di splicing
2. Splicing: In questo meccanismo di “taglia e cuci”, presente solo nelle cellule eucariote, vengono rimossi alcuni tratti del pre-mRNA chiamati introni e sono collegate insieme le sezioni rimanenti, chiamate esoni. Gli esoni sono la parte del gene che serve per la codifica delle proteine. Il meccanismo dello splicing dell’RNA dipende in modo cruciale da quella che è stata chiamata la regola GT −AG: quasi sempre gli introni cominciano con GT (o GU se ci si riferisce all’RNA) e terminano con AG.
Grazie allo splicing, gli esoni vengono ad essere congiunti nello stesso ordine in cui si trovavano nel DNA. In questo modo si mantiene la collinearità fra gene e proteina, relativamente ai singoli esoni e alle parti corrispondenti della catena proteica, ma le distanze nei geni non corrispondono alle distanze nella proteina: la lunghezza del gene è definita dalla lunghezza dell’iniziale pre-mRNA invece che dalla lunghezza dell’mRNA. Notiamo che i geni eucarioti non sono necessariamente interrotti. Alcuni corrispondono direttamente alla proteina prodotta, come nei geni procarioti. Nel Saccharomyces cerevisiae, per esempio, molti geni non sono interrotti. In eucarioti evoluti, molti geni sono interrotti e gli introni sono molto spesso più lunghi degli esoni, creando geni che sono molto più grandi delle loro regioni codificanti. Tanto più il genoma diventa grande, tanto più gli introni sono estesi mentre gli esoni sono sequenze più brevi.
10
Il risultato della fase di splicing è l’mRNA.
3. Traduzione: Raggiunto il citoplasma l’mRNA viene tradotto nello specifico linguaggio delle proteine. Questo processo richiede una complessa serie di operazioni dovendosi trasferire l’informazione scritta nella sequenza dei quattro nucleotidi dell’mRNA, nella specifica sequenza dei venti possibili amminoacidi3 delle proteine. Per ovviare a questa
necessità il codice dovrà essere basato su triplette di nucleotidi (ovvero, tre nucleotidi adiacenti). Ciascuna tripletta è chiamata codone e codifica per un amminoacido. Dal momento che ci son 43 = 64 codoni e solamente 20 amminoacidi, il codice è ridondante
in quanto singoli amminoacidi possono corrispondere anche a due o più triplette diverse. Va, tuttavia, osservato che un codice così fatto non è mai ambiguo, in quanto la stessa tripletta codifica sempre per un unico amminoacido.
Nel citoplasma l’mRNA forma un complesso con il ribosoma (una struttura composta da proteine ed RNA). Poiché i trinucleotidi e gli amminoacidi sono elementi strutturalmente incongruenti, sorge il problema di come ogni particolare codone possa trovare corrispondenza in un determinato amminoacido. Questo compito è affidato alla molecola tRNA (RNA di trasferimento), che traduce il linguaggio dei singoli nucleotidi in quello degli amminoacidi grazie a due sue proprietà fondamentali:
1. rappresenta un singolo amminoacido al quale si lega in modo covalente;
2. contiene una sequenza di tre nucleotidi, l’anticodone, complementare al codone
che rappresenta il suo amminoacido. L’anticodone permette al tRNA di riconoscere il codone. È importante osservare il fatto che esiste almeno un tRNA per ogni amminoacido.
Per la sua struttura il tRNA può portare ciascun amminoacido al ribosoma e riconoscere un codone nel mRNA; l’amminoacido portato dal tRNA è aggiunto alla nascente proteina.
Da notare che, benché esistano 64 codoni, il numero di molecole di tRNA caratterizzate da codoni differenti è inferiore: esistono infatti solo 30 tipi di tRNA citoplasmatici e 22 tipi di tRNA mitocondriali. L’uso di tutti e 64 i codoni sui ribosomi citoplasmatici e mitocondriali è tuttavia possibile perché le regole di appaiamento tra basi sono meno
3 sono i “mattoni” delle proteine. Vengono classificati in base alla natura dei loro gruppi laterali. Il nome di ogni
11
rigide quando si tratta di riconoscimento tra codone e anticodone. L’ipotesi dell’imprecisione dell’accoppiamento (wobble hypothesis) afferma che l’appaiamento tra codone e anticodone segue le normali regole A − U, G − C per le basi nelle prime due posizioni di un codone, ma che nella terza posizione si potrebbe verificare una “incertezza” e che quindi è ammesso anche l’appaiamento G − U. La traduzione continua finché si incontra un codone di terminazione e cioè UAA, UAG, UGA nel caso degli mRNA codificati nel nucleo; UAA, UAG, UGA o AGG nel caso degli mRNA codificanti nei mitocondri (si ricorda che il codone d’inizio è quasi sempre AUG).
Il termine della traduzione è la parte finale dell’espressione del gene e il prodotto è una proteina, la sequenza della quale corrisponde alla sequenza codificata dell’mRNA.
12
Capitolo 2
Nel seguente capitolo verrà fornita, in primis, una breve descrizione del batterio Listeria
monocytogenes, successivamente si passerà ad analizzare nel dettaglio la descrizione della pipeline
che ha portato all’identificazione degli SNPs per il batterio considerato.
2.1
Listeria monocytogenes
Listeria monocytogenes è la specie di batteri patogeni che causa l’infezione della listeriosi. È un
batterio facoltativamente anaerobico, capace quindi di sopravvivere sia in presenza che in assenza di ossigeno. È in grado di crescere e riprodursi all’interno di cellule ospiti ed è uno dei patogeni di origine alimentare più virulenti, responsabile negli USA di all’incirca 1600 infezioni e 260 morti all’anno.
Figura 9: Listeria Monocytogenes visto tramite microscopio elettronico.
Listeria monocytogenes è un batterio Gram positivo (rimane colorato di blu o viola dopo aver
subito la colorazione di Gram), asporigeno (cioè che non genera spore, ovvero cellule avvolte da una membrana che le protegge dagli agenti atmosferici i quali potrebbero danneggiarne il contenuto), aerobio-anaerobio facoltativo. Il microrganismo cresce in un range di temperatura molto largo (tra i +3 ºC e i 45 ºC) con un optimum tra i 30 ºC e i 38 ºC. Presenta buona resistenza a varie condizioni di pH (tra 4,4 e 9,6) e temperatura, caratteristiche che lo rendono un potenziale contaminante di alimenti, anche se conservati in frigorifero.
