1
Ai miei genitori e a mio fratello
sempre vicini in ogni traguardo della mia vita.
2
«I bambini che sono in grado di sentire imparano a
parlare senza particolare sforzo. Catturano al volo le
parole che cadono dalle labbra altrui, così come sono,
felicemente, mentre i piccoli bambini sordi devono
intrappolarle attraverso un processo lento e spesso
doloroso. »
(Helen Keller, La storia della mia vita, 1903)
«La scienza non è affatto noiosa come molti credono . Io
non saprei immaginare la mia vita senza: è eccitante la
scoperta e il lavoro per arrivarci. A un giovane oggi direi
"se non hai paura, studia la chimica e ti divertirai un
mondo"»
3
Indice
Premessa ... 5
Capitolo 1 Anatomia e fisiologia dell’udito ... 6
1.1 L’Orecchio esterno ... 6
1.2 L’Orecchio medio ... 7
1.3 L’Orecchio interno e Sistema vestibolare ... 9
1.4 I recettori dell’udito ... 11
1.5 Come avviene la trasmissione di un’onda sonora ? ... 14
Capitolo 2 Che cos’è la sordità ... 20
2.1 Definizione ... 20
2.2 Tipi di ipoacusie ... 20
2.3 Origine e causa della sordità ... 22
2.4 Ototossicità da farmaci ... 23
Capitolo 3 Sordità genetiche o ereditarie ... 37
3.1 La nascita della genetica ... 37
3.2 Cenni di genetica ... 39
Capitolo 4 Metodo di studio della genetica medica ... 44
4.1 Alberi Genealogici ... 44
4.2 Forme sindromiche e Forme non-sindromiche ... 47
Capitolo 5 Mutazioni ricorrenti dei geni nelle forme non sindromiche ... 50
5.1 Geni che causano sordità non sindromica autosomica recessiva ... 50
5.2 Geni che causano sordità non sindromica autosomica dominante ... 56
4
Capitolo 6 Inquadramento diagnostico delle
ipoacusie genetiche ... 63
Capitolo 7 Apparecchiature usate per l’indagine molecolare della sordità ereditaria ... 71
7.1 Polymerase chian reaction (PCR)... 71
7.2 Sequenziamento del DNA ... 74
Conclusioni ... 80
Bibliografia ... 81
Ringraziamenti ... 85
5
Premessa
La scelta da parte mia di questa tesi è data, in primis, sicuramente dal fatto che
sono sorda, come anche mio fratello. La causa sembrerebbe ereditaria.
Tante sono state le ricerche fatte dalla mia famiglia, la sicurezza però non c'è o
perlomeno non è ancora chiaro, con certezza quale fattore l'abbia determinata.
Ricercando il materiale per la tesi e affrontando il problema mi sono resa conto
di quanto complicato e difficile sia dare delle risposte nette!
La scelta di questa tesi deriva anche dal fatto che i docenti, incontrati nel mio
percorso universitario, hanno fatto crescere in me, il desiderio di approfondire
6
1.
ANATOMIA E FISIOLOGIA DELL’UDITO
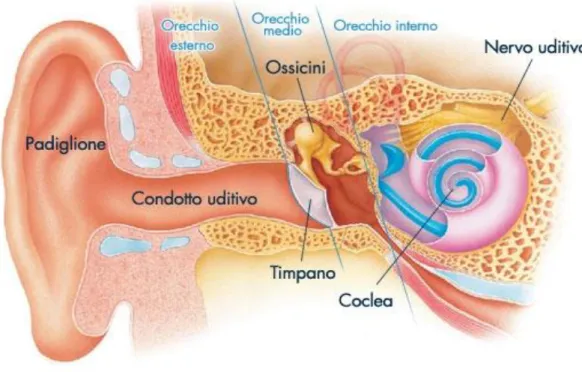
Il nostro organo uditivo è costituito da tre parti essenziali: Orecchio esterno
Orecchio medio Orecchio interno
Figura 1 Organizzazione generale dell’orecchio esterno, medio e interno. Fonte: audika.it
1.1 L’orecchio esterno
L’ orecchio esterno comprende il padiglione auricolare di cartilagine elastica, che circonda il condotto uditivo esterno proteggendolo e fornendo sensibilità direzionale all' orecchio, impedendo o favorendo il passaggio del suono attraverso la membrana timpanica o timpano. Quest’ultima è una lamina sottile, estremamente delicata di tessuto connettivo che segna il confine tra orecchio
7
esterno ed orecchio medio (Martini et al., 2010). Il condotto contiene, al suo interno, ghiandole ceruminose che secernono un materiale ceroso e al suo esterno, numerosi piccoli peli che si proiettano nel canale impedendo l’accesso a particelle estranee o insetti. Inoltre il cerume deposto sulla parete del condotto rallenta la crescita dei microorganismi e riduce in tal modo il rischio di contrarre infezioni (Fig. 1).
1.2 L’orecchio medio
L’orecchio medio è descritto da uno spazio pieno d’aria, definito cavità timpanica, che contiene gli ossicini dell’udito (Fig. 1). La cavità timpanica è separata dal canale uditivo esterno tramite la membrana del timpano, ma comunica con il rinofaringe attraverso la tuba uditiva. La tuba uditiva, chiamata anche tuba di Eustachio o tuba faringotimpanica, equilibra la pressione presente nella cavità dell’orecchio medio con la pressione atmosferica esterna ed ambedue le pressioni devono risultare uguali sui due lati della membrana timpanica.
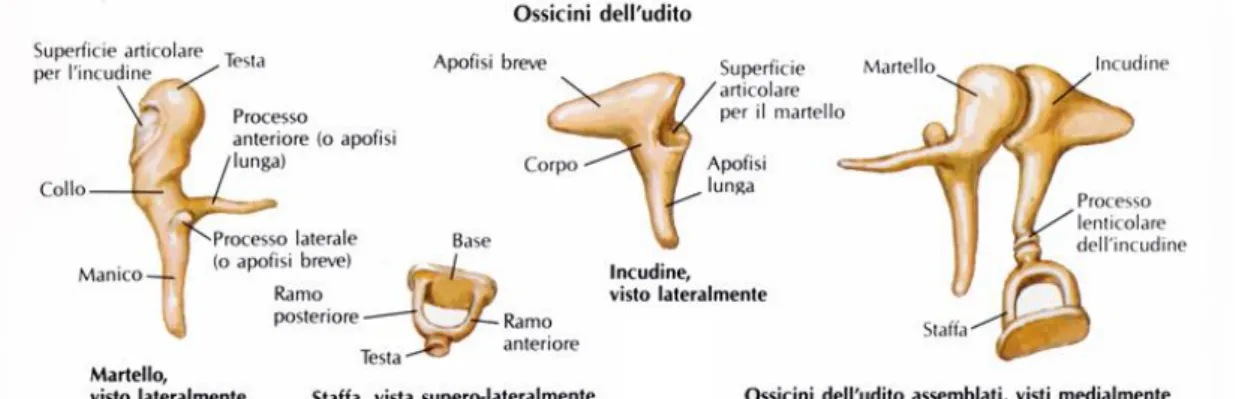
L’orecchio medio contiene la catena degli ossicini (rappresentano i più piccoli segmenti ossei del corpo umano), che connettono la membrana timpanica con il complesso recettoriale dell’orecchio interno (Fig. 2).
Figura 2 Gli Ossicini dell’udito: separati e assemblati. Fonte: medicinapertutti.altervista.org
8
La catena degli ossicini è costituita dal martello la cui superficie laterale è connessa in tre punti alla superficie interna della membrana del timpano; dall’incudine che è l’ossicino intermedio e connette la faccia mediale del martello alla staffa, la cui base occlude completamente la finestra ovale (un’apertura nella parete ossea della cavità dell’orecchio medio) (Fig. 1 e 2). Tra la base della staffa e il margine osseo della finestra ovale è teso il legamento
anulare.
La vibrazione del timpano converte le onde sonore in arrivo in movimenti meccanici. Gli ossicini dell’udito conducono poi questi movimenti alla staffa, che invia le vibrazioni ai liquidi dell’orecchio interno. Grazie alle connessioni tra i tre ossicini, il movimento del timpano diretto verso l’interno determina un moto oscillante a livello della staffa. L’entità del movimento aumenta proporzionalmente dalla membrana del timpano alla finestra ovale: la membrana risulta più ampia della finestra ovale. Questo processo di amplificazione produce una deviazione relativamente potente della staffa a livello della finestra ovale e permette di percepire suoni anche molto deboli, ma può diventare un problema in caso di rumori particolarmente forti. Per ovviare a questo problema, all’interno della cavità timpanica, ci sono due piccoli muscoli che hanno il compito di proteggere il timpano e gli ossicini da movimenti particolarmente violenti in seguito a situazioni molto rumorose:
Il muscolo tensore del timpano (Fig. 3) è accolto in un canale osseo situato al di sopra della tuba uditiva dalla cui apertura esce il tendine del muscolo; il tendine attraversa il cavo del timpano fino a prendere inserzione sulla radice del manico del martello. Contraendosi, esso porta medialmente il martello, irrigidendo il timpano e riducendo cosi l’escursione del movimento. Il muscolo tensore del timpano è innervato da fibre motrici della branca mandibolare del nervo trigemino.
