UNIVERSITÁ DI PISA
Facoltà di Scienze Matematiche, Fisiche e Naturali
Corso di Laurea Magistrale in
Biotecnologie Molecolari ed Industriali
Tesi di Laurea Magistrale
Espressione e assemblaggio delle proteine di AAV:
localizzazione ed incapsidazione del DNA in
Saccharomyces cerevisiae.
Relatore: Candidato:
Dott.ssa Tiziana Cervelli Filippo Cipriani
Dedica
Dedico il mio percorso di studi e questa tesi ai miei genitori Piero e Anna e a mio fratello Andrea come gesto di Gratitudine per l’esempio che mi danno ogni giorno. “Le persone sono esseri umani qualunque e tuttavia sono in grado di compiere azioni straordinarie. Molta gente comune vive la propria vita normale in modo eroico. Se il mondo venisse a sapere del loro valore verrebbero chiamati eroi e diventerebbero famosi, ma resterebbero comunque persone normali che hanno agito eroicamente.”
Indice:
Riassunto ...………...……...1
Abstract ...………...……...3
1.0 INTRODUZIONE 5
1.1 La terapia genica ...……….5
1.2 Il virus Adeno-associato di tipo 2 (AAV2) ……….12
1.2.1 Il ciclo vitale e d’infezione del Virus Adeno-associato ……….
14
1.2.2 Il Genoma di AAV2 ………
19
1.2.3 Regolazione e localizzazione delle componenti virali in relazione alla formazione delle VLPs ………
23
1.2.4 Incapsidazione del DNA nei virus Adeno-associati………...
26
1.2.5 Produzione di vettori basati sul virus Adeno-associato ………...
29
1.3
Il lievito Saccharomyces cerevisiae ………..33
1.3.1 Il lievito S.cerevisiae in campo virologico ………...
35
3.1 Ceppi di Saccharomices Cerevisiae ………..38
3.1.1 Ceppo RSY12-Rep68 ………
38
3.1.2 Ceppo BY4743 ………...
39
3.2 Terreni di coltura per il lievito ………..40
3.3
Plasmidi utilizzati ………..43
3.3.1 pAAVpokURA ……….
43
3.3.2 pESCGALVP2,3GALVP1KM ………...
44
3.3.3 pG.Rep68 & pG.Rep68-Hyg ……….
45
3.3.4 pFA6a-hphNT1 ………...
46
3.4 Trasformazione di S.cerevisiae con DNA plasmidico ……….47
3.5 Gene targeting ………..49
3.6 Crescita in terreno induttivo ed estrazione proteica ………51
3.7 Estrazione e purificazione delle VLPs ………...53
3.8 Protocollo d’isolamento dei nuclei ……….55
3.10 Estrazione e purificazione del DNA a basso peso molecolare …...60
3.11 Analisi dell’incapsidazione del DNA nelle VLPs ………61
3.12 Immuno blot ……….63
4.0 RISULTATI 65
4.1 Ingegnerizzazione dei ceppi utilizzati nella caratterizzazione delle
Vps ………65
4.1.1 Ceppo RSY12-Rep68 ………
65
4.1.2 Ceppo BY4743 ………...
68
4.2 Confronto tra RSY12-Rep68 e BY4743 ………71
4.3 Immuno blot ………...73
4.3.1 Studio di RSY12-Rep68 ………
73
4.3.2 Studio di BY4743 ………...
75
4.3.3 Studio della cinetica della formazione delle VLPs ………
76
4.4 Isolamento dei nuclei e localizzazione di Vp1,2,3 ………..80
4.5 Analisi dell’estrazione e della purificazione delle VLPs ………..83
5.0 DISCUSSIONE 87
6.0 CONCLUSIONI E PROSPETTIVE FUTURE 91
7.0 RIFERIMENTI BIBLIOGRAFICI 92
Riassunto
Il punto di partenza di questo progetto di tesi si basa sugli studi effettuati sul lievito S. cerevisiae come modello per la produzione di vettori derivati dal virus Adeno Associato (AAV) per la terapia genica. AAV è un virus difettivo, non patogeno della famiglia dei parvovirus che dipende dalla co-infezione di un virus Helper (come adenovirus o Herpes simplex) per una sua replicazione produttiva. Il genoma di AAV consiste di un DNA a singolo filamento contenente due open reading frames (ORF) fiancheggiate da due sequenze invertite ripetute dette ITR. Le due ORF codificano per le proteine Rep: Rep40, 52, 68 e 78, necessarie per la replicazione e le proteine Cap, Vp1, 2 e 3, necessarie per la formazione del capside. In laboratorio, è stato dimostrato che il lievito è in grado sostenere la replicazione di vettori AAV ricombinanti (rAAV) e la formazione di capsidi.
Lo scopo di questa tesi è pertanto quello di caratterizzare ulteriormente la formazione dei capsidi sia andando a studiare la localizzazione delle proteine del capside Vp1, Vp2 e Vp3, sia sviluppando una nuova metodica di investigazione che riveli con più facilità la produzione di vettori virali in S.cerevisiae. Un altro aspetto che abbiamo studiato è stata l’incapsidazione del DNA.