Per il seguente lavoro di tesi, si aveva a disposizione 3+3=6 campioni del batterio (in particolare 3 isolati differenti con le relative copie) sui quali era già stato eseguito il sequenziamento tramite il metodo Illumina. Per una migliore comprensione è stata riportata la tabella 1 dove si elencano i nomi dei batteri con le relative copie; ognuno di quegli elementi è stato completamente analizzato secondo i metodi descritti nei paragrafi successivi [4].
13
Isolato Copia LIST1 LIST63 LIST2 LIST64 LIST3 LIST65
Tabella 1: Elenco dei batteri analizzati con le relative copie.
2.2
Analisi computazionale su sequenze di DNA
Prima di addentrarsi nello studio della pipeline dell’analisi genetica, è bene dire che il processo di elaborazione digitale, che porta dalla lettura grezza delle sequenze di DNA all’identificazione di varianti genetiche, è un processo molto complesso e variabile. La complessità del workflow (o pipeline) è dovuta principalmente all’eventualità di possibili errori nelle fasi di lettura e di allineamento, che avvengono con una probabilità maggiore nei nuovi strumenti NGS rispetto ai sequenziatori pre-NGS.
La continua evoluzione dei metodi e delle tecnologie hardware e software del settore, inoltre, impone frequenti cambiamenti nei formati di dati standard e negli strumenti software che filtrano e analizzano le sequenze. Lo sviluppo di un workflow ben definito e automatizzato per l’analisi dei dati genetici è diventato di fondamentale importanza negli ultimi anni.
La figura 10, rappresentata in maniera poco dettagliata (in quanto le operazioni sono molto variabili), è una pipeline tipica per l’analisi delle sequenze esoniche.
In verde sono mostrati i processi, mentre in blu i dati di output/input. Se si considera il diagramma come uno modello layer a strati, i dati di output generati da uno strato, rappresentano i dati di input per lo strato successivo.
All’interno di questa tesi verrà approfondito ogni stadio dell’analisi, senza entrare troppo in dettagli specifici dipendenti dalla piattaforma o da particolari necessità.
Per ora ci si limita semplicemente a dire che la figura 10 mostra una pipeline per il sequenziamento del DNA utilizzata per identificare alcune varianti somatiche.
La prima fase, che prende il nome di Base calling è altamente specifica e collegata alla procedura di sequenziamento usata per generare sequenze di reads.
14
Figura 10: Pipeline utilizzata per l’identificazione degli SNPs.
La seconda fase, quella di Variant Calling, allinea le sequenze di reads al genoma di riferimento (in questo caso genoma umano), per ricostruire un completo assemblaggio delle sequenze e dedurre tutte le variazioni dovute al filtraggio.
Infine, l’ultimo step (Variant Filtering), salva queste variazioni utilizzando più fonti esterne come riferimenti [5].
La seguente pipeline è stata riportata come esempio generico per il sequenziamento del genoma umano, tuttavia in questa tesi, si prenderà in considerazione (come già accennato precedentemente) lo studio e l’analisi del genoma del batterio Listeria monocytogenes.
2.3
Sequenziamento Next-Generation
Il NGS (Next-generation sequencing) è un nome scelto per indicare tutte quelle piattaforme di sequenziamento, e le relative tecnologie, nate dopo il 2005, che hanno rivoluzionato il processo di sequenziamento permettendo la parallelizzazione dello stesso, a beneficio di costi e prestazioni. La crescita esponenziale nella velocità di sequenziamento delle macchine NGS, capaci di generare molti milioni di sequenze lette per ogni esecuzione, ha spostato il collo di bottiglia dalla generazione delle sequenze, alla gestione ed analisi dei dati.
La velocità di esecuzione del sequenziamento per queste tecnologie, va però a discapito dell’accuratezza nel basecalling e nell’allineamento delle reads, svantaggio a cui si rimedia con letture ripetute e appositi processi computazionali nelle successive fasi di analisi.
2.4
Base calling & FastQC
Il base calling è un processo ad opera dello strumento di sequenziamento NGS, che associa ad ogni nucleotide letto un valore di probabilità per ogni base azotata.
15
Spesso la stessa sequenza viene letta più volte per ovviare alla mancanza di accuratezza delle letture e a valori non soddisfacenti di probabilità. Il formato dei dati di output più diffuso tra le piattaforme NGS è il FASTQ, un formato testuale di cui esistono diverse versioni.
Proprio a causa di questa imprecisione, risulta necessario compiere alcuni controlli di qualità che confermino che i frammenti siano stati effettivamente letti correttamente.
Esistono numerosi programmi utilizzati per questo tipo di analisi, e uno dei più completi è proprio FastQC. Questo software permette una visualizzazione delle problematiche introdotte dal sequenziamento, con dettaglio sufficiente per spiegare anche le possibili cause della scarsa affidabilità dei dati [6].
Il programma riceve in ingresso un file nel formato .fastq in cui è stato raccolto l’elenco delle sequenze.
Questo programma valuta differenti aspetti per il controllo della qualità complessiva dei frammenti, e passa in rassegna: 1) la qualità della lettura delle singole basi, calcolata con opportuni algoritmi; 2) la verosimiglianza dell’assegnamento di ciascuna base della sequenza al nucleotide scelto; 3) le duplicazioni delle sequenze e alcune considerazioni sulle loro lunghezze. Per ogni voce, poi, il programma, secondo opportuni standards, assegna automaticamente un simbolo che indichi il livello di qualità associato:
ottima qualità del dataset rispetto al parametro in esame; qualità scarsa che necessita di verifiche;
qualità del tutto insufficiente: è necessario prestare attenzione. 1.1 FASTQ
FASTQ è diventato negli anni un formato molto comune per la condivisione di dati genetici di sequenziamento, poiché combina sia la sequenza di basi, che un quality score associato ad ogni base nucleica, ovvero un punteggio di attendibilità sulla lettura di quella base all’interno della sequenza.
Il formato FASTQ nasce come un’estensione del formato FASTA, in quanto aggiunge specifiche informazioni sull’attendibilità della lettura, rappresentando così la sequenza a disposizione con un livello di dettaglio maggiore, ma senza pesare sulla dimensione dei dati.