Il muscolo stapedio (Fig. 3) è contenuto nell'eminenza piramidale della parete mediale del cavo del timpano. Il tendine esce dalla parete e si termina inserendosi sulla testa della staffa. E' innervato dal nervo faciale. Contraendosi,
9
tira indietro la testa della staffa, determinando così un leggero spostamento della base di questo ossicino verso il cavo del timpano (Martini et al., 2010).
Quando il sistema uditivo è stimolato da un suono troppo intenso, si attua un riflesso protettivo, chiamato riflesso di attenuazione, che consiste nella contrazione dei muscoli stapedio e (in misura minore) tensore del timpano. Entrambi i muscoli agiscono in reciproca opposizione: il loro accorciamento conferisce alla catena degli ossicini una maggiore rigidità, e ciò comporta una consistente riduzione della trasmissione dei suoni a bassa frequenza. La funzione principale di questo riflesso è la protezione delle membrane e dei recettori dell’orecchio interno da stimolazioni eccessive che potrebbero danneggiarli. Questo meccanismo consente inoltre di filtrare i suoni a bassa frequenza in ambiente rumoroso, migliorando cosi la percezione dei suoni alle frequenze di comunicazione verbale. Infine è stato visto che il meccanismo si attiva mentre si parla, per attenuare la percezione della propria voce; in questo caso l’attivazione dipende da segnali nervosi trasmessi dai centri che controllano la vocalizzazione.
1.3 L’Orecchio interno e Sistema vestibolare
L’orecchio interno è costituito dalla coclea, che contiene i recettori del sistema uditivo, e dall’ utricolo, dal sacculo e dai canali semicircolari, che contengono i recettori del sistema vestibolare. Tutte insieme coesistono di una parte ossea esterna e di una parte membranosa interna.
Orecchio interno
L’orecchio interno è costituito dalla coclea, dall’utricolo, dal sacculo e dai canali semicircolari che contengono i recettori del sistema vestibolare.
10
Figura 3 Orecchio interno. Fonte: Carbone et al., 2008
La coclea ha una struttura ossea a spirale avvolta attorno ad una struttura conica ossea definita modiolo. Nell’uomo il numero dei giri intorno al modiolo è di due giri e tre quarti, circa trentacinque mm di lunghezza. Elicotrema è il punto apicale della spirale dove sono in comunicazione: la scala vestibolare e la scala timpanica.
L’interno della coclea è suddiviso da due membrane, la membrana di Reissener o vestibolare e la membrana basilare che suddividono la coclea in tre compartimenti contenenti i fluidi (Fig. 4):
11
1. Il dotto o scala vestibolare contiene la perilinfa e si trova a diretto contatto con la staffa attraverso la finestra ovale. È la prima componente che riceve l’onda pressoria. Alla sua estremità troviamo la membrana di Reissener che separa il dotto vestibolare dal dotto cocleare .
2. Il dotto cocleare o scala media contiene l’endolinfa che presenta una concentrazione di potassio relativamente alta e una concentrazione relativamente bassa di sodio, esattamente l’opposto dei fluidi extracellulari. Questa componente ionica serve a far funzionare i recettori. Il dotto cocleare è separato dal dotto timpanico dalla membrana basilare, sulla quale si trova l’organo del Corti, che contiene le cellule recettoriali del sistema uditivo.
3. Il dotto timpanico o scala timpanica contiene la perilinfa ed è separato dall’orecchio medio da una membrana che ricopre la finestra rotonda, e comunica con il dotto vestibolare a livello dell’elicotrema (Carbone et al.,2008).
1.4 I recettori dell’udito
Le cellule ciliate recettoriali, che trasformano i suoni in impulsi nervosi, vengono anche chiamate cellule capellute, poiché presentano, al palo apicale, lunghe sterocilia e sono disposte su una fila più interna, vicina al modiolo e su tre file esterne. Sono circondate da cellule di sostegno e controllate da fibre afferenti. Le sterocilia delle cellule ciliate attraversano delle fenditure della lamina reticolare, e quelle più lunghe sono inserite in una sottile membrana gelatinosa, la membrana tectoria, che copre parzialmente l’organo dei Corti (Fig. 5).
12
Figura 5 Organo del Corti: membrana tectoria ribaltata (color verde), le cellule ciliate
IHC e OHC (color azzurro) e membrana basilare. Fonte :cell.com/cell_picture_show-hearing
L’organo del Corti accoglie circa 16,000 cellule ciliate ordinate in una fila di cellule interne (IHC) e tre file di cellule ciliate esterne (OHC). Le OHC fungono da amplificatori cocleari per conto della membrana basilare dato che la resistenza viscosa all’interno della coclea dissiperebbe l’energia del suono e con questo amplificano il movimento della membrana basilare. Invece le IHC rilevano queste vibrazioni e attivano i neuroni afferenti.
La membrana tectoriale si adagia sulla superficie apicale dell’organo del Corti ed è connessa al fascetto di stereociglia delle cellule ciliate esterne. Ogni cellula ciliata forma dei legami forti con le cellule di sostegno, le quali a loro volta aderiscono a turno a livello della loro superficie basale ad un'altra matrice extracellulare, la membrana basilare.
Ogni cellula ciliata ospita sulla sua superficie apicale, i fascetto di ciglia, dove sono raggruppate una dozzina di stereociglia. Quest’ultime sono disposte in fila
13
di altezza decrescente, dove le stereociglia più lunghe sono affiancate al chinociglio (Fig. 6).
Figura 6 La freccia indica il chinociglio presente nel Sacculo di rana. Fonte: unipv.it/
Il chinociglio è un ciglio singolo assonemale vale a dire che la sua struttura interna è formata da un complesso molto organizzato di microtubuli che causano e coordinano il movimento di tale microvillo. E’ interessante conoscere la genesi dei fascetti di ciglia e l’importanza che il chinociglio assume per una buona funzionalità di tali fascetti ciliari: la superficie apicale di ciascuna cellula è ricoperta da microvilli che si allungano formando stereociglia di ugual misura; in questa fase un singolo chinociglio è situato al centro della superficie apicale della cellula. In seguito il chinociglio si sposta alla periferia della cellula e le stereociglia vicine al chinociglio iniziano ad allungarsi. Dopodiché interrompono la crescita, ma si sviluppano in larghezza aggiungendo filamenti di actina. I filamenti si allungano alla base per formare piccole radici; le estremità basali delle stereociglia acquistano una forma sottile. Infine le stereociglia riprendono l’allungamento per crescere fino alla lunghezza finale. Dopo la maturazione del fascetto di ciglia, in alcune cellule il chinociglio scompare. Secondo degli studi morfologici, il chinociglio viene ritenuto fondamentale perché un suo deficit può generare un’orientazione sbagliata dei fascetti di ciglia. Per questa ragione il chinociglio è importante per lo sviluppo della polarità e il giusto orientamento del fascetto di ciglia. I filamenti
14
extracellulari collegano le stereociglia e il chinociglio in un unico fascetto e contribuiscono al raggruppamento dei meccanismi passivi. Mentre ponti proteici o tip links si localizzano tra le stereociglia e fungono da molle che trasmettono l’energia meccanica di flessione delle stereociglia ai canali di meccano trasduzione (MET), la loro distruzione o la loro mancanza elimina la
meccanosensitivita’ delle cellule ciliate. Nelle stereociglia sono presenti filamenti β e γ actina che formano legami
crociati con le proteine globulari: esparina, fimbrina 1 e fimbrina T. Queste proteine servono a far deflettere le stereociglia durante la depolarizzazione e l’actina gioca un ruolo essenziale nel mantenere la rigidità delle ciglia. Ciò avviene sia nelle cellule IHC e nelle OHC. Nelle stereociglia alcuni dei filamenti di actina formano piccole radici che radicano le stereociglia in una rete di actina specializzata, il piano cuticolare (Cuticolar plate). In questo punto la tropomiosina e la spectrina , radunate intorno alle piccole radici, hanno capacità elastiche e facilitano la giunzione tra la membrana laterale delle HC e le cellule di supporto.
1.5 Come avviene la trasmissione di un’onda sonora?
L’onda sonora arriva alla membrana del timpano (Fig. 7), ed il suo movimento provoca lo spostamento degli ossicini uditivi. Il movimento della staffa a livello della finestra ovale determina un’onda di pressione sulla perilinfa del dotto vestibolare. L’onda pressoria provoca la distorsione della membrana basilare rivolta verso la finestra rotonda del dotto timpanico. La vibrazione della membrana basilare determina la vibrazione delle cellule capellute contro la membrana tectoria, ovvero la stimolazione delle cellule capellute e il rilascio dei neurotrasmettitori. Le informazioni riguardanti la regione stimolata e l’intensità di stimolazione vengono veicolate dalla branca acustica (cocleare) del VII paio di nervi encefalici (nervo stato acustico).