Il lavoro di tesi è stato eseguito sul ceppo di lievito aploide, RSY12, e sul ceppo diploide, BY4743. In laboratorio era stato costruito in precedenza il ceppo RSY12 contenente il gene Rep68 integrato nel genoma codificante per le proteine di replicazione di AAV, che è stato trasformato con il plasmide pESCVP2,3VP1KM, per l’espressione di Vp1, Vp2, Vp3. Il ceppo diploide, ingegnerizzato allo stesso modo dell’aploide, è stato costruito durante questo lavoro di tesi al fine di avere un ceppo di lievito in grado di crescere più velocemente dell’aploide. Analisi mediante Western blot ha mostrato che l’espressione di Vp1, 2 e 3 avviene nella giusta proporzione come avviene nell’aploide.
Per determinare la localizzazione intracellulare di Vp1, 2 e 3 nel ceppo RSY12, è stato messo a punto uno specifico protocollo di frazionamento che permette di caratterizzare la ripartizione delle proteine investigate nei vari compartimenti subcellulari. Analisi mediante Western blot delle proteine dei compartimenti
lievito. Per verificare l’effettiva formazione di capsidi virali in vivo, smentendo ogni possibile dubbio sul fatto che questi si potessero formare durante le procedure di estrazione proteiche, é stato ideato e sviluppato un sistema Immuno blot che consente lo studio di una cinetica che mette in rapporto la produzione delle proteine virali con la formazione dei capsidi interi attraverso ibridazioni con anticorpi che riconoscono discriminatamente epitopi delle proteine libere ed epitopi delle proteine incapsidate. I risultati finora ottenuti su entrambi i ceppi indicano che con questo sistema è possibile capire se le cellule esprimono Vp1, 2 e 3 e se le tre proteine si assemblano correttamente.
I due ceppi di lievito sono stati trasformati con il plasmide pAAVpokURA contente il marker di selezione URA3 fiancheggiato dalle ITR del virus. Dai cloni ottenuti sono state purificate le VLPs ed è stata analizzata la presenza del ssDNA all’interno dei capsidi.
Abstract
The starting point of this thesis project is based on studies carried out in the yeast S. cerevisiae as a model for the production of vectors derived from Adeno Associated Virus (AAV) for gene therapy. AAV is a defective virus, non-pathogenic, belonging to the parvovirus family of Dependovirus because its productive replication depends on co-infection with a helper virus (such as adenovirus or Herpes simplex). The AAV genome consists of a single-stranded DNA containing two open reading frames (ORF) flanked by two inverted repeated sequences such ITR. The two ORF encodes the Rep proteins, Rep40, 52, 68 and 78, necessary for the replication and Cap proteins, Vp1, 2 and 3, necessary for the formation of the capsid. In the laboratory, it has been shown that the yeast is able to support the replication of recombinant AAV vectors (rAAV) and the formation of capsids. The purpose of this thesis is therefore to further characterize the formation of capsids going to study the localization of capsid proteins Vp1, Vp2 and Vp3, and developing a new and easy method of investigation that reveals the production of viral vectors in S. cerevisiae. Another aspect that we have studied has been the encapsidation of the DNA. The thesis work has been performed on the haploid yeast strain, RSY12, and the diploid strain, BY4743. The RSY12 strain containing the gene Rep68 integrated into the genome coding for the proteins of replication of AAV, which has been transformed with the plasmid pESCVP2, VP1KM, for the expression of Vp1, Vp2, Vp3 had been previously constructed in laboratory. The diploid strain, engineered in the same way of haploid strain, was constructed during this thesis work in order to obtain a yeast strain able to grow faster than haploid. Analysis by western blot showed that the expression of Vp1, 2 and 3 takes place in the right proportion as occurs in the haploid strain. To determine the intracellular localization of Vp1, 2 and 3 in RSY12 strain, it has been developed a specific fractionation protocol that allows to characterize the distribution of the proteins investigated in various subcellular compartments. Western blot analysis of protein subcellular compartments showed a homogeneous presence of Vp1, Vp2, Vp3 within the yeast. To verify the formation of viral capsids in vivo, denying any possible doubt that these could be formed during protein extraction
the study of kinetics of the expression of the viral proteins and the formation of capsids through the hybridization with antibodies which recognize epitopes of proteins indiscriminately free and epitopes of proteins incapsidate. The results obtained from both strains indicate that with this system it is possible to understand whether the cells express Vp1, 2 and 3 and if the three proteins are assembled correctly. The two yeast strains are transformed with the plasmid pAAVpokURA containing the selection marker URA3 flanked by the ITR. VLPs were purified from the clones obtained and analyzed for the presence of ssDNA into capsids.