Grazie all’estrema semplicità del formato, il FASTQ è ampiamente utilizzato per l’interscambio di dati; tuttavia il FASTQ, anche se nato come evoluzione del FASTA, continua a soffrire dell’assenza di una definizione chiara e non ambigua, mancanza che ha portato all’esistenza di molte varianti incompatibili tra loro.
16
Per ogni insieme di frammenti, pertanto, si propone una valutazione complessiva della qualità, con le indicazioni della bontà di lettura secondo i differenti parametri. Le analisi vengono compiute o sulla qualità complessiva delle diverse sequenze, o sulla valutazione della qualità media tra tutte le sequenze nella lettura delle diverse basi.
A causa del processo di lettura, infatti, la qualità varia a seconda di quante basi del frammento siano già state lette (in particolare, le prime basi lette vengono identificate univocamente, mentre più il processo va avanti, più la qualità si abbassa).
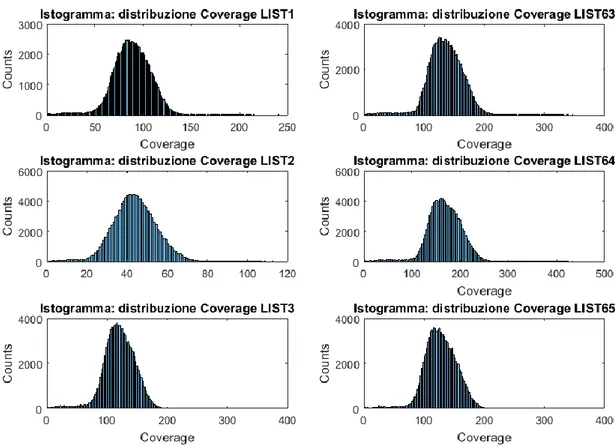
La Figura 11 propone, a titolo di esempio, la valutazione complessiva del dataset relativo al batterio LIST1 (Listeria) utilizzato per le successive analisi.
Figura 11: Valutazione della qualità complessiva del dataset relativo al batterio LIST1.
Per ragioni di brevità, si è scelto di non riportare completamente l’analisi dettagliata della qualità relativa a tutti i campioni del batterio Listeria, come da schema riassuntivo proposto in Figura 11.
Poiché però si è parlato di qualità delle sequenze, risulta perlomeno utile prendere in considerazione il diagramma di figura 12, dove viene mostrata un’overview dell’intervallo dei valori di qualità per tutte le basi in ogni posizione nel file FastQ.
17
Figura 12: FastQC: qualità per ogni base della sequenza Per ogni posizione nella read è riportato un box plot che ha come elementi:
• La linea centrale rossa che rappresenta la mediana. • I box gialli mostrano il range interquartile.
• La linea blu rappresenta la qualità media.
Sull’asse y del grafico è presente il quality score: più alto è questo valore e migliore è la chiamata della base. Sullo sfondo si possono distinguere tre colori che corrispondono a una chiamata molto buona della base (in verde), una chiamata di qualità rivedibile (arancione) e una di scarsa qualità (rosso). Nella maggior parte delle piattaforme che eseguono il sequencing, la qualità della chiamata si degrada con l’andare del processo: di conseguenza, è abbastanza comune vedere che verso la fine della read, le chiamate cadano nella zona arancione/rossa.
Tendendo in considerazione quanto detto poco sopra, questi risultati portano a concludere che il dataset inerente al batterio preso come esempio (LIST1), possiede complessivamente una buona qualità: come si può notare, tutte le reads cadono nella zona verde, la quale implica una qualità elevata. Tale procedimento è stato ripetuto per tutti gli altri campioni, nei quali si sono ottenuti risultati che hanno permesso di procedere verso lo step successivo.
2.5
Controllo di qualità - Trimmomatic
Il passo successivo della sequenza prende in considerazione il controllo di qualità delle reads. In base ai risultati ottenuti con FastQC, è possibile decidere di ripulire il dataset delle reads, per facilitare e rendere più accurate le analisi successive. Le possibili operazioni sono:
18
• Scartare le estremità di bassa qualità. • Scartare le reads con bassa qualità media.
• Scartare le reads che dopo le operazioni di trimming rimangono troppo corte. • Rimuovere gli adattatori (adapters).
Al fine di eseguire una o più delle procedure elencate sopra, risulta necessario applicare un processo che prende il nome di “trimming delle reads”, che consiste fondamentalmente nel tagliare i frammenti. Esistono due metodologie di trimming che si possono utilizzare:
1) Trimming statico: si tagliano tutte le reads nello stesso punto;
2) Trimming dinamico (flexible): le reads vengono tagliate sia dall’estremità 5’ che dall’estremità 3’, finché la qualità resta sotto un valore di soglia definito. Le reads, quindi, non avranno più tutte la stessa lunghezza. Il vantaggio di questa seconda tecnica è quello di ottenere reads più corte, ma di maggiore qualità media.
Nel seguente lavoro di tesi si è scelto di utilizzare il software Trimmomatic [7], uno strumento
capace non solo di leggere e tagliare i dati FASTQ prodotti da Illumina, ma anche di rimuovere gli adattatori.
Infatti, quando i dati sono sequenziati tramite Illumina, a questi vengono aggiunti degli adattatori per i frammenti, in modo tale che si aggancino alle “beads”. Se questi adattatori non vengono rimossi, possono causare un assemblaggio non corretto o altri problemi. Inoltre, la qualità delle sequenze dipende anche dalla lunghezza delle reads, e le zone che possiedono un basso quality score, possono essere tagliate utilizzando Trimmomatic.
Le immagini che seguono mostrano un esempio di quello che accade alle reads una volta subìto il processo di trimming. In particolare, risulta evidente come e quanto la lunghezza delle reads nell’estremità destra sia stata notevolmente ridotta, dando luogo a una distribuzione più uniforme del quality score.
19
Figura 13: Quality score delle reads prima del trimming
20
2.6
Allineamento al genoma di riferimento – BWA
Dopo aver raccolto, sequenziato i frammenti di DNA e verificato la qualità della lettura, si procede con l’allineamento delle reads al genoma di riferimento.