15
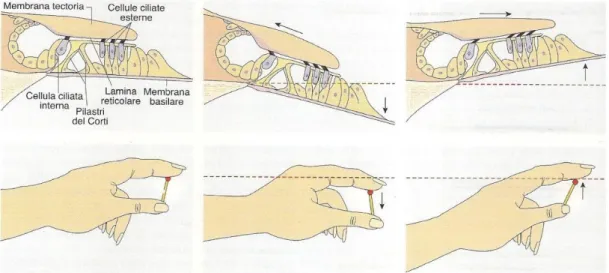
Movimento delle stereociglia L’onda pressoria nell’endolinfa fa flettere sia la membrana basilare che la membrana tectoria; poiché queste membrane sono connesse in due punti
Figura 8 Movimento sterocilia. Fonte: Carbone et al., 2008
diversi del modiolo, lo spostamento verso il basso (fase di compressione dell’onda sonora) determina uno spostamento tra le due membrane: la membrana basilare si sposta verso destra rispetto alla membrana tectoria, e di
16
conseguenza le stereociglia si flettono verso sinistra (Fig. 8); quando le membrane si spostano verso l’alto (fase di ridotta intensità dell’onda sonora), le cellule ciliate si spostano verso sinistra e quindi le stereociglia si flettono verso destra (Fig. 8).
Struttura meccano-elettrica delle cellule ciliate
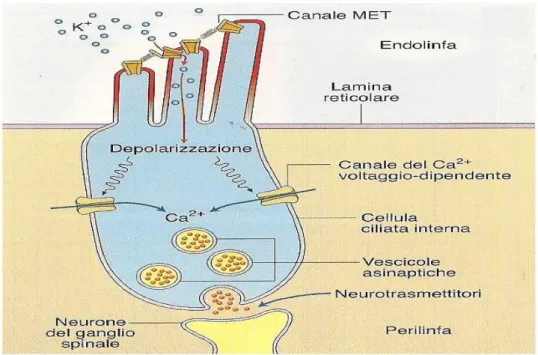
Le cellule recettoriali ciliate sono meccanorecettori, cioè recettori sensibili agli stimoli meccanici che sono inserite sulla membrana basale. Nella parte superiore della cellula si trova il fascetto di ciglia che è costituito da sterocilia ricche in actina che contengono canali di meccano trasduzione in corrispondenza dei loro apici. Le stereociglia, immerse nell’endolinfa, sono organizzate in fila di altezza decrescente e ogni punto apicale della sterocilia presenta un canale cationico TRPA1, generalmente definito trasduttore meccanico (MEchano Trasducer, MET), appartenente alla famiglia di canali TRP (Transient Receptor Potential). I canali sono collegati da ponti proteici che esercitano una tensione più o meno forte a seconda degli stimoli sonori: aumentando la tensione, tengono aperti i canali e viceversa, diminuendo la tensione, questi si chiudono. Questo processo avviene perché le stereociglia sono immerse nell’endolinfa dove è presente un’alta concentrazione di K+ e una bassa concentrazione di Na+ per effetto di meccanismi di trasporto attivo delle cellule della stria vascolare; presenta, quindi, un potenziale endococleare di circa +80/+90 mV. Di conseguenza il potenziale di riposo delle cellule ciliate, che è di circa 45/70 mV rispetto agli altri liquidi extracellulari, è di circa -125/-160 mV rispetto all’endolinfa. Questo valore elevato consente di ottenere risposte recettoriali molto rapide ed efficienti. Quindi avendo alte concentrazioni di K+ nell’endolinfa e in assenza di stimoli sonori si attua una tensione dei ponti con moderato flusso di ioni K+ verso l’interno delle stereociglia. Questo flusso di ioni K+ che entra causa una lieve depolarizzazione che determina l’apertura di un numero limitato dei canali al calcio
voltaggio-17
dipendenti che si trovano nella membrana del corpo cellulare con conseguente ingresso nella cellula di un limitato numero di ioni Ca2+ che provocano un rilascio moderato di neurotrasmettitori (glutammato) nel vallo sinaptico. I neurotrasmettitori, quindi, generano una scarica tonica di potenziali d’azione nella fibra sensitiva (Fig. 9). Quando si ha lo spostamento verso il basso della membrana basilare si ha la flessione delle stereociglia verso sinistra (in direzione opposta rispetto allo stereociglio più lungo) che fa diminuire la tensione dei ponti proteici e di conseguenza diminuisce il numero dei canali MET aperti ed il flusso di ioni K+, ciò causa una iperpolarizzazione della membrana, chiusura dei canali del Ca2+, diminuzione del rilascio di neurotrasmettitore e diminuzione della frequenza di scarica nella fibra sensitiva (Fig. 10.A). Al contrario, lo spostamento verso l’alto della membrana basilare causa la flessione delle stereociglia verso destra, cioè verso lo stereociglio più lungo, induce a un aumento della tensione dei ponti proteici di collegamento, e di conseguenza aumenta il numero dei canali MET aperti e il flusso di k+ entro la cellula con conseguente apertura dei canali del Ca2+ che a loro volta attivano le vescicole a rilasciare il neurotrasmettitore nel vallo sinaptico ed aumentare così la frequenza di scarica nella fibra sensitiva (Fig. 10.B).
18
Figura 9 Cellula ciliata in stato di riposo.
Figura 10 Cellula ciliata iperpolarizzata (A) e Cellula ciliata depolarizzata (B) Fonte: Carbone et al., 2008
La membrana basilare è costituita da fibre di dimensioni diverse: sono più corte e di maggiore spessore nella parte iniziale (in corrispondenza della finestra ovale), ed aumentando progressivamente di lunghezza e calano di spessore procedendo verso l’apice della coclea. Quindi la membrana è più rigida nella parte iniziale e diventa progressivamente più elastica, quindi varia anche la frequenza di risonanza, da circa 20.000 Hz nella parte iniziale fino a circa 20 Hz nella parte apicale della coclea. Quando l’onda raggiunge la porzione di
19
membrana caratterizzata da una frequenza di risonanza pari alla frequenza dell’onda, quella porzione di membrana entra in risonanza, cioè si genera una vibrazione molto più ampia, che disperde completamente l’energia vibratoria e quindi l’onda si arresta; vengono perciò attivate maggiormente le cellule ciliate che si trovano in quella posizione della membrana basilare. I suoni a frequenza più elevata fanno vibrare la parte iniziale della membrana basilare, quelli a frequenza intermedia la parte centrale e quelli a frequenza più bassa la parte apicale. Quindi i suoni che sentiamo vengono normalmente scomposti dalla membrana basilare nei singoli suoni puri che li compongono; ciascun suono puro fa entrare in risonanza un punto specifico della membrana basilare, e di conseguenza attiva maggiormente le cellule ciliate che si trovano in quel punto ed a livello recettoriale avremo l’attivazione simultanea di recettori specifici per i singoli suoni puri che lo compongono. Il sistema uditivo, inoltre, quantifica il livello sonoro dei singoli suoni puri: un suono puro di bassa intensità provoca una piccola fluttuazione della parte di membrana basilare che lo codifica, che fa deflettere di poco le sterocilia che si trovano in quel punto; di conseguenza si attivano solo alcune cellule ciliate ed aumenta di poco la frequenza di scarica nelle fibre sensitive. Se lo stesso suono puro aumenta di intensità, l’oscillazione della porzione di membrana risonante diventa più ampia, quindi si genera una flessione maggiore delle sterocilia; di conseguenza si attivano maggiormente le cellule ciliate che erano già state attivate quando l’intensità era minore e aumenta maggiormente la frequenza di scarica nelle fibre sensitive ad esse collegate e inoltre vengono attivate altre cellule ciliate che fanno aumentare la frequenza di scarica in altre fibre sensitive, determinando una sommazione degli impulsi lungo le vie afferenti (Carbone et al.,2008).
20
2.
CHE COS’È LA SORDITÀ
2.1 Definizione
“ sordità s. f. [dal lat. surdĭtas -atis, der. di surdus «sordo»]. – Diminuzione della funzione uditiva, unilaterale o bilaterale, congenita o acquisita, distinta in base all’entità della menomazione in lieve, media, medio-grave, grave e gravissima; a seconda della sede della lesione (malformativa, infiammatoria, tossica, traumatica, neoplastica, ecc.) che ne è causa, in s. dell’orecchio esterno, dell’orecchio medio, dell’orecchio interno, del nervo acustico, delle vie nervose; con criterio fisiopatologico, in s. di trasmissione da lesioni dell’orecchio esterno e dell’orecchio medio, in s. neurosensoriale (in passato definita di percezione) da lesioni dell’orecchio interno o del nervo acustico o delle vie acustiche nervose centrali anche di natura traumatica (s. da rumore), e in s. miste determinate da cause che agiscono contemporaneamente sui meccanismi di trasmissione e neurosensoriali. La S. verbale è una rara sindrome neurologica determinata da lesioni cerebrali generalmente di natura vascolare, eccezionalmente traumatica o neoplastica, caratterizzata dall’incapacità del paziente di comprendere e ripetere ciò che ascolta.”