1.0 Introduzione
1.1 La terapia Genica
La terapia genica è un approccio innovativo per curare e prevenire malattie (Morgan e Anderson, 1993). In particolare, la terapia genica consiste nel trasferimento di uno o più geni sani in una cellula malata, al fine di curare una patologia causata dall’assenza o dal difetto di uno o più geni (mutati). Le prime applicazioni della terapia genica erano previste per il trattamento di pazienti con malattie ereditarie monogeniche con eredità recessiva (perciò per caratteri mendeliani) come fibrosi cistica, distrofie muscolari, malattie da accumulo lisosomiale, emofilia e molte altre. Successivamente questo approccio è stato esteso anche alle patologie non mendeliane, molto più comuni nella popolazione, come malattie cardiovascolari, tumori (Liu et al., 2010; Mahvi et al., 1997; Okada, 2010; Kawashima e al., 2010), infezioni da HIV (Applegate et al., 2010; Di Giusto et al., 2010; Symonds et al., 2010) e altre patologie in cui non si va a sostituire un gene difettivo ma se ne introduce uno che possa indurre un fenotipo terapeutico. Una volta individuata la funzione genica da ristabilire è importante scegliere l’adeguato oligonucleotide terapeutico da trasfettare nella cellula bersaglio. Gli oligomeri terapeutici possono essere geni codificanti proteine, DNA non codificanti, DNA e RNA decoys etc... Il gene terapeutico oltre ad essere specifico per la cellula bersaglio, deve essere adeguato alla tipologia di terapia genica utilizzata.
Esistono due tipologie di terapia genica, quella delle cellule germinali e quella delle cellule somatiche:
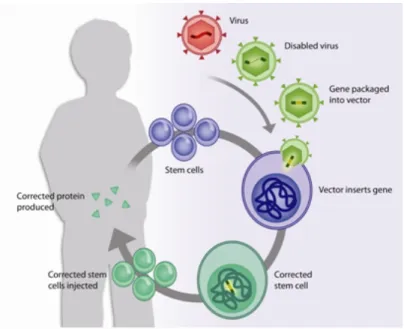
1. La prima strategia presuppone di trasfettare le cellule della linea germinale come spermatozoi ed ovociti o le cellule staminali totipotenti dei primissimi stadi di sviluppo dell’embrione (alla fase di 4-8 cellule), ma attualmente essa non viene messa in pratica sia per ragioni tecniche e, soprattutto, per i grandissimi dilemmi etici che solleva (Fig. 1).
Fig. 1: Terapia genica con cellule staminali.
2. La seconda tipologia invece si propone di modificare solamente le cellule somatiche, senza intaccare quindi la linea germinale; oggigiorno è la via più studiata. La terapia genica delle cellule somatiche, a sua volta, viene suddivisa in due gruppi: la terapia genica ex vivo e quella in vivo (Fig. 2).
La terapia genica ex vivo:
Questa strategia consiste nel prelievo delle cellule somatiche dal paziente per essere messe in coltura in laboratorio e successivamente trasfettate con il gene d’interesse inserito tramite l’apposito vettore, infine vengono reinfuse o reimpiantate nel corpo del soggetto; è una procedura lunga e costosa ma permette di selezionare ed amplificare le cellule d’interesse ottenendo un’ efficienza molto alta del processo.
La terapia genica in vivo:
Questa strategia viene applicata in tutti quei casi in cui le colture non possono essere messe in coltura o prelevate e reimpiantate, come le cellule del sistema nervoso o dell’apparato cardio-respiratorio e della maggior parte degli organi interni. In questo caso il gene, o l’oligonucleotide d’interesse viene inserito nell’organismo tramite un opportuno vettore direttamente per via locale o sistemica. Questo modello terapeutico possiede un’elevata “compliance” ed è molto economico, tuttavia attualmente è di difficile applicazione (Giacca M. 2010).
Fig. 2: Rappresentazione schematica terapia genica in vivo & ex vivo.
Un aspetto fondamentale nell’attuazione della terapia genica è la scelta del metodo di trasferimento del materiale genetico terapeutico all’interno delle cellule bersaglio attraverso l’uso di vettori. I vettori usati devono essere selettivi, entrare facilmente nelle cellule target, e permettere l’espressione del transgene in modo opportuno. Esistono due tipi di vettori per il trasferimento: non virali e virali (Boulaiz et al., 205; Kay 2011).
Il trasferimento genico non virale:
Le metodologie adottate per trasferire il DNA nelle cellule senza ricorrere a virus comprendono: l’iniezione di DNA nudo, la tecnica di elettroporazione, l’inserimento tramite liposomi, l’inserimento attraverso l’uso di polimeri cationici ed il bombardamento tramite particelle (Gene gun) (Ledley, 1994).
L’iniezione di DNA nudo è la procedura più lineare e più semplice ed inoltre permette di trasferire costrutti genici di grandi dimensioni. Consiste nell’iniettare il gene terapeutico sotto forma di plasmide direttamente nella cellula mediante l’utilizzo di una micropipetta. Lo svantaggio di questa tecnica risiede nel fatto che l’iniezione deve essere effettuata singolarmente per ogni cellula ed il rendimento della metodica risulta piuttosto basso (Sloane et al., 2009).
L’elettroporazione consiste nell’inserimento di molecole di DNA all’interno delle cellule attraverso i pori della membrana cellulare che vengono aperti, per un brevissimo tempo mediante piccole e ripetute scariche elettriche. Anche se la tecnica ha un’alta efficienza di trafeszione molte cellule non sopravvivono al trattamento (Chang 1991).