L’esigenza di un allineamento locale efficace, ha portato allo sviluppo di numerose tecniche di allineamento migliori della semplice ricerca di stringhe, con la valutazione in sequenza di tutto il genoma. Il numero di operazioni per compiere questa ricerca, infatti, è molto elevato e dell’ordine di O(LgLrNr) con Lg lunghezza del genoma, Lr lunghezza dei frammenti e Nr
numero di frammenti da analizzare. Tale valore, nel caso del genoma umano, risulta molto alto contando all’incirca 3.3B di basi.
A partire dagli anni 2000 sono stati sviluppati numerosi algoritmi di allineamento locale. Alcuni di questi sono basati sulla scansione di tutto il genoma (come MAQ); altri, invece, sulla definizione della trasformata di Burrows-Wheeler (come SOAP2, Bowtie o BWA).
La trasformata di Burrows-Wheeler, oltre ad essere molto adatta alla compressione di stringhe e pertanto utilizzata in programmi come bzip2, è assai efficace per l’allineamento di brevi sequenze al genoma, soprattutto perché consente di memorizzare sinteticamente molte caratteristiche delle stringhe da analizzare, che altrimenti occuperebbero un’eccessiva quantità di memoria.
In questa sezione si prende in considerazione la tecnica del Burrows-Wheeler Alignemnt (BWA) [8], sviluppata da Heng Li e Richard Dublin che permette di allineare brevi frammenti
alla sequenza del genoma umano, consentendo mismatches e introduzione di spazi. Questo algoritmo permette di allineare efficacemente i frammenti al genoma; dalla simulazione condotta dagli stessi H. Li e R. Durbin e riportata nel dettaglio in [9] sull’allineamento di frammenti lunghi 30 basi, si ottiene, per l’allineamento single-end, un’accuratezza dell’80.6%, risultato migliore degli altri algoritmi basati sulla trasformata di Burrows-Wheeler. Fondamentalmente il pacchetto software BWA è formato da tre algoritmi:
• BWA-backtrack • BWA-SW • BWA-MEM
Il primo di questi è stato creato per delle sequenze di reads in grado di contenere fino a 100bp, mentre gli altri due analizzano sequenze che variano da 70bp fino a 1Mbp.
BWA-MEM e BWA-SW condividono infatti caratteristiche simili, ma BWA-MEM, l’ultimo creato, è generalmente consigliato per queries specifiche per il fatto che risulta più veloce e accurato, motivo per cui è stato scelto nel seguente lavoro di tesi.
Una volta lanciato BWA-MEM, questo genera in output un file di tipo .sam (sequence alignment
map), che contiene tutte le informazioni sul procedimento di allineamento e sul risultato.
21
compone. Esiste, infatti, una prima parte di intestazione (header) e una seconda in cui sono presentati nel dettaglio i risultati della procedura (alignment).
Figura 15: Esempio di file .sam La sezione di intestazione comprende generalmente:
• la definizione dell’indice di riferimento dell’allineamento (@HD);
• alcune informazioni sulla sequenza di riferimento, come la localizzazione sul genoma e la sua lunghezza (@SQ);
• alcune caratterizzazioni dei frammenti allineati, come l’identificativo del centro che li ha prodotti e la piattaforma con cui sono stati generati (@RG);
• le caratteristiche del tool che è stato utilizzato per l’allineamento, come il nome del software e la versione (@PG).
La sezione di allineamento contiene, invece, undici campi obbligatori che includono non solo delle informazioni generali sull’alignment, ma anche sulle caratteristiche dettagliate dei frammenti e della sequenza.
L’ultimo campo, infine, è un codice di qualità identificato con la codifica ASCII, che valuta nel complesso l’esito del processo di allineamento. Data la complessità del formato, esistono numerosi tool (samtools) che permettono l’estrazione di informazioni rilevanti dai file .sam e li convertono poi in formati più leggibili: ad esempio, il formato .bed che contiene semplicemente l’elenco dei frammenti allineati e la loro localizzazione sul genoma di riferimento.
2.6.1
Trasformata di Burrows – Wheeler (BWT)
La trasformata di Burrows-Wheeler[10] (abbreviata con BWT) è un algoritmo utilizzato in
innumerevoli applicazioni per la compressione dei dati.
La trasformata, che costituisce gran parte dell'algoritmo, fu sviluppata da Wheeler nel 1983 e viene attualmente utilizzata come primo stadio di trasformazione della stringa da comprimere in molti programmi di compressione commerciali, primo tra tutti bzip2.
22
La trasformata BWT, prima di iniziare la fase di decodifica, deve necessariamente avere a disposizione l'intera sequenza codificata e, se applicata ad una stringa di caratteri, non effettua una vera e propria compressione, ma permuta in maniera reversibile l'ordine dei caratteri stessi. Data quindi una sequenza 𝑥1 𝑥2… 𝑥𝑁 di lunghezza N, se ne creano N - 1 permutazioni,
ciascuna delle quali è uno shift ciclico della sequenza originale che prende il nome di rotazione. In pratica, si ottiene la i-esima rotazione concatenando la sottostringa degli ultimi caratteri con la sottostringa dei primi N - i, ottenendo le seguenti permutazioni della stringa di partenza:
𝑥𝑁−𝑖+1𝑥𝑁𝑥1𝑥2… 𝑥𝑁−𝑖 ∀𝑖 = 1,2, … , 𝑁 − 1
Si riordinano poi le N sequenze in ordine lessicografico. Il codificatore invia la sequenza di lunghezza N, che si compone prendendo l'ultima lettera di ogni rotazione. La ricostruzione della stringa può essere assicurata in due diversi modi. Nel primo modo, il codificatore invia la posizione occupata dalla sequenza originale nella lista ordinata di rotazioni. Nel secondo modo, invece, il codificatore aggiunge un carattere speciale alla fine della stringa prima di effettuare la trasformazione di Burrows-Wheeler.
La sequenza così trasformata è strutturata in modo che si ottiene una compressione particolarmente efficiente. Viene presentato, ora, un esempio di trasformata di Burrows-Wheeler, per renderne più facile la comprensione.