2.2 Tipi di ipoacusie Tre tipi di perdite uditive:
• Ipoacusia trasmissiva: L'ipoacusia trasmissiva o di trasmissione si ha quando l'abbassamento o la perdita dell'udito sono causati da un danno localizzato nell'orecchio esterno o nell'orecchio medio. La funzione principale di questa porzione di orecchio è la trasmissione meccanica del suono
• Ipoacusia neurosensoriale: La parte colpita è la porzione interna dell'orecchio, nello specifico la coclea od il nervo acustico.
21
• Perdita di udito mista: si riferisce a una combinazione di ipoacusia trasmissiva e neurosensoriale. Questo significa che ci possono essere danni a livello dell'orecchio esterno o medio e dell'orecchio interno (coclea) o del nervo acustico.
• Ipoacusia improvvisa: perdita di udito rapida e inaspettata. Le cause di questo disturbo possono essere molteplici: eventi traumatici (quali fratture o esplosioni a distanza ravvicinata), infezioni virali (parotite, morbillo, varicella, rosolia), infezioni batteriche (labirintite), sclerosi multipla, di vascolare o tossica (es. da farmaci).
In base alla classificazione stabilita dal Bureau International d’Audiophonologie, a seconda dell’entità della perdita uditiva espressa in decibel (dB: unità di misura di attenuazione), la sordità può essere:
• Lieve (da 30 dB a 40 dB di perdita uditiva): solo la voce bisbigliata non viene percepita.
• Moderata (da 41 a 60 dB di perdita uditiva): la voce emessa a livello di normale conversazione non viene udita perfettamente; ad intensità superiore la persona percepisce i suoni, ma ha una certa difficoltà a distinguere le parole. In particolare se il deficit uditivo è presente in un bambino alla nascita o nel primo periodo di vita, l’acquisizione del linguaggio senza la protesi acustica avverrà in modo limitato e sempre con un certo ritardo nel tempo.
• Severa (da 61 a 90 dB di perdita uditiva): un individuo con questa sordità percepisce solo alcuni suoni delle parole anche se pronunciate a intensità elevata.
• Profonda (superiore 91 dB di perdita uditiva): vengono percepiti solo i suoni più gravi e intensi aventi una notevole componente vibratoria, come il rombo del motore, lo sbattere della porta e pochi altri. La parola non viene assolutamente udita per cui senza un ausilio protesico associato alla lettura delle
22
parole sulle labbra non è possibile alcuna forma di apprendimento del linguaggio verbale.
2.3 Origine e causa della sordità
La sordità può sopraggiungere in qualsiasi momento. Le cause che la determinano sono molteplici. Nelle ipoacusie trasmissive la sordità può essere dovuta all’ostruzione del condotto uditivo esterno, alla rottura traumatica della membrana timpanica, a infiammazioni acute e croniche della cavità timpanica, ad otosclerosi su base ereditaria, a insufficienza respiratoria nasale da adenoidi, a tumori rinofaringei, alla deviazione del setto nasale o a malformazioni.
Mentre la sordità neurosensoriale può insorgere:
• prima della nascita, prenatale (origine ereditaria, virale, tossica, ecc.);
• alla nascita, perinatale (asfissia, ittero, ecc.);
• nei primi mesi di vita, neonatale (meningite, encefalite, malattie infettive virali, ecc.) ;
• nel corso degli anni (trauma cranico, intossicazioni, forme virali, presbiacusia, ecc.).
Il bambino sordo dalla nascita (sordità preverbale) non è in grado di sviluppare il linguaggio in modo normale senza una adeguata terapia protesico/riabilitativa.
Il bambino diventato sordo verso i 3/4 anni (sordità periverbale) perde quasi completamente il linguaggio se non viene tempestivamente protesizzato.
La persona diventata sorda dopo la completa acquisizione della parola (sordità postverbale) conserva pressoché inalterato il proprio patrimonio linguistico; ciò che viene compromessa, se non vengono presi opportuni provvedimenti, è la
23
comunicazione verbale, con inevitabili conseguenze sul piano sociale e psicologico.
2.4 Ototossicità da farmaci
Nei paesi sviluppati, almeno il 60-70% dei casi di ipoacusia è dovuto a cause genetiche mentre la rimanente parte è dovuta a cause di natura ambientale quali infezioni durante la gravidanza, traumi, farmaci. Un caso particolare di ipoacusia che deve essere preso in considerazione è quello conseguente all’uso di farmaci ototossici, in quanto questi farmaci possono causare in forma reversibile o irreversibile una piccola o grande perdita di udito. L’ototossicità di questi farmaci induce una lesione reversibile o non delle strutture dell’orecchio interno sia della porzione vestibolare che di quella cocleare. Quando è interessata la coclea si ha perdita uditiva di tipo neurosensoriale, bilaterale e simmetrica. Può essere congenita se la madre ha praticato terapie con farmaci ototossici durante la gravidanza.
Gli aminoglicosidi (AGs) sono una classe di antibiotici molto potenti, ad ampio spettro e particolarmente efficaci nel trattamento di malattie infettive. Il capostipite della classe degli aminoglicosidi è la Streptomicina (Fig. 11): è stato il primo antibiotico amminoglicosidico ad essere scoperto ed è stato anche il primo antibiotico attivo contro il Mycobacterium tubercolosis. La Streptomicina è stata isolata da Selmon Waksman (1943) da colture di Streptomices griseus. Successivamente ne furono isolati
altri (kanamicina, gentamicina e
24
tobramicina), i quali stabilirono definitivamente l’efficacia terapeutica di questa classe di antibiotici nei confronti delle infezioni indotte da batteri gram -. Nel 1970 si ottennero per via semisintetica, la dibecacina, l’amikacina e la netilmicina, composti attivi su ceppi resistenti ai primi aminoglicosidi e con migliori profili tossicologici. Ai giorni d’oggi, nove AGs (streptomicina, neomicina, tobramicina, kanamicina, paromomicina, spectinomicina, gentamicina, netilmicina, e amikacina) sono ufficialmente approvati dalla Food and Drug Administration [FDA],(Drew R.H., 2011).
Gli AGs, sono una classe di antibiotici, che presentano nella loro struttura due o più aminozuccheri uniti attraverso un legame glicosidico ad un nucleo esoso (aminociclitoli), che generalmente si trova in posizione centrale. Gli
aminozuccheri (Fig. 12) più importanti presenti nelle diverse strutture sono: - streptosio, ribosio, glucosamina , purpurosamina, neosamina B (Fig.
12) O HOH2C OH OH O H O OH NH2 NH-CH CH3 CH3 O OH OH NHCH3 O H C H3 O OH R R' O H R''-H2C ribosio purpurosamina garosamina R=R' OH; R'' = NH2 6-Glucosamina R'=R''OH; R = NH2 2-Glucosamina R=R'' OH; R' = NH2 3-Glucosamina R=NH2; R'=OH; R''=NH2 Neosamina B Figura 12
L’aminociclitolo può essere una streptidina (nella streptomicina) o una 2-deossisteptamina (negli altri AGs) (Fig. 13).