I liposomi sono vescicole sferiche la cui parete è composta da un doppio strato fosfolipidico; usando liposomi cationici è possibile far complessare ad essi il DNA, che a pH fisiologico presenta una carica negativa. Il complesso DNA-liposoma può fondersi con la membrana cellulare ma nella maggior parte dei casi viene internalizzato tramite endocitosi. Questo processo è a bassa efficienza in quanto si è visto che solo lo 0,1% del DNA introdotto viene espresso. Per ovviare a ciò nei liposomi sono stati inseriti altri elementi come proteine ed anticorpi che possano aumentare l’efficacia della procedura minimizzando la degradazione del DNA e facilitando il corretto direzionamento della vescicola.
L’uso dei polimeri cationici è molto simile all’utilizzo dei liposomi cationici. I polimeri cationici sono dotati di molteplici cariche positive che interagiscono con il DNA provocandone la condensazione e proteggendolo nella traslocazione verso il nucleo. I complessi DNA-policatione vengono internalizzati dalla cellula per endocitosi e possono essere attivamente indirizzati verso specifiche linee cellulari o tessuti utilizzando anticorpi o altre molecole direzionali.
Il gene gun prevede l’utilizzo di particolari strumenti elettrici o ad alta pressione, dette pistole geniche che permettono di inviare nella cellula particelle microscopiche d‘oro o di tungsteno ricoperte di DNA. Al momento non esistono studi sull’uomo di questa metodica ma solo su animali (Li et al., 2008; Li et al., 2009).
Vantaggi dei vettori non virali:
- Possibilità di trasferire diversi tipi di molecole e molecole di DNA molto grandi; - Certezza di non generare nuovi virus patogeni;
- Riduzione del rischio di reazione immunitaria;
Svantaggi dei vettori non virali:
- Scarsa efficienza sia di trasduzione sia di integrazione che non consentono effetti duraturi;
- Se integrati possono a loro volta dare fenomeni di mutagenesi inserzionale.
Il trasferimento genico virale:
Questa tecnica si basa sull’utilizzo di opportuni virus ricombinanti e sfrutta la loro capacità di infettare diversi tipi di cellule e di veicolare il DNA che contengono all’interno della cellula. Rispetto ai sistemi di trasferimento non virali, hanno un’efficienza nettamente maggiore. Tuttavia i virus che possono essere utilizzati devono avere alcune caratteristiche:
- Le particelle virali ricombinanti devono essere difettive rispetto alla replicazione; - I virus non devono possedere alcune qualità indesiderate, quali la capacità di
produrre dei composti tossici o l’attivazione del sistema immunitario;
- Le dimensioni spaziali devono essere sufficienti all’inserzione del gene terapeutico;
I virus attualmente utilizzati come vettori per la terapia genica sono: • I retrovirus
• I lentivirus • Gli adenovirus
• I virus adeno-associati • Gli herpesvirus
I retrovirus hanno la capacità d’inserire il gene terapeutico in modo stabile nel genoma dell’ospite attraverso un’integrazione random, possiedono un ampio tropismo d’infettività ed una relativa facilità di manipolazione del genoma virale. Tuttavia questi virus riescono ad infettare soltanto le cellule in divisione.
I lentivirus, appartenenti alla famiglia dei retrovirus, possiedono un’elevata capacità d’inserimento nel genoma dell’ospite, infatti riescono ad infettare sia le cellule in divisione che quelle non in divisione. Tuttavia sono difficili da produrre e sono considerati potenziali oncogeni perché s’integrano in maniera random nel genoma
Gli adenovirus sono considerati sicuri (non integrano il loro genoma), facilmente manipolabili, stabili, e sono capaci di infettare anche cellule quiescenti. Un importante vantaggio dei vettori basati su adenovirus è la capacità di veicolare inserti di grosse dimensioni (36 Kb) e possono essere prodotti con alti titoli. Purtroppo la loro espressione è transiente e inducono un’alta risposta immunitaria.
I virus adeno-associati possiedono i seguenti vantaggi: integrazione sito specifica ed espressione stabile nel tempo, infettano le cellule in divisione e non, sono stabili, hanno un ampio tropismo cellulare, bassa immunogenicità e non patogenicità. Tuttavia l’uso di questi virus come vettori per la terapia genica è limitato da alcuni svantaggi: limitato spazio di inserimento per i transgeni (4,7 Kb), difficile generazione di alti titoli virali, il vettore ricombinante inoltre non ha sempre un’integrazione sito specifica e talvolta resta in forma episomiale.
Gli herpes simplex virus, grazie alla loro naturale capacità di stabilire infezioni latenti nei neuroni, vengono utilizzati per il trasporto di geni nel Sistema Nervoso Centrale, e inoltre possono trasportare geni di grandi dimensioni; ma possono indurre importanti effetti citotossici.