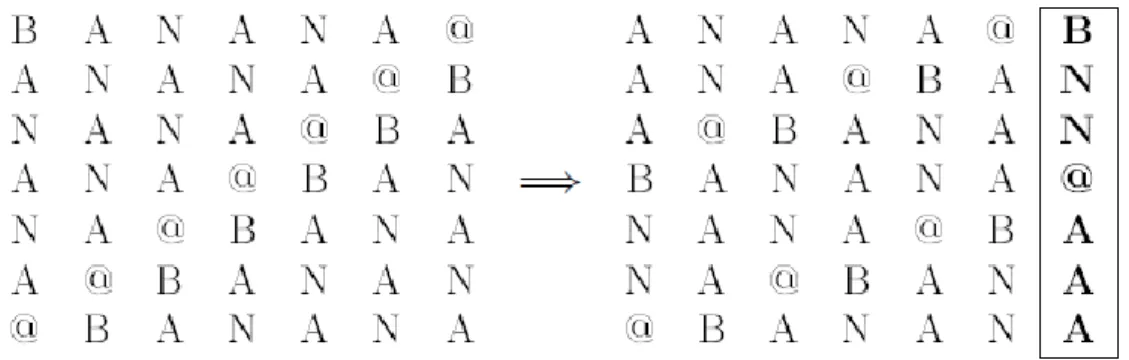
Si prenda in considerazione la stringa campione “BANANA”. Si aggiunga ad essa un carattere finale facendo in modo che quest'ultimo non compaia all'interno del testo, al fine di distinguere l'ultimo carattere, quando si ricostruisce la stringa. Otteniamo una nuova stringa BANANA@, che costituirà la stringa di input.
Si costruisce, ora, una tabella con tutte le possibili rotazioni della stringa, ed infine si riorganizzano in ordine alfabetico le sue righe, come mostrato in figura 16.
Figura 16: Esempio di trasformata di Burrows-Wheeler: Passo (1): elenco di tutte le rotazioni possibili sulla stringa “BANANA@”; Passo (2): riordinamento delle righe in ordine alfabetico.
La stringa in output sarà l'ultima colonna della tabella riordinata alfabeticamente, cioè “BNN@AAA”, stringa che contiene l'ultimo carattere di ognuna delle rotazioni effettuate. Viene descritto ora l'algoritmo inverso.
23
Si consideri una tabella vuota con righe e colonne pari al numero dei caratteri della stringa permutata. Conoscendo soltanto l'informazione della stringa permutata, è possibile ricostruire facilmente la stringa originale. L'ultima colonna mostra, infatti, quali siano i caratteri del file originale. Basterà soltanto riordinarli alfabeticamente, per ottenere quella che nella precedente figura 16 riordinata, costituiva la prima colonna. A questo punto, affiancando l'ultima colonna alla prima, si ottengono le coppie di caratteri successivi del file originale. Tali coppie, vengono ordinate alfabeticamente: in questo modo si ottiene la prima e la seconda colonna della figura 17. Affianchiamo a queste, di nuovo, l'ultima colonna ottenendo le triple di caratteri successivi del file originale. Continuando in questo modo, possiamo ricostruire l'intera stringa. A questo punto, la colonna contenente il carattere che indica la fine del testo come carattere iniziale rappresenta la stringa ordinata.
Figura 17 : Ricostruzione della stringa originale dalla stringa “BNN@AAA” trasformata attraverso il metodo BWT. Ad ogni passo si crea una nuova tabella riordinando le righe della tabella corrente ed aggiungendo ad esse la stringa “BNN@AAA” come
prima colonna.
2.7
Alignment Processing - PICARD
Come è stato descritto poco sopra, il file di tipo .sam, prodotto da BWA, deve essere manipolato in modo tale da poterne estrarre delle informazioni specifiche.
A tal fine, viene utilizzato il software PICARD-tools [11], che consiste in un insieme di linee di
comando scritte in codice Java, adatto per la manipolazione di dati HTS (High-Throughput Sequencing) e formati come SAM/BAM/CRAM e VCF.
Vengono elencati di seguito i tools utilizzati per questa fase di alignment processing: • PICARD - SortSam
• PICARD – MarkDuplicate • PICARD – BuildBamIndex
24
Proprio come suggerisce il nome, il modulo SortSam ordina e indicizza un file di tipo SAM, producendo in output un file di indici in formato .bam ( la versione binaria e compressa del SAM).
A questo punto, l’applicazione “Mark Duplicate Reads”, è stata utilizzata per contrassegnare e rimuovere le reads duplicate (duplicati ottici e artefatti di PCR) presenti nel file BAM.
Eseguendo, infine, l’ultimo tool (Build Bam Index), questo genera un file “.bai” che è formato sostanzialmente da un indice del file BAM. Tale procedimento permette una ricerca più veloce dei dati nel file BAM stesso e svolge, in poche parole, la stessa funzione dell’indice in un database.
2.8
Variant Calling – GATK
La chiamata delle varianti, altrimenti detta variant calling, è la fase dell'analisi bioinformatica che segue quella dell'alignment. Infatti, una volta che le sequenze dei geni sono state ricostruite tramite l'allineamento delle reads ottenute nel sequenziamento, occorre individuare tutti i punti nei quali i geni differiscono dalle sequenze del genoma di riferimento (reference sequences). Queste varianti saranno naturalmente numerosissime e in massima parte irrilevanti, trattandosi, per lo più, di semplici polimorfismi alla base delle differenze interindividuali.
Il variant calling viene eseguito in automatico grazie all'ausilio di un software (nel seguente lavoro di tesi è stato utilizzato GATK) e, una volta terminata l'operazione, il risultato viene generalmente salvato in un file VCF (Variant Call Format).
Uno dei problemi maggiori di un'operazione di variant calling è dato dalla difficoltà nel riuscire a distinguere le varianti vere da quelle irreali, dovute ad artefatti del sequenziamento o ad errori nella fase di alignment. È poi dalla qualità del risultato di tale operazione che dipende la probabilità di identificare, o meno, la mutazione-malattia; oppure, nel caso degli studi di popolazione, la possibilità di determinare in modo affidabile la frequenza allelica dei polimorfismi.
Tuttavia, non è così semplice identificare queste varianti: infatti, volendo scendere più nel dettaglio, ci sono tre fattori che complicano maggiormente tale processo:
1. La presenza di indels (inserzioni/delezioni), che possono essere erroneamente scambiati per varianti di singolo nucleotide (SNV).
2. Errori di sequenziamento dovuti ad artefatti della PCR (Polymerase Chain Reaction), i quali sono meno frequenti laddove si usino sistemi NGS basati sulle paired-end reads. 3. Variabilità della qualità del sequenziamento in corrispondenza delle estremità delle
reads.