25 OH N H OH OH O H N H NH2 N H2 NH NH OH NH2 OH OH O H N H2 NH2 OH OH O H N H2
Streptidina Streptamina 2 Deossi-streptamina
Figura 13
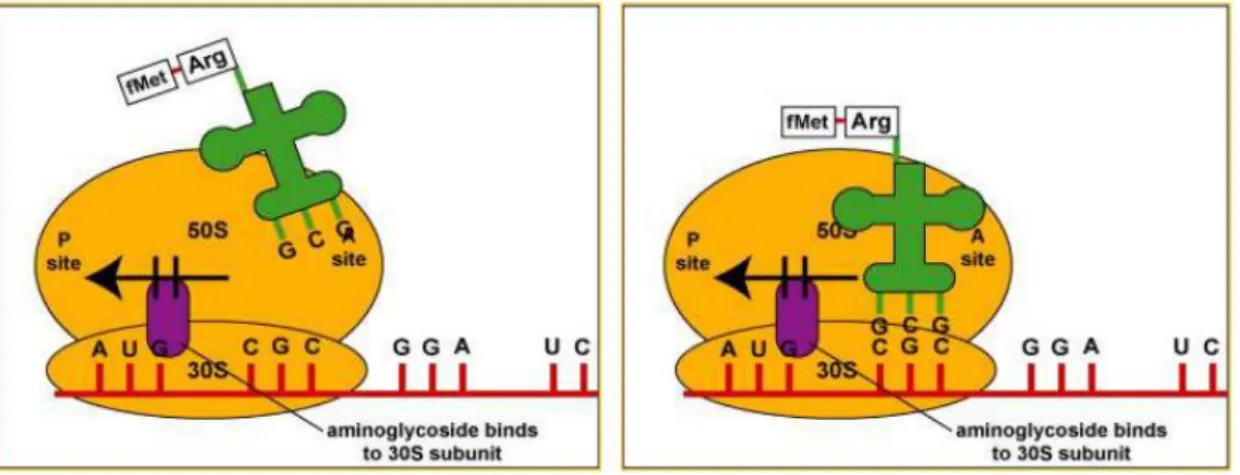
Meccanismo d’azione degli AGs
Gli aminoglicosidi si legano alla subunità 30S ribosomiale e provocano la distorsione del sito accettore interferendo con il corretto inserimento delle molecole degli aminoacil-tRNA durante l’allungamento della catena peptidica. Si assiste, quindi, all’inibizione della sintesi proteica mediante blocco del complesso d’inizio ed errata lettura per distorsione della subunità ribosomiale. L’accumulo di proteine non funzionali, detti “monosomi streptomicinici”, danneggia le membrane cellulari causando l’espulsione di ioni K+ e di aminoacidi con lisi cellulare. Gli AGs interrompono l’ulteriore traduzione e provocano un’interruzione prematura della sintesi. Per di più inducono un’errata lettura dello stampo di RNA messaggero (mRNA), incorporando aminoacidi errati nelle catene polipeptidiche. Agiscono perciò, sul mRNA, che non è più in sintonia con le informazioni ricevute dal DNA, per cui sceglie degli aminoacidi errati e ne risulta un peptide anomalo. Le proteine aberranti così prodotte, possono essere inserite nella membrana cellulare, determinando un’alterata permeabilità e un ulteriore stimolo al trasporto degli aminoglicosidi. Il passaggio di queste molecole polari attraverso la membrana dei batteri gram- è un processo autoindotto, che implica la distruzione dei ponti di Mg²+ e Ca²+ presenti tra le molecole di lipopolisaccaride (LPS carica negativa) e porta alla formazione di fori nella parete cellulare. Il successivo trasporto degli aminoglicosidi attraverso la membrana citoplasmatica è dipendente dal trasporto elettronico e dall’energia termica della fase I (EDP-I). L’energia per questo processo è fornita dal gradiente elettrochimico trasmembrana ed il trasporto è accoppiato ad una pompa protonica. Pertanto il trasporto è inibito da riduzione
26
del pH, dall’anaerobiosi e da iperosmolarità, mentre è aumentato da farmaci attivi sulla parete come vancomicina e penicilline, così che questo incremento può essere alla base del sinergismo. Una volta entrati nel citosol, gli AGs legano una sequenza di rRNA nella subinità 30S che è vicina al sito di riconoscimento codone-anticodone dove si lega l’aminoacil-t-RNA (sito A). L’AGs legato stabilizza l’interazione t-RNA-mRNA nel sito A attraverso la ridotta dissociazione del tRNA, che interferisce con lo stadio di correzione che assicura la fedeltà della traduzione (Karimi R., et al.,1994).
Figura 14 Fonte: ordinefarmacistinapoli.it
In definitiva il legame aminoglicoside-ribosoma causa tre effetti:
Previene la formazione del complesso di inizio della sintesi proteica (legame m-RNA-t-RNA e associazione della subunità 50S);
Blocca l’elongazione della catena nascente interrompendo il processo di controllo traduzionale (lettura errata o terminazione precoce);
La proteina nativa aberrante può essere inserita nella membrana cellulare, di cui altera la permeabilità ed al tempo stesso incrementa il trasporto e l’accumulo dell’aminoglicoside.
Farmacocinetica degli AGs
La struttura molecolare degli AGs conferisce proprietà basiche, elevata solubilità in acqua e di conseguenza idrofilia. Come risultato, l’assorbimento
27
enterale è scarsa e sono generalmente somministrati per via parenterale o topica (Silva et al., 2007). Dopo la somministrazione per via parenterale, il picco plasmatico è compreso tra 30 e i 90 minuti (Drew R.“Aminoglycosides,” 2011); presenta basso legame con le sieroproteine; distribuzione tissutale variabile con accumulo nella perilinfa e nell’endolinfa dell’orecchio interno e nella corticale renale; il metabolismo è minimo, circa il 99% di AGs somministrati vengono eliminati, inalterati, nel tubulo prossimale per filtrazione glomerulare (Al-Amoud et al.,2002). Gli AGs hanno un’emivita di circa 2-2,30 ore, ma il tempo si prolunga nei neonati e nelle persone che presentano funzionalità renale ridotta.
Il percorso degli AGs nelle cellule ciliate
Le cellule ciliate esterne sono colpite prima ed in misura più rilevante delle cellule ciliate interne.
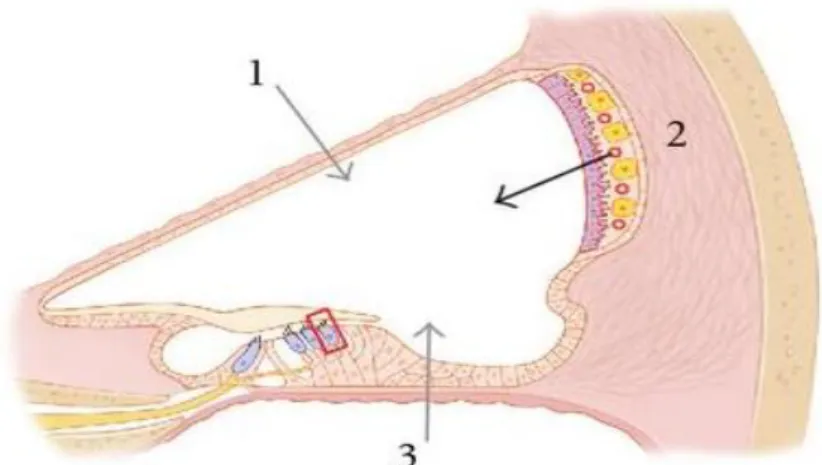
I possibili siti d’ingresso degli AGs nell’orecchio interno sono tre (Fig. 15):
1. Tramite la membrana di Reissner 2. Tramite la stria vascolare
3. Tramite la membrana basilare
28
A livello delle cellule ciliate, gli AGs possono attraversare la cellula capelluta in tre modi (Fig. 16) :
- Canali maccanotrasduttori situati nelle stereociglia (A)
- Endocitosi sulle membrane apicali o basolaterali (A, B, o C)
- Recettori che determinano variazioni transitorie di potenziale, TRP (A, B, o C) - Recettori per l’ATP (A)
L’AG giunto nella cellula capelluta può causare
danni, in modo diretto o non, dapprima induce una confusione delle stereociglia e, infine, termina con la morte cellulare per apoptosi (Abi-Hachem R.N. et al.,2010). L'esposizione ad AG porta alla compromissione della traduzione e l'inibizione della sintesi delle proteine all'interno dei mitocondri. Si pensa, inoltre, che l'inibizione della sintesi proteica mitocondriale porti ad una diminuzione dell’ATP (Guan M.X.,2011). Con questa diminuzione viene compromessa l’integrità mitocondriale e ciò predispone ad una fuoriuscita di citocromo C e successiva attivazione delle cascate apoptotiche.
Generalmente le vie apoptotiche sono due: 1. Di tipo intrinseco: è la via principale apoptotica indotta da AGs. Può
essere innescata da vari fattori di danno, tipo danno al DNA, stress citotossico. In pratica avviene la formazione di grossi pori sulla membrana del mitocondrio che porta alla fuoriuscita del citocromo c che è in grado di attivare una pro-caspasi 9 d’inizio che fa partire la cascata delle pro-caspasi. A livello del mitocondrio c’è la possibilità di modulazione attraverso la produzione di proteine prodotte da particolari geni, della famiglia BCL-2 che formano dei dimeri che si inseriscono nella membrana del mitocondrio. Queste proteine possono essere pro-apoptopiche (BaX, DP5, Bad) e anti-apoptopiche (Bcl-2 e Bcl-XL). A seconda della quantità e del tipo di proteine che sono espresse dalla cellula in quel momento la bilancia si può spostare verso l’apoptosi oppure
Figura 16 Fonte: Huth M.E. et al.,2011
29
verso la sopravvivenza della cellula. Il dimero BaX/BaX è pro-apoptotico, mentre il dimero Bcl-2/Bcl-2 è anti-apoptotico.
2. Di tipo estrinseco: è mediata dai recettori di morte. Appartengono alla famiglia dei recettori per il fattore di necrosi tumorale (TNF). Ci sono due tipi di recettore: il Fas e il TNFR-1. Il Fas lega Fas-L (ligando), che causa la trimerizzazione del recettore che quindi si attiva, e si lega alla proteina FADD che a sua volta attiva la pro-caspasi 8 che è una delle caspasi d’inizio della cascata. Anche il TNF-1 si lega al recettore che subisce una trimerizzazione, per cui si modifica e si lega alla proteina TRADD che attiva a sua volta un’altra proteina FADD che infine attiva la pro-caspasi 8 che è una delle caspasi d’inizio della cascata.
Il meccanismo con cui si induce il danno da AGs, è probabilmente mediato dalla liberazione dei radicali liberi dell’ossigeno (ROS). Più precisamente aumenta la concentrazione delle specie reattive dell’ossigeno nei mitocondri delle cellule ciliate cocleari (Clerici et al.,1995) ed in quelli delle cellule renali (Walkers,1987; Yang et al.,1995).