L’effettiva importanza della terapia genica
Le sperimentazioni cliniche della terapia genica sono ufficialmente iniziate nel 1990. Il gruppo di ricerca del Dott. F.W. Anderson tentò di curare un paziente, afflitto da un difetto enzimatico che causava una grande immunodeficienza, utilizzando una terapia genica; il tentativo però non ottenne il risultato sperato (Blaese R.M. et al., 1995). Oggi si sa che il vettore che venne utilizzato per il trasferimento genico non può garantire un’espressione del gene trasferito persistente nel tempo, di conseguenza si ha una spiegazione razionale della ragione del fallimento di questo tentativo. In seguito sono stati creati vettori più efficienti e sono state condotte nuove sperimentazioni cliniche. Si segnala che la maggior parte di questi studi sono stati eseguiti su animali, alcuni sono trial clinici in fase 3 (inferiore al 4%) e molti non superano la fase 1 in cui lo scopo della ricerca non è quello di studiare gli effetti terapeutici di un trasferimento genico, ma quello di testarne la tossicità. Più del 70% di questi studi sono effettuati utilizzando vettori virali, una chiara indicazione che
questi sistemi di distribuzione sono considerati migliori degli altri metodi non virali (Vannucci et al 2013).
In particolare, tra i vettori virali, i virus adeno-associati grazie ai propri vantaggi risultano essere i migliori candidati. Essi presentano uno specifico tropismo per le cellule quiescenti, in particolare per le cellule muscolari lisce, striate e cardiache, per gli epatociti, per le cellule neuronali della retina e del sistema nervoso centrale. La capacità di trasdurre efficacemente nel tessuto muscolare scheletrico (Clark et al., 2002; Fischer et al., 1997; Snyder et al., 1997) è stata sfruttata per il trattamento dell’emofilia B. Ad oggi i virus adeno-associati vantano circa 100 sperimentazioni di terapia genica per lo più per la cura di malattie monogenetiche (Grieger e Samulski2012). L’ambito più studiato è quello relativo alle malattie genetiche, grande interesse è rivolto anche all’ambito delle malattie neurodegenerative (Ad es. distrofia muscolare di Duchenne), malattie cardiovascolari, tumori e a seguire una serie di altri campi dove la terapia genica cerca un utilizzo.
Guardando indietro, il percorso della terapia genica è lastricato di successi, ma anche d’incidenti gravi, e citando il famoso editoriale Nature “la terapia genica ha da tempo perso la sua innocenza” (Hollon, 2000). Tuttavia ad oggi si è ben consapevoli dei progressi fatti in termini di sicurezza e tollerabilità dei vettori virali; una prova è arrivata a Giugno scorso quando l’agenzia europea per i medicinali ha raccomandato l’approvazione di un farmaco terapico-genico basato su un vettore adeno-associato per il trattamento di una rara malattia genetica ereditaria, la deficienza della lipoproteina lipasi (Gaudet et al., 2012).
1.2 Il virus Adeno-associato di tipo 2 (AAV2)
Il virus Adeno-associato è un piccolo virus di dimensioni di 18-30 nm costituito da un capside icosaedrico privo d’involucro lipidico contenente DNA a singolo filamento di 4,7 Kb. I virus Adeno-associati appartengono alla famiglia dei Parvovirus che sono tra i più piccoli virus identificati in natura, ed al genere Dependovirus. I Parvovirus sono agenti patogeni e sono capaci di replicarsi autonomamente nella cellula ospite; contrariamente i Dependovirus sono non-patogeni e non essendo in grado di replicarsi autonomamente dipendono dalla co-infezione di un “Helper Virus”, che può essere un adenovirus (Ad), un herpes simplex virus (HSV) o un papilloma virus (HPV) (Geoffroy et al., 2005). Adenovirus (Ad) per esempio, si pensa che sia il virus helper naturale dell’AAV, in quanto AAV è stato spesso trovato come contaminante delle colture di Ad (Ni et al., 1998). Nonostante l’alta siero-prevalenza di AAV nella popolazione umana (approssimativamente l’80% è positivo per AAV2) il virus non è stato collegato a nessuna patologia umana, infatti non è in grado di provocare malattie, ma causa solamente una leggera risposta immunitaria. Ad oggi oltre 100 varianti sono state isolate da colture cellulari provenienti da tessuti umani e non umani, e circa 14 sierotipi sono stati ben caratterizzati. Tutti i sierotipi conosciuti possono infettare le cellule provenienti da molteplici tipi di tessuti. La specificità di tessuto è determinata anche dal tipo di capside. Questa caratteristica insieme ad altre guida la scelta del vettore adeno-associato più specifico da utilizzare in terapia genica. Ad oggi non è ancora possibile definire quale sia il sierotipo specifico per ogni tessuto, poiché si continua ad isolarne sempre di nuovi (Schultz et al., 2008). Un vettore genico contenente le ITRs di un certo sierotipo può essere impacchettato all’interno di un capside di un altro sierotipo, producendo un vettore virale ricombinante di pseudo-tipo (Rabinowitz et al., 2004). Ovviamente dopo la costruzione del primo vettore nel 1984, i vettori virali di AAV2 hanno suscitato grande interesse per l’impiego in terapia genica grazie alla loro mancanza di patogenicità, alla larga gamma di specie che sono capaci di infettare e alla loro capacità di stabilire un’espressione transgenica a lungo termine (Berns, 1990; Carter, 2004; Wu, 2006).