Alcuni tra i software di Variant Calling più utilizzati sono ATLAS 2, SOAPsnp, VarScan e GATK.
Il tool utilizzato nel seguente lavoro di tesi, che oltretutto risulta anche essere quello più sfruttato per l’identificazione e l’analisi delle varianti, è fornito dal software GATK (Genome
25
Analysis Toolkit) [12], lo stesso utilizzato nel progetto 1000 Genome Project e in The Cancer
Genome Browser.
Proprio come PICARD e BWA, il software GATK è formato da numerosi tools che hanno come obiettivo primario quello di identificare le varianti. I tools presi in considerazione sono:
• GATK – HaplotypeCaller • GATK - SelectVariants • GATK – VariantFiltration
Il primo di questi, HaplotypeCaller, prende in input un file di tipo .bam restituendo in uscita un file VCF (Variant Call Format), ed è in grado di chiamare SNPs e indels contemporaneamente, tramite l’assemblaggio locale de novo di aplotipi (combinazioni di varianti alleliche) in una regione attiva. In altre parole, ogni volta che il programma incontra una regione che mostra segni di variazione, scarta le informazioni di mappatura esistenti e ricompone completamente le reads in quella regione.
Poiché spesso un file VCF contiene molti campioni e/o varianti, risulterà necessario creare un sottoinsieme di questi/e, in modo tale da facilitare i processi di analisi che seguiranno.
Il tool SelectVariant può essere utilizzato per questo proposito. Questo sottoinsieme di varianti, viene selezionato in base a:
• Criteri specifici che garantiscono l’inclusione (nel sottoinsieme) imponendo delle soglie relative a certi valori di annotazione, ad esempio: “DP > 1000” (profondità del coverage maggiore di 1000x) “AF < 0.25” (siti con frequenza allelica minore di 0.25).
• Tracce di concordanza o discordanza al fine di includere o escludere le varianti che sono presenti anche in altri callsets.
• Criteri come il loro tipo (ad esempio solo indels), lo stato di filtraggio, allelicità e così via…
L’ultimo tool preso in esame, VariantFilatration, è stato progettato per un filtraggio “pesante” delle varianti chiamate, ed è basato su determinati criteri. Questa operazione viene fatta andando a modificare alcuni campi all’interno della stringa che lancia il programma.
Il file restituito è sempre un formato VCF, ma questa volta si tratterà di un VCF “hard-filtered”.
2.8.1
File VCF
Il file VCF [13] è un formato standardizzato generico, utilizzato per la memorizzazione della
maggior parte delle varianti genetiche esistenti, tra cui SNPs, indels e varianti strutturali, associate ad annotazioni libere.
Tale tipo di file si divide principalmente in due parti: • Una sezione di header
26
L’intestazione (header) fornisce dei cosiddetti “meta-dati” i quali descrivono il corpo del file stesso. Ogni linea di meta-dati comincia con il delimitatore ##, mentre la linea di definizione della struttura comincia con il carattere #.
Le meta-informazioni possono essere usate per descrivere il mezzo con cui è stato creato il file, la data di creazione, la versione della sequenza di riferimento, i software usati e tutte le informazioni rilevanti sulla storia del file stesso.
Nel codice di esempio mostrato in figura sottostante, è illustrato un file VCF con varie meta informazioni e quattro diverse varianti. Gli header obbligatori sono il ## fileformat e la linea di definizione dei campi #CHROM; le altre linee sono informazioni sul file e sulla sequenza da cui sono state estrapolate le varianti.
Figura 18: Esempio di un formato valido VCF
Il corpo (body) del VCF segue l'intestazione, ed è sostanzialmente suddiviso in otto colonne. Nella tabella sottostante vengono elencati i nomi delle colonne contenute nel corpo del file con la relativa descrizione.
27
Nome
Descrizione
1 CHROM
Nome della sequenza (tipicamente un cromosoma) nella quale è stata chiamata la variazione. Questa sequenza è di solito conosciuta come 'sequenza di riferimento ', ovvero la sequenza nella quale si possono
vedere le variazioni del nostro campione. 2 POS Posizione della variazione nella sequenza data. 3 ID Identificatore della variazione.
4 REF La base di riferimento (o basi nel caso di un indel) in una data posizione in una sequenza di riferimento. 5 ALT La lista degli alleli alternativi in quella posizione
6 QUAL Rappresenta il "Quality Score" associato all'inferenza di un dato allele. 7 FILTER È un flag che indica quale insieme di filtri la variazione ha superato. 8 INFO Lista di parole chiave (campi) che descrivono la variazione. Campi multipli sono separati sa un “;” con valori opzionali. 9 FORMAT Lista (opzionale) di campi che descrivono il campione. + SAMPLES Per ogni campione descritto nel file, sono forniti i valori per i campi elencati in FORMAT.
Tabella 2: descrizione di tutti i campi del formato VCF.
Una volta ottenuto il file VCF, questo viene caricato su un file di tipo .xlsx (Excel) per permettere una migliore visualizzazione dei dati contenuti.
La figura 19 mostra un esempio di estrazione dei dati raw per il campione LIST1. Come si può notare, tutti i campi presenti rispecchiano la struttura tipica del file VCF.
Figura 19 : esempio di un file VCF trattato con Excel.
La parte di analisi dati che viene presentata nel capitolo 4, verte principalmente sullo studio del campo INFO del file VCF. Tale campo contiene al suo interno fino a 14 parametri utilizzati da
28
GATK per la chiamata delle varianti. Nonostante non siano visibili tutti (per ragioni di spazio), i parametri contenuti vengono indicati come segue:
AC, AF, AN, DP, FS, MLEAC, MLEAF, MQ, QD, SOR, BaseQRankSum, ClippingRankSum, ReadPosRankSum, MQRankSum.
La maggior parte di questi, i primi 10 solitamente, è comune a tutti gli SNPs, mentre gli ultimi 4 elencati, non possono essere calcolati per i siti omozigoti; cioè tali parametri compaiono soltanto nei siti eterozigoti, ovvero dove esistono diversi alleli alternativi allineati sul riferimento.
Al fine di favorire una migliore comprensione di ciò che si andrà ad analizzare, di seguito vengono elencati i parametri [14] e una loro breve descrizione:
• Allele Count (AC) – è il numero di alleli chiamati in un campione.