Normalmente, la cellula si protegge dall’accumulo di ROS con produzione intrinseca di anti ossidanti come il glutatione (Gutteridge et al.,2000). Quando la formazione di ROS, travolge la capacità intrinseca di questi meccanismi di riparazione e di protezione, la cellula viene sottoposta al
processo dell’apoptosi (Jeong S.W. et al.,2010). L’AG forma un complesso con un metallo di transizione, in questo caso, il
ferro, secondo la reazione di Fenton:
30
Fe²
++ H
2O
2Fe
3++ HO° +HO
¯catalizzando così la formazione dei ROS / RNS compresi l'anione radicale superossido, perossido d’idrogeno, radicale idrossile, l'anione perossinitrito, etc. Questi reagenti attivano le proteine chinasi JNK (c-Jun N-terminal chinasi) e una loro stimolazione determina l’attivazione, nel nucleo, di fattori di trascrizione di geni implicati nella via della morte cellulare; questi prodotti sono poi trasferiti ai mitocondri dove promuovono il rilascio di citocromo c (Cyt c). Nel citosol, il Cyt c determinerà l'attivazione di una serie di caspasi seguita da apoptosi attraverso quello che viene definito come morte cellulare caspasi-dipendente.
Neutralizzazione delle specie reattive dell’ossigeno
Com’è stato finora descritto gli AGs formano complessi con il ferro catalizzando, quindi, la formazione di ROS. Per evitare questi danni ossidativi e neutralizzare le specie reattive dell’ossigeno e far si che la reazione di Fenton non avvenga , è utile usare degli agenti chelanti il ferro.
N-acetilcisteina (NAC) è un farmaco usato comunemente dai pazienti come mucolitico, ma è anche un ottimo antiossidante e non dimostra effetti collaterali ototossici.
Una miriade di altri agenti con questa capacità sono D-metionina (Met-D), acido α - lipoico (α – LA), vitamine come α - tocoferolo (vitamina E), vitamina C, nonché gli estratti di erbe Gingko biloba. L' ormone melatonina, normalmente secreto dalla ghiandola pineale (o epifisi), ha anche attività antiossidante (Kim J.B. et al.,2009).
Nel complesso gli antiossidanti attenuano i danni ototossici indotti da AGs. Tuttavia, la maggior parte di questi agenti non ha indotto una protezione completa da questa ototossicità (Conlon B.J. et al 2000) e gli effetti del trattamento a lungo termine restano ancora da studiare.
31 Ototossicità e predisposizione genetica
Alti dosaggi di AGs somministrati per lunghi periodi, causano danni a livello vestibolare e cocleare. Nondimeno, anche l’assunzione di piccole dosi di AGs risulta dannosa se esiste una certa predisposizione genetica. In presenza della mutazione A1555G, il gene, che risulta essere il più coinvolto nella predisposizione genetica alla ototossicità, è l’rRNA 12S che diviene il principale bersaglio degli antibiotici AGs con conseguente inibizione parziale della sintesi proteica e riduzione delle normali funzioni cellulari. In pratica il bersaglio di tali farmaci, come abbiamo detto prima, è l’RNA ribosomiale dei batteri e la mutazione A1555G fa si che l’RNA mitocondriale risulti simile a quello di questi agenti infettati. La mutazione A1555G dell’rRNA 12S determina una ipoacusia che può apparire a qualsiasi età, spontaneamente o dopo assunzione di AGs.
Il cisplatino (cis-DDP) è un chemioterapico ampiamente utilizzato nel trattamento dei tumori testicolari e tumori ovarici ed è anche ampiamente utilizzato per il trattamento del carcinoma della vescica, del polmone, dell’esofago e dello stomaco. Nonostante il suo successo, il cisplatino ha diversi svantaggi, che comprendono gravi effetti collaterali tra cui nefrotossicità, neurotossicità, ototossicità, nausea e vomito (Giaccone G. 2009). Questi effetti tossici limitano la dose che può essere somministrata ai pazienti.
Il cisplatino è una piccola molecola costituita da uno ione platino centrale circondato da quattro ligandi, due gruppi amminici e due cloridrici; quando questi ultimi sono disposti in posizione cis,
la molecola ha attività chemioterapica, se sono in posizione trans non ha attività (Goodsell, 2006). Nel 1965, nel corso di alcuni saggi in vitro conseguiti per valutare gli effetti del campo magnetico sulla crescita di Escherichia coli, Rosenberg e i suoi studiosi notarono che la forma dei batteri da cilindri divenivano filamentosi (Fig. 18):
32
Figura 18 Scanning electron micrograph of wild-type Escherichia coli treated with cisplatin. Fonte: personal.bgsu.edu
essi erano cioè aumentati di volume senza tuttavia dividersi (Rosenbreg et al., 1965). Gli elettrodi inibivano la divisione di E. coli e ne inducevano una crescita filamentosa. Fu cosi scoperta una nuova classe di agenti antimorali in grado di inibire la divisione cellulare.
L’ototossicità del cisplatino è dose e frequenza dipendente, si manifesta con una perdita uditiva inizialmente alle alte frequenze per poi progredire verso quelle della parola ed è spesso accompagnata da tinnito (Rademaker-Lakhai et al., 2006). Nell’orecchio interno il cisplatino ha principalmente effetto sull’organo del Corti, sulle cellule del ganglio spirale e sulla stria vascolare causando alterazioni morfologiche e morte cellulare.
Farmacocinetica
Il cisplatino è somministrato per via endovenosa, si concentra nel fegato, nei reni, nell’intestino tenue e crasso dell’uomo e degli animali; apparentemente il farmaco ha una limitata penetrazione nel sistema nervoso centrale. I livelli plasmatici diminuiscono in modo bifasico con un’iniziale emivita di 20-50 minuti e un’emivita finale di 58-72 ore. Più del 90% della quantità di platino somministrata si lega alle proteine nella fase di post-somministrazione. Il
33
cisplatino viene escreto principalmente nell’urina attraverso la filtrazione glomerulare.
Ancora non è chiaro il meccanismo con cui il cisplatino diffonde attraverso la membrana cellulare (Fig. 19): potrebbe essere trasportato per trasporto passivo, trasporto attivo mediato da ATP (Ivy et al., 2013), e tramite il trasportatore ad alta affinità per il rame (CTR-1). CTR- 1 è coinvolto nella resistenza ai trattamenti chemioterapici in alcune forme di tumore; di fatto vi è un’alterazione dei livelli di espressione di alcune proteine trasportatrici di ioni rame.
Figura 19 Schematic depiction of cisplatin uptake and efflux and binding to nuclear DNA. Fonte: Cepeda et al.,
34
Ottimi leganti del platino sono gli aminoacidi contenenti zolfo, ovvero la metionina e la cisteina che sono ottimi leganti anche per lo ione rame nello stato di ossidazione +1, cioè nella sua forma ridotta, che è la forma in cui esso viene importato nelle cellule da CTR-1 ed esportato dal citoplasma attraverso le Cu-ATPasi di Menkes e Wilson.
Una volta che cisplatino è nel citoplasma può essere inattivato dal glutatione (GSH), oppure può legarsi al suo principale bersaglio cellulare, il DNA nucleare.
L’effetto tossico del cisplatino si manifesta attraverso diverse vie:
- Induce la formazione dei radicali liberi dell’ossigeno (ROS) nelle cellule cocleari (Clerici et al. 1995) come l’anione superossido ed il monossido di azoto (NO) che si forma per merito dell’enzima ossido nitroso sintetasi (NOS). L’incremento della formazione dei ROS è conseguenza della riduzione di antiossidanti cellulari (superossido dismutasi, catalasi, e glutatione perossidasi e reduttasi). La riduzione avviene tramite il legame tra i gruppi sulfidrilici degli enzimi con il cisplatino, e come conseguenza della riduzione del rame e selenio che sono importanti per il funzionamento della superossidodismutasi e della glutatione perossidasi (DeWoskin et al., 1992).
- Si lega al DNA formando diversi tipi di legami: legami crociati intermolecolari tra basi di eliche parallele; legami crociati
intramolecolari tra basi di una stessa elica; legami tra il DNA e le proteine ad esse associate (DNA polimerasi, DNA topoisomerasi, proteine istoniche; Fig. 20).
35
Figura 20
- Studi su cellule coltivate in vitro hanno dimostrato che il più frequente addotto bivalente formato dal farmaco e responsabile dell’azione tossica è costituito dal legame tra le purine adiacenti della stessa elica di DNA, con preferenza per le guanine (Fig. 21). In questo caso, il legame avviene
36
in corrispondenza dell’azoto in posizione 7 a causa della sua natura
nucleofilica. Infatti, il cisplatino quando entra nella cellula, a causa della bassa concentrazione di ioni cloruro cede quest’ultimi diventando un potente elettrofilo che può interagire con i vari residui nucleofili delle macromolecole cellulari.
Entrambe le vie d’azione del cisplatino portano alla morte cellulare per apoptosi o per necrosi, ma i meccanismi molecolari con i quali il chemioterapico altera la funzionalità delle cellule uditive non è stata ancora completamente chiarito.
37
3.
SORDITÀ GENETICHE O EREDITARIE
3.1 La nascita della genetica
Il padre fondatore “morale” della genetica è Gregor Mendel (1822-1884), monaco agostiniano che nel 1856, nell’abazia di Brünn, allora in Austria (oggi è Brno, nella repubblica Ceca) iniziò a fare incroci delle piante di pisello.