Il sierotipo 2:
Il sierotipo più studiato è il sierotipo 2 (AAV2). Le sue caratteristiche lo rendono un ottimo candidato per la creazione di vettori virali: studi recenti hanno dimostrato che il sierotipo 2 (AAV2) riesce a discriminare le cellule tumorali da quelle sane, uccidendo selettivamente solo quelle malate.
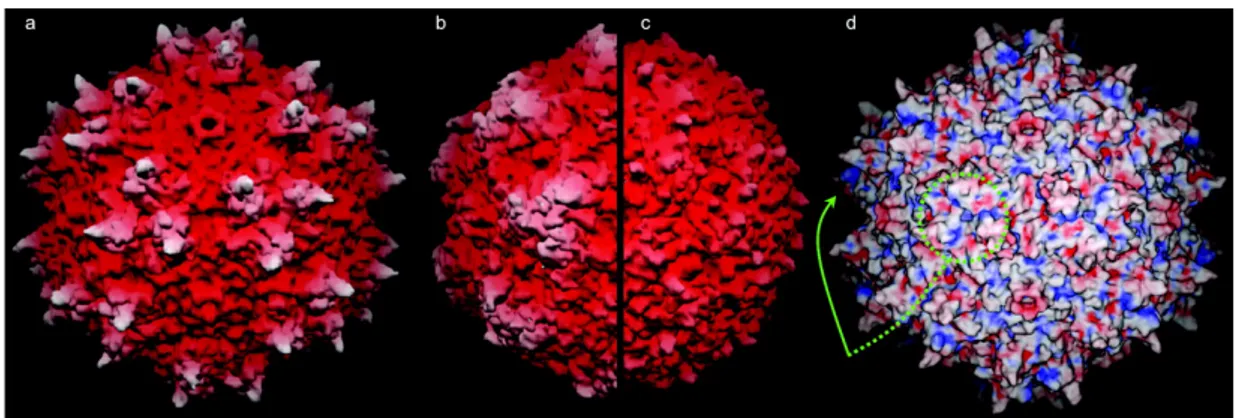
La superficie di AAV2 (Fig. 3) ha una topologia più caratteristica di quella del Parvovirus o del Densovirus. Le caratteristiche più importanti sono il “triplice clustering” dei picchi intorno ad ogni asse di rotazione della struttura icosaedrica; al centro di ogni picco è presente un Subloop della proteina capsidica VP2 (residui 348-379) ed una serie di loop di subunità vicine interagiscono intimamente. Le strutture che formano queste interazioni tra le subunità vicine sono uniche per AAV2 (Qing Xie et al., 2002).
Fig. 3: La tolopologia superficiale colorata in base alla distanza dal centro virale, ed il
potenziale elettrostatico di superficie indicando che il recettore putativo dei siti di legame è carico positivamente. Fonte (Qing Xie et al., 2002).
1.2.1 Il ciclo vitale e d’infezione del Virus Adeno-associato
Il ciclo vitale di AAV presenta diverse fasi d’infezione. Per prima cosa le particelle di AAV devono attaccarsi alla membrana cellulare della cellula che stanno infettando prendendo contatti multipli con recettori di membrana glicoproteici (HSPG) ed altri co-recettori necessari sia per l’internalizzazione che per gli eventi successivi di processamento (Ad esempio alcuni co-recettori indicati sono: l’integrina αvβ5, il fattore di crescita dei fibroblasti, il fattore di crescita degli epatociti per il siero AAV2; 2,3 O-like acido sialico per AAV4; 2,3 N-like acido sialico e recettore del fattore di crescita delle piastrine per AAV5; 2,3 e 2,6 N-like acido sialico per AAV1 e AAV6; (Goncalves, 2005; Stieger, 2009; Summerford, 1999). È proprio grazie a questa diversità dei recettori cellulari in grado di riconoscere e legare particelle virali che è possibile un’infezione da parte di AAV su una così ampia varietà di tipi cellulari (Summerford and Samulski, 1998).
All’interno della cellula le particelle virali vengono processate ed infine sono veicolate all’interno del nucleo; il trasporto all’interno del nucleo avviene grazie alle componenti proteiche del capside che inducendo cambi conformazionali riescono a sfruttare un meccanismo di endocitosi interno per passare da un compartimento cellulare all’altro. Si ritiene che l’ambiente fortemente acido dell’endosoma provochi un cambiamento conformazionale del capside che porta all’esposizione di proteine segnale di localizzazione nucleare come la fosfolipasi A2 (Girod et al., 2002). Le particelle virali una volta rilasciate dall’endosoma possono essere degradate dal proteosoma o finalmente entrare nel nucleo.
Fig. 4: Schema infezione di AAV (Shultz and Chamberlain, 2008).