• Allele Frequency (AF) - è la misura della frequenza relativa di un allele in un locus genico nella popolazione.
• Allele Number (AN) – è il numero totale di alleli nel genotipo chiamato.
• Depth of Coverage (DP) - numero di volte che un nucleotide è stato "letto" durante il sequenziamento. È una misura di ridondanza. All'aumentare del coverage aumenta il grado di confidenza nel risultato del sequenziamento.
• Fisher Strand (FS)- L’annotazione “Fisher Strand” si riferisce a uno dei numerosi metodi che permettono di valutare se c’è un bias di strand nei dati. Questo metodo utilizza il Test esatto di Fisher per determinare se c’è un bias di strand nel filamento positivo o negativo del DNA di riferimento.
• Maximum Likelihood Expectation for the Allele Counts (MLEAC) per ogni allele alternativo, nello stesso ordine di come è elencato.
• Maximum Likelihood Expectation for the Allele Frequency (MLEAF), per ogni allele alternativo, nello stesso ordine di come è elencato.
• RMSMapping Quality (MQ) – Questo parametro fornisce una stima della qualità della mappatura delle reads che supportano una chiamata variante.
Vengono prodotti sia i dati grezzi (somma di quadrati e numero delle reads totali) che il valore quadratico medio (RMS). I dati grezzi vengono utilizzati per calcolare con precisione l’errore quadratico medio quando si prende in considerazione più di un campione.
• Quality by Depth (QD) - QD è il punteggio di qualità (QUAL) normalizzato per l’allele depth (AD) per una data variante. Per un singolo campione, HaplotypeCaller calcola il QD utilizzando il rapporto QUALAD .
• Strand Odds Ratio (SOR): Tale parametro rappresenta uno dei numerosi metodi che permettono di valutare se esiste un bias di strand nei dati.
Il bias di strand è un tipo di errore nel sequenziamento in cui un filamento di DNA è favorito rispetto a un altro, e può provocare una non corretta valutazione della quantità di prove osservate per un allele rispetto all’altro.
29
Questo parametro, quindi, è una forma aggiornata del Fisher Strand Test, e tiene conto della grande quantità di dati nelle situazioni di alto coverage.
• BaseQRankSum: La colonna BaseQRankSum contiene una valutazione dei punteggi di qualità nelle reads che hanno una chiamata variante, la quale viene paragonata con il punteggio di qualità dell’allele di riferimento. Le varianti per le quali non è stato chiamato un corrispondente allele di riferimento, non possiedono un valore di BaseQRankSum. Allo stesso modo, non viene calcolato alcun valore per gli alleli di riferimento.
• ClippingRankSum - Questo parametro testa se i dati che supportano l’allele di riferimento mostrano più o meno un base clipping rispetto a quelli che supportano l'allele alternativo. Il risultato ideale è un valore prossimo allo zero, il che indica che c’è poca o nessuna differenza. Un valore negativo indica che le reads che supportano l’allele alternativo possiedono più basi hard-clippled, rispetto a quelle che supportano l’allele di riferimento. Al contrario, un valore positivo indica che le reads che supportano l’allele alternativo hanno un minor numero di basi hard-clipped rispetto a quelle che supportano l’allele di riferimento. Trovare una differenza statisticamente significativa in entrambi i casi, suggerisce che il processo di sequenziamento e/o la mappatura, possono essere stati affetti da un errore o da un artefatto.
• ReadPosRankSum – Esegue un test per vedere se c’è una evidenza di bias nella posizione degli alleli (all’interno delle reads che li supportano), tra gli alleli di riferimento e quelli alternativi.
Vedere un allele solo vicino alle estremità delle reads è indicativo di un errore, perché è proprio lì che i sequenziatori tendono a fare il maggior numero di errori. Tuttavia alcune varianti, situate in prossimità dei bordi della regione sequenziata, saranno necessariamente coperte dagli estremi della reads, in modo tale che non si possa semplicemente fissare una soglia assoluta di “distanza minima dalla fine delle reads”. Questo è il motivo per cui viene usato un rank sum test, cioè per valutare se c’è una differenza nel modo in cui l’allele di riferimento e l’allele alternativo sono supportati. Il risultato ideale è un valore prossimo allo zero, il che indica che c’è poca o nessuna differenza nella posizione in cui si trovano gli alleli rispetto alle estremità delle reads. Un valore negativo indica che l'allele alternativo si trova alle estremità delle reads più frequentemente rispetto all'allele di riferimento. Viceversa, un valore positivo indica che l'allele di riferimento si trova alle estremità delle reads più spesso rispetto all'allele alternativo.
• MQRankSum – Questa annotazione confronta la qualità di mappatura delle reads che supportano l’allele di riferimento con quelle che supportano l’allele alternativo. Anche in questo caso, il risultato ideale è un valore prossimo allo zero, il che indica che c’è poca o nessuna differenza. Un valore negativo indica che le reads che supportano l’allele alternativo hanno un quality score di mappatura più basso rispetto a quelli che supportano l’allele di riferimento. Al contrario, un valore positivo indica che le reads che supportano
30
l’allele alternativo hanno un quality score di mapping più alto rispetto a quelle che supportano l’allele di riferimento.
31
Capitolo 3
Nel seguente capitolo vengono descritti e spiegati nel dettaglio quali sono i test statistici utilizzati per analizzare i dati ottenuti. In particolare, ciò che si andrà a fare, sarà analizzare dal punto di vista statistico, alcuni parametri degli SNPs trovati (ad esempio il coverage), i cui risultati saranno esposti nel capitolo 4.
3.1
Analisi della Varianza (ANOVA)
L’analisi della varianza è un metodo sviluppato da Fisher, che è fondamentale per l’interpretazione statistica di molti dati biologici ed è alla base di molti disegni sperimentali. L’analisi della varianza [15] (in inglese: Analysis of Variance, abbreviata con l’acronimo ANOVA) è utilizzata per testare le differenze tra medie campionarie e per fare questo si prendono in considerazione le rispettive varianze.
Il principio alla base di questo test è quello di stabilire se due o più medie campionarie possono derivare da popolazioni che hanno la stessa media parametrica. Quando le medie sono solamente due è indifferente usare questo test o il t-test, mentre si deve necessariamente utilizzare l’ANOVA quando le medie sono più di due, o quando si vuole suddividere la variabile di raggruppamento in più variabili, per eliminare eventuali fonti di variazione oltre a quella prodotta dal fattore di cui si vuole valutarne l’effetto.