Mendel fece esperimenti per dieci anni, utilizzando circa 30·000 piante conteggiando
scrupolosamente i risultati ottenuti e così scoprì le regole fondamentali che governano la trasmissione dei caratteri ereditari .
Mendel seguì il seguente procedimento sperimentale:
Come materiale di studio scelse le piante di pisello (Pisum sativatum) Puntualizzò la sua attenzione su sette coppie di caratteri unitari, cioè con caratteri che si presentavano solo con due forme alternative facilmente distinguibili (seme liscio, seme rugoso)
Selezionò delle linee pure
Incrociò piante di linea pura che differivano solo per un carattere o per due caratteri
Le leggi mendeliane:
I legge di Mendel: incrociando una linea pura dominante ed una recessiva si ottengono solo discendenti dominanti (uniformità degli ibridi).
II legge di Mendel: ogni individuo possiede due copie di ogni fattore e che esse si separano (si segregano) durante la formazione dei gameti. Questa legge è denominata Legge della Segregazione degli ibridi.
Figura 22 Gregor Mendel Fonte: primolevimarino.it
38
III legge di Mendel: dall’incrocio di due individui che differiscono per due o più caratteri, ognuno di questi si trasmette ai discendenti seguendo la 1ª e la 2ª legge, indipendentemente dagli altri caratteri.
Per spiegare i risultati ottenuti, Mendel avanzò alcune ipotesi, poi rivelatesi corrette:
Mendel chiamò i fattori ereditari, i caratteri ereditati dai genitori. I geni esistono in forme alternative (gli alleli).
Ogni organismo possiede due copie di ogni fattore (due alleli per ogni gene) per ogni carattere ereditato e ciascun fattore (ogni allele) deriva da uno dei due genitori.
Le due copie di un fattore (i due alleli di un gene) si separano (si
segregano) durante la meiosi, così che i gameti possiedono un'unica copia (un
solo allele) per ogni carattere.
L’unica cosa che non seppe spiegare fu la natura fisica cioè i cromosomi perché non erano stati ancora scoperti. Nel 1866 i suoi dati vennero pubblicati sugli atti della Società dei Naturalisti di Brünn in un breve lavoro intitolato: “Esperimenti sugli ibridi vegetali” che conteneva , espresse chiaramente, le leggi e i principi. Sfortunatamente la rivista aveva scarsa diffusione e Mendel non era un noto professore universitario, per cui i risultati da lui ottenuti furono a lungo disconosciuti o svalutati dalla Scienza. Solo 35 anni dopo, nel 1900, quando Mendel era ormai morto da 16 anni, tre botanici, l’olandese De Vries (1848-1935), il tedesco Correns (1864-1933) e l’austriaco Tschermack (1871-1962), indipendentemente l’uno dall’altro, arrivano, alle stesse conclusioni di Mendel, e concordemente attribuirono il merito della corretta interpretazione e della chiara e precisa enunciazione delle leggi a colui che da allora è riconosciuto come il fondatore della Genetica. Possiamo pertanto definire la Genetica come la scienza che studia le leggi della Conservazione, della Variazione, dell’Espressione e della Trasmissione delle informazioni scritte in un codice chimico che permettono la realizzazione dei caratteri ereditari, cioè delle
39
informazioni genetiche necessarie per lo sviluppo delle strutture e delle funzioni di tutti gli esseri viventi.
Figura 23 L’origine del DNA. Fonte: memorydr.com
3.2 Cenni di Genetica
I caratteri ereditari di ciascun individuo provengono dal corredo cromosomico trasmessogli dai genitori. In ciascun cromosoma si trovano disposti longitudinalmente i fattori dei caratteri detti geni di provenienza materna e paterna (Fig. 23); essi sono responsabili della trasmissione di un determinato carattere ereditario. Più precisamente può essere definito come l’unità di informazione contenete il progetto per la sintesi di una proteina. Il progetto complessivo di un organismo è costituito dall’insieme dei progetti coordinati di tutti i geni (Genoma) che contribuiscono alla realizzazione di tutte le sue strutture e funzioni.
Il gene è contenuto nel locus e la variante del gene si chiama allele .
Si dice omozigote il soggetto che possiede nel proprio corredo cromosomico due geni uguali (uno paterno e uno materno) siano essi responsabili della
40
trasmissione di un determinato carattere normale o patologico esempio aa o AA; si dice eterozigote quando i due alleli sono diversi (Aa).
Il genotipo di un individuo è dato dal suo corredo genetico, è ciò che è "scritto" nel DNA contenuto nel nucleo di tutte le sue cellule ed è quindi immutabile. Il
fenotipo, invece, è l'insieme dei caratteri che l'individuo manifesta; dipende dal
suo genotipo, dalle interazioni fra geni e anche da fattori esterni e dunque può variare.
Per mutazione si intende ogni cambiamento raro, grande o piccolo, che si verifica all’interno del materiale genetico e che non sia causato da ricombinazione genetica.
Il termine mutazione fu introdotto nel 1896 dal botanico olandese de Vries, il quale studiando la pianta ornamentale Oenothera lamarckiana (Fig. 24), originaria dall’ America e inselvatichita nei giardini olandesi, trovò che presentava numerose piccole variazioni ereditarie che secondo la sua interpretazione davano origine a nuove piccole specie.
Ricercatori successivi dimostrarono che le supposte mutazioni di de Vries erano modificazioni fenotipiche dovute ad altri eventi quali la ricombinazione o condizioni citogenetiche particolari come la poliploidia o la aneuploidia.
Le mutazioni si dividono in geniche, cromosomiche e genomiche. Mutazione geniche: consistono in una modificazione della struttura del gene e
sono dovute a cambiamenti nelle sequenze nucleotidiche. Se il cambiamento riguarda solo una coppia di nucleotidi vengono denominante Mutazioni Puntiformi. Si suddividono in : Sostituzioni (un nucleotide viene sostituito da uno diverso)
Vi sono due tipi di scambio:
41
- la transizione, quando una base purinica viene sostituita con l’altra purinica (da A a G o viceversa) o una pirimidina viene sostituita con l’altra pirimidina (da T a C o viceversa).
- la trasversione, quando la purina viene scambiata con una pirimidina o viceversa. Sempre se avvengano in un esone, le mutazioni per sostituzione di una base possono avere a livello proteico effetti diversi ed essere cosi classificati:
1. Mutazioni missenso: formazione di una tripletta o codone che specifica un amminoacido diverso da quello originario.
2. Mutazioni non-senso: sono dovute alla sostituzione di una base che porta alla formazione di uno dei tre codoni (UAG;UGA;UAA) che non specificano alcun amminoacido e costituiscono un segnale stop della sintesi proteica.
3. Mutazioni neutre: sono dovute alla formazione di una tripletta diversa dalla prima che codifica per un amminoacido diverso. Tale sostituzione non altera la funzionalità della proteina specificata dal gene in quanto i due amminoacidi, pur diversi, sono chimicamente equivalenti. (cambiamento della tripletta AGC ad AAG che porta alla sostituzione della Lisina con Arginina. Entrambi questi amminoacidi sono basici e hanno proprietà simili).
4. Mutazioni silenti: sostituzione di una base che non determina alcun cambiamento a livello fenotipico in quanto la tripletta esprime il solito amminoacido (da AGG a AGA viene codificata sempre l’Arginina).
Delezione (un nucleotide viene perso) e Inserzione (viene aggiunto). Le
mutazioni per delezione o inserzione di un nucleotide provocano lo scivolamento della cornice di lettura dell’informazione ereditaria (frameshift), cioè una lettura fuori fase in quanto il messaggio viene letto continuamente senza interruzioni. Si avrà, quindi, una modificazione di tutte le triplette, e pertanto verranno inseriti amminoacidi diversi da quelli originali con grandi e gravi effetti sulla funzionalità della proteina (Fig. 25).
42
Figura 25 Esempi di mutazioni geniche per inserzione o delezione di una o due basi
Mutazioni cromosomiche: consistono in alterazioni della struttura dei cromosomi, che a loro volta sono suddivisibili in :
Delezioni e Duplicazioni, che sono alterazioni quantitative.
Inversioni e Traslocazioni, che sono alterazioni qualitative o
riordinamenti di struttura.
Mutazioni genomiche: consistono in variazioni di numero dei cromosomi. Si suddividono in:
Euploidie: consistono nella variazione del corredo rispetto a quello
normale per interi assetti aploidi in memo o in più. Aneuploidie: consistono nella variazione del corredo dovuta a perdita o a
43
La sordità è il deficit neonatale più frequente, dato che 1/1000 - 1/700 neonati ha una sordità profonda o grave. Attualmente, nei paesi industrializzati, il 60-80% delle sordità precoci ha un'origine genetica. Di questo il 30% fa parte di una condizione sindromica, mentre il 70% è costituito da forme non sindromiche, in cui il deficit uditivo è l'unico segno clinico. Sono state riscontrate mutazioni in più di 100 geni che sono causa di ipoacusia non sindromica. Le sordità non sindromiche, presentano una grande eterogeneità genetica. I diversi loci cromosomici delle forme non sindromiche di sordità ereditaria sono indicati con l’acronimo DFN (“deafness”) seguito dalla lettera A o B a seconda della modalità di trasmissione rispettivamente, autosomica dominante o recessiva. Le lettere A o B sono seguite da un numero che indica
l’ordine cronologico di scoperta dei geni associati al locus. Fino ad oggi sono stati però individuati 41 loci associati ad ipoacusia a
trasmissione autosomica dominante (DFNA), e 46 loci a trasmissione autosomica recessiva (DFNB), ma la continua evoluzione delle tecniche di biologia molecolare aggiorna continuamente tale lista (http://webh01.ua.ac.be/hhh/;http://davinci.crg.es/deafness/). Sono stati
identificati anche geni ad ereditarietà X-linked e mitocondriali. (Fonte: www.aou-careggi.toscana.it)
44
4.