Quando la particella virale è nel nucleo si formano gli intermedi di replicazione a doppio filamento di DNA virale. Le proteine Rep (Rep78, Rep68, Rep52, Rep40) che sono coinvolte nella replicazione del DNA sono le prime ad essere tradotte, successivamente vengono codificate dal gene Cap le tre proteine che compongono il capside (Vp1, Vp2, Vp3). Quando tutte le componenti virali sono presenti nello stesso ambiente nucleolare (dove avviene l’evento di packaging) in concentrazione opportuna avviene la sintesi della progenie in particelle contenenti DNA a singolo filamento e quindi l’assemblaggio completo dei virioni; infine a questo segue il rilascio delle particelle virali complete dalla cellula infettata.
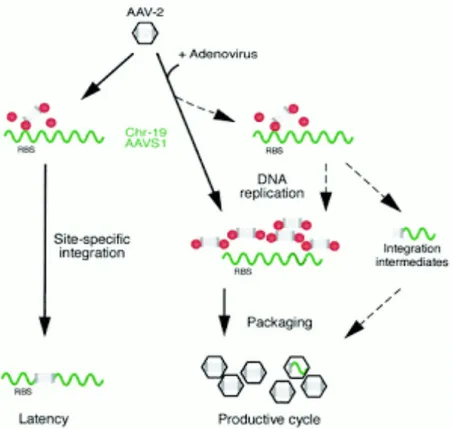
Tuttavia il ciclo vitale di AAV è un ciclo bifasico, ovvero una volta che il virus è all’interno del nucleo può andare incontro ad una fase latente o litica (Fig. 5); ciò dipende dallo stato fisiologico della cellula e se l’ambiente permette una corretta replicazione di AAV.
Fig. 5: Rappresentazione schematica del ciclo bifasico di AAV.
Il virus AAV va incontro alla fase litica, ovvero la fase durante la quale si replica, solo quando la cellula viene infettata sia da AAV che da un “virus helper”
(Figura 5 identificato come Adenovirus).
L’infezione da parte di un virus helper e stress genotossici causati da diversi agenti chimico/fisici sono fattori che creano un ambiente molto permissivo per la replicazione di AAV all’interno della cellula ospite. La conversione del genoma virale a singolo filamento (ss) nella forma trascrizionalmente attiva a doppio filamento (ds) avviene secondo un meccanismo detto “Strand displacement” (Fig. 6) grazie alla struttura ad hairpin delle ITRs che fungono come stampo per la replicazione. A partire dal 3’OH inizia la sintesi del filamento complementare per azione delle polimerasi della cellula ospite, si forma una molecola a doppio filamento in cui una delle due terminazioni è covalentemente unita (Goncalver, 2005). A questo punto è necessaria una reazione di taglio da parte di Rep78 o Rep68 a livello della sequenza
trs (terminal resolution site). Il DNA che si forma rappresenta un intermedio che può
ciclo di replicazione (Hong et al., 1994). Questo processo è chiamato “risoluzione terminale”, ed è un meccanismo unico usato dalla famiglia dei Parvovirus per mantenere l’integrità delle sequenze terminali (Goncalves, 2005). In caso di mancato taglio della sequenza trs l’allungamento procede formando un dimero che può avere una configurazione sia testa-testa che testa-coda. Avvenuta la sua replicazione il dsDNA si separa di nuovo e forma ssDNA con le ITRs alle estremità (le proteine Rep40/52 sono coinvolte in questo processo), le nuove copie sono impacchettate nei capsidi virali oppure nuovamente replicate. Tuttavia si possono però formare anche delle strutture intermedie di dsDNA, ad esempio nel caso in cui la forma dsDNA persiste nel nucleo può verificarsi la formazione di concatenameri lineari o circolari dovuti dall’unione di due o più monomeri (Schultz and Chamberlain 2008).
Fig. 6: Schema della replicazione di AAV.
Infine la fase litica prevede che il virus così replicato venga assemblato nelle particelle virali che daranno origine alla lisi cellulare ed al rilascio di un altissimo numero di particelle virali (Berns et al., 1995; Agrawal et al., 2002; Mori et al., 2002).
AAV va incontro alla fase latente quando infetta la cellula in assenza dell’helper. In queste condizioni il genoma di AAV non viene replicato ma è integrato
nel genoma della cellula ospite in uno specifico locus sul cromosoma 19 (Fig. 7), grazie all’azione delle proteine Rep con un meccanismo ancora non del tutto noto (Huser et al., 2010; Huser et al., 2003; Huser and Heilbronn, 2003; Surosky et al., 1997; Laughlin et al., 1986).
1.2.2 Il Genoma di AAV2
Il genoma di AAV è composto da DNA a singolo filamento lungo circa 4,7 Kb ed è organizzato in due grandi “open reading frames” (ORF), Rep e Cap, in parte sovrapposte (Fig. 8), fiancheggiate da due sequenze terminali ripetute ed invertite, dette ITR (inverted terminal repeat). Le ITRs formano una struttura secondaria a forma di T e sono necessarie per regolare la replicazione, l’integrazione e l’incapsidamento del virus (Mansilla-soto et al., 2009)
Fig. 8: Rappresentazione del genoma di AAV e dei prodotti proteici.