Grazie allo “Statistics and Machine Learning Toolbox™” fornito da MatLab, è possibile eseguire l’analisi della varianza (ANOVA) di tipo 1, di tipo 2 e di tipo N; l’analisi multivariata della varianza (MANOVA); e l’analisi di covarianza (ANCOVA). Tuttavia nel seguente lavoro di tesi si è scelto di prendere in considerazione solo l’ANOVA di tipo 1.
3.1.1 One-way ANOVA
Tramite il comando anova1di MatLab, è possibile lanciare la funzione che permette di eseguire
l’analisi di varianza di tipo 1. Lo scopo di tale analisi è quello di determinare se i dati provenienti da diversi gruppi hanno una media comune. Cioè, l’ANOVA di tipo 1, permette di scoprire se i diversi gruppi di una variabile indipendente, hanno effetti diversi sulla variabile di risposta y. Questo tipo di analisi (ANOVA-1) è un caso speciale del modello lineare, ed è esprimibile mediante la seguente equazione:
32
dove 𝑦𝑖𝑗 è un’osservazione, nel quale i rappresenta il numero di osservazioni e j un diverso
gruppo, 𝛼𝑗 è la media della popolazione, 𝜀𝑖𝑗 è il “residuo” o errore sperimentale con media zero
e varianza costante.
L’analisi della varianza esegue un test per verificare le differenze delle medie tra i gruppi, scomponendo la varianza totale in due componenti:
• Varianza interna ai gruppi (anche detta Varianza Within) definita come: 𝑦𝑖𝑗 − 𝑦̅
• Varianza tra i gruppi (Varianza Between) uguale a: 𝑦̅ − 𝑦̅; dove 𝑦𝑗 ̅ è il campione medio 𝑗
del gruppo j e 𝑦̅ è il campione medio totale.
In altre parole, ANOVA separa la somma totale dei quadrati (SST) in una somma di quadrati dovuta all’effetto tra gruppi (SSR) e una somma di errori quadratici (SSE), per cui:
∑ ∑(𝑦𝑖𝑗 − 𝑦̅)2 𝑗 𝑖 ⏟ 𝑆𝑆𝑇 = ∑ 𝑛𝑗 𝑗 (𝑦̅ − 𝑦̅)𝑗 2 ⏟ 𝑆𝑆𝑅 + ∑ ∑(𝑦𝑖𝑗 − 𝑦̅ )𝑗 2 𝑗 𝑖 ⏟ 𝑆𝑆𝐸
Dove 𝑛𝑗 è la grandezza del campione per il j-esimo gruppo.
A questo punto l’ANOVA confronta la varianza tra gruppi con la varianza interna ai gruppi. Se il rapporto della varianza interna ai gruppi con la varianza tra gruppi risulta essere significativamente alto, allora si può concludere che le medie dei gruppi sono significativamente differenti tra loro. Questo rapporto si può misurare usando un test statistico che ha una distribuzione F con (k-1, N-1) gradi di libertà.
𝐹 = 𝑆𝑆𝑅 𝑘 − 1 ⁄ 𝑆𝑆𝐸 𝑁 − 𝑘 ⁄ = 𝑀𝑆𝑅 𝑀𝑆𝐸 ~ 𝐹𝑘−1,𝑁−𝑘
Dove MSR e MSE sono quadrati medi della regressione e dell’errore, k è il numero di gruppi e
N è il numero totale di osservazioni. Se il p-value per la statistica F è minore del livello di
significatività, il test rifiuta l'ipotesi nulla che le medie di tutti i gruppi sono uguali e conclude che almeno una media di un gruppo è diversa dalle altre. I livelli di significatività più comuni sono 0.05 e 0.01.
Affinché l’ANOVA possa essere eseguita è necessario che:
• Gli elementi che costituiscono i vari gruppi non siano oggetto di una particolare selezione, ma siano stati assegnati a caso (random).
• I campioni devono essere tra loro indipendenti, ovvero i dati osservati in un campione non devono essere influenzati da quelli osservati in un altro campione.
33
Quindi, prima di eseguire tale test, occorre verificare che le varianze dei vari gruppi siano omogenee.
3.2 Test di Bartlett
Prima di eseguire l’analisi della varianza, occorre verificare che le varianze dei vari gruppi siano omogenee (in statistica si parla di: omoschedasticità delle varianze).
Tra i vari test di omogeneità, è possibile utilizzare il cosiddetto Test di Bartlett [15], nel quale i dati
presi in considerazioni sono non bilanciati, il che significa che i gruppi scelti hanno numerosità diverse.
Tale tipo di test, in particolare, viene utilizzato per verificare se più campioni di dati provengono da una distribuzione normale e possiedono la stessa varianza (ipotesi nulla), andando in contrasto con l’ipotesi alternativa che, almeno due dei campioni di dati, non hanno varianza uguale.
La statistica del test è esprimibile mediante la seguente formula: 𝑇 = (𝑁 − 𝑘) ln 𝑠𝑝 2− ∑ (𝑁 𝑖 𝑘 𝑖=1 − 1) ∗ 𝑙𝑛 𝑠𝑖2 1 + (3(𝑘 + 1)) [(1 ∑ (𝑁 1 𝑖 − 1)) − 1 𝑁 − 𝑘 𝑘 𝑖=1 ] dove:
• 𝑠𝑖2 è la varianza dell’ i-esimo gruppo
• N è il numero totale dei campioni
• 𝑁𝑖 è la grandezza del campione dell’i-esimo gruppo • 𝑘 è il numero di gruppi
• 𝑠𝑝2 è la varianza combinata
La varianza combinata viene definita come segue: 𝑠𝑝2 =
∑𝑘 (𝑁𝑖
𝑖=1 − 1)𝑠𝑖2
(𝑁 − 𝑘)
La statistica segue la distribuzione di 𝜒2 con p-1 gradi di libertà.
Il test di Bartlett è potente, ma seriamente sensibile alla non normalità della distribuzione e i risultati del test vanno considerati con una certa cautela.
D’altro canto non ci sono procedure particolarmente adatte nel caso di distribuzioni che si discostano sensibilmente dalla normalità.