METODO DI STUDIO DELLA
GENETICA MEDICA
I principali metodi di studio della genetica umana sono: Metodo statistico
Alberi genealogici
I due metodi consentono di individuare se un carattere è ereditario, se è autosomico o legato al sesso, quale dei due fenotipi alternativi è dominante e quale è recessivo, se un carattere è monofattoriale o determinato da più geni e cosi via. Essi differiscono per l’applicabilità, infatti il metodo statistico viene usato per lo studio di caratteri con fenotipi alternativi comuni, cioè per caratteri così detti fisiologici, nei quali tutte le forme alternative non sono nocive e non vengono quindi selezionate negativamente, ad esempio il colore degli occhi, gruppi sanguigni.
4.1 Alberi Genealogici
Gli alberi genealogici “dall’inglese pedigree” si usano invece per caratteri rari, principalmente patologici. Viene definito tipo di indagine “in verticale” nel senso che trovato un probando affetto da una certa malattia di cui non si conoscono le caratteristiche ereditarie, si cerca di risalire, attraverso interviste ai familiari viventi, al fenotipo di tutti i parenti per il maggior numero possibile di generazioni, in modo da costruire un albero genealogico il più completo e veritiero possibile.
1) Malattia autosomica recessiva: si ha di solito una frequenza non molto elevata di individui affetti, qualche generazione in cui la malattia non si manifesta, distribuzione in misura simile della malattia tra maschi e femmine, comparsa della malattia in un individuo con genitori sani (che sono portatori sani della malattia, cioè eterozigoti).
45
2) Malattia autosomica dominante: per le quali la malattia, una volta comparsa nell’albero, si manifesta in tutte le generazioni, all’incirca nella metà dei figli di entrambi i sessi di un genitore malato.
46
3) Malattia legate al cromosoma X (Diaginiche) recessive: si ha una maggiore frequenza di maschi malati che sono di solito figli di genitori entrambi sani (la madre sarà portatrice o eteroziogote). Tali figli affetti, di solito trasmettono la malattia ai nipoti attraverso le figlie femmine che risultano essere sane-portatrici.
4) Malattia legate al cromosoma X (Diaginiche) dominanti: i maschi affetti sposati a donne normali hanno tutte figlie affette e tutti figli sani, mentre donne affette sposate con uomini sani hanno metà figli affetti sia di sesso maschile che femminile.
47
5) Malattia legata al cromosoma Y (Olandriche): di facile identificazione perché la malattia si trasmette sempre e comunque da padre a figlio.
4.2 Forme ereditarie sindromiche e Forme non-sindromiche
Forme sindromiche (30%): la perdita d’udito si associa ad altri sintomi e/o manifestazioni cliniche. La sordità può essere un sintomo rilevante o avere un’importanza secondaria. Sono elencate brevemente alcune condizioni sindromiche in cui l’ipoacusia è un sintomo importante per la diagnosi.
SINDROME DI ALPORT
La sindrome, descritta da Alport nel 1927, associa glomerulonefrite progressiva, ematuria intermittente e deficit uditivo neuro-sensoriale. Sono state descritte almeno 6 forme cliniche distinte. La più frequente è la forma dominante legata al cromosoma X. La sindrome di Alport è presente in almeno nel 1% dei soggetti affetti da sordità ed ha una frequenza di 1/5000 nati.
SINDROME BRANCHIO-OTO-RENALE (BOR)
La sindrome BOR fu descritta nel 1975 da Melnick. I pazienti presentano anomalie degli archi branchiali, difetti auricolari ed uditivi, anomalie renali di vario tipo. La Sindrome è trasmessa come carattere autosomico dominante con espressività variabile.
SINDROME DI JERVELL/LANGE-NIELSEN
Questa sindrome è caratterizzata da sordità neuro-sensoriale profonda associata ad una anomalia elettrocardiografia, intervallo Q-T lungo, che è causa di episodi ripetuti di sincope o morte improvvisa. La malattia è trasmessa come carattere autosomico recessivo, ha una frequenza 1/100.000 ed interessa lo 0,25% dei soggetti audiolesi.
SINDROME DI PENDRED
E’ una sindrome autosomica recessiva che interessa almeno il 10% dei soggetti affetti da sordità. Nel 70% dei casi è presente gozzo tiroideo, associato a
48
normale o ridotta funzione della ghiandola. In più del 50% dei casi è documentabile un deficit di organificazione dello iodio mediante il test al perclorato. La sordità è congenita, variabile, lentamente progressiva ed interessa in prevalenza i toni alti. Il gene responsabile della malattia, denominato PDS, è localizzato sul cromosoma 7 e codifica per un trasportatore dello iodio.
SINDROME DI NORRIE
La sindrome di Norrie fu descritta da questo autore nel 1927. E’ definita anche displasia oculo-acustico cerebrale, a causa del coinvolgimento di questi tre apparati. E’ una patologia recessiva legata al cromosoma X. La malattia ha una espressività completa nei maschi. Le femmine eterozigoti sono clinicamente indenni anche se possono presentare alcune anomalie retiniche all’esame del fondo oculare.
SINDROME DI TREACHER-COLLINS
La sindrome, definita anche disostosi mandibulo-facciale, si caratterizza per anomalie del massiccio facciale e delle orecchie che delineano un aspetto del volto caratteristico. La malattia si trasmette come carattere autosomico dominante ed ha espressività variabile.
SINDROME DI USHER
Questa sindrome è caratterizzata dall’associazione di retinite pigmentosa e sordità. Sono descritte tre forme cliniche distinte, tutte trasmesse come carattere autosomico recessivo.
SINDROME DI WAARDENBURG
Questa sindrome è molto caratteristica ed è trasmessa come carattere autosomico dominante ad espressività variabile. I sintomi principali sono costituiti da: aumento della distanza dell’angolo interno degli occhi (telecanto) ed anomalie di pigmentazione dei capelli, dell’iride e della cute. Spesso i due occhi hanno un colore differente (eterocromia dell’iride). La malattia ha una
49
espressività molto variabile. Al momento sono stati identificati cinque geni responsabili della malattia.
SINDROMI MITOCONDRIALI
Sono sindromi causate da un difettoso funzionamento dei mitocondri. La malattia interessa gli apparati ad alto consumo energetico quali il cervello, la retina, l’orecchio interno, i muscoli ed il pancreas. Pertanto la sintomatologia è caratterizzata da sintomi variamente associati. La sintomatologia più evidente interessa il sistema nervoso centrale.
Forme non sindromiche (70%): la perdita d’udito è l’unico sintomo presente e possono essere trasmesse come carattere autosomico recessivo, autosomico dominante, legato al cromosoma X, o mitocondriale
50
5.
MUTAZIONI RICORRENTI DEI GENI
NELLE FORME NON-SINDROMICHE
5.1 Mutazioni ricorrenti dei geni che causano sordità non- sindromica autosomica recessiva
Classificazione in ordine di ALTA frequenza dei geni che causano l’ARNSHL sono:
GENE LOCUS N° di mutazioni Funzione nel processo uditivo GJB2 DFNB1 >220 Omeostasi dello ione
SLC26A4 DFNB4 44 Omeostasi dello ione
MYO15A DFNB3 28 Cellula ciliata, proteina motrice OTOF DFNB9 26 Esocitosi delle vescicole
CDH23 DFNB12 21 Cellula ciliata, proteina di adesione TECTA DFNB21 10 Proteina della matrice extracellulare ESPN DFNB36 6 Cellula ciliata, formazione citoscheletro PCDH15 DFNB23 5 Cellula ciliata, proteina di adesione MYO7A DFNB2 5 Cellula ciliata, proteina motrice GJB6 DFNB1 4 Omeostasi dello ione
MYO6 DFNB37 3 Cellula ciliata, proteina motrice MYO3A DFNB30 3 Cellula ciliata, proteina motrice
GJB3 2 Omeostasi dello ione
GENE GJB2
Gap Junction Protein Beta 2
(Gene GJB2) è il gene che codifica per la connessina 26 e si trova sul braccio lungo del cromosoma 13q11-q12. La
connessina 26 è una proteina presente sulla membrana cellulare dove è coinvolta nella formazione delle gap junction, giunzioni che mettono in comunicazione cellule contigue permettendo il passaggio di piccole molecole e ioni. Come nel caso in cui arriva la stimolazione sonora le cellule ciliate