Fonte: http://hvd.ens-lyon.fr
Il gene Rep codifica per una famiglia di quattro proteine non strutturali con regioni
codificanti sovrapposte (Rep78, Rep68, Rep52 e Rep40), che partono da due promotori, mappati alle posizioni 5 e 19, chiamati p5 e p19 (Cervelli, 2011; Glauser et al., 2005; Goncalves, 2005). È stato dimostrato che tutte e quattro le proteine Rep sono capaci di legare ATP e di possedere attività elicasica (Zhou et al., 1999; Brister and Muzycza, 1999; Wu et al., 1999).
Rep78 e Rep68, sono proteine tradotte da un unico mRNA trascritto a partire dal
interfacce di riconoscimento che sono RBE (Rep Binding Element) e trs (Terminal Resolution Site) (Fig. 9). Rep68 e Rep78 sono inoltre necessarie per l’integrazione sito-specifica, per il metabolismo del DNA e per la regolazione trascrizionale dei promotori virali e cellulari (Dutheil et al., 2000; Kotin et al., 1990; Surosky, 1997; Ward, 1994; Weitzman, 1994; Van Vliet et al., 2008).
Rep52 e Rep40, sono proteine tradotte da un unico mRNA trascritto a partire dal
promotore p19, sono più piccole di Rep78 e Rep68, e sono coinvolte nella formazione e nell’accumulo del genoma virale a singolo filamento di DNA a partire dall’intermedio replicativo a doppio filamento (Van Vliet et al., 2008). In particolare, le proteine Rep52 e Rep40 sono richieste per la traslocazione del DNA a singolo filamento nei capsidi preformati vuoti; la funzione elicasica svolta da Rep52 e Rep40 ha una direzione 3’-5’ ed è essenziale per l’inserimento del genoma nel capside che avviene appunto in direzione 3’-5’ (King JA et al., 2001). Un modello proposto per l’inserimento del genoma, è che il capside sia immobilizzato da un complesso contenente l’elicasi e che un motore molecolare inserisca il DNA a singolo filamento dentro il capside formato. È stato dimostrato che Rep52 ha un’attività ATPasica costitutiva che è attiva in assenza delle molecole di DNA (Smith RH et al., 1998).
Studi in vitro hanno dimostrato che sia Rep78 che Rep68 da sola se over-espressa è sufficiente per la replicazione del DNA di AAV; Rep52 e Rep40 non sono richieste per la replicazione ma migliorano l’efficienza di accumulo della progenie del DNA a singolo filamento (HÖlscher C. et al., 1995). I pre-mRNA dei geni Rep sono soggetti a splicing alternativo e questa è la ragione per cui l’estremità N terminale delle proteine Rep differiscono. In presenza di “virus helper”, Rep78 e Rep68 sono richieste per la replicazione del DNA di AAV, il controllo trascrizionale, lo splicing alternativo di AAV, il packaging del DNA virale e l’integrazione sito-specifica nel cromosoma 19 in cellule umane (Im and Muzyczka, 1990; Stracker et al., 2004). È stato dimostrato anche che l’espressione delle proteine Rep inibisce la propagazione di un virus ibrido Adenovirus/Simian virus40, Papillomavirus bovino, virus umano dell’immunodeficienza e herpesvirus; inibisce la trascrizione e arresta le cellule nella fase S (Labow and Berns, 1988; Marcello et al., 2000). Sembra che la proteina Rep interagisca con una varietà di proteine cellulari e del “virus helper” anche se il
significato funzionale di queste interazioni non è ancora chiaro (Nash et al., 2009). Come succede per altri virus a DNA, si ritiene che AAV interagisca o modifichi le proteine della cellula ospite per svolgere il suo ciclo d’infezione.
Il gene Cap codifica per tre proteine strutturali che compongono il capside: Vp1 (87
KDa), Vp2 (73 KDa) e Vp3 (62 KDa) dal promotore mappato in posizione 40 (p40). Tutte e tre le proteine sono tradotte da un unico pre-mRNA. Dopo che questo viene sintetizzato, può avvenire lo splicing alternativo in due modi diversi dando origine a due trascritti di lunghezza differente:
§ Un trascritto lungo 2,6 kb che possiede il canonico AUG come sito d’inizio della traduzione e codifica per la proteina virale Vp1;
§ Un trascritto lungo 2,3 kb che possiede un codone d’inizio non canonico ACG che viene usato per tradurre Vp2 anche se con bassa efficienza, e un dominio canonico AUG che guida la sintesi molto efficiente di Vp3 favorita anche dalla presenza di una regione Kozak. È stato anche dimostrato che nello splicing è favorita la formazione del prodotto più piccolo (Becerra et al., 1988; Chapman et al., 1993; Van Vliet et al., 2008).
Le tre proteine strutturali del capside differiscono solo nella porzione N-terminale e si assemblano formando una struttura a simmetria icosaedrica T1 costituita da circa 60 subunità. Un ruolo fondamentale è rivestito dal rapporto delle tre proteine che compongono il capside per favorire un corretto packaging e una corretta infettività del virus. Il rapporto tra le proteine della particella virale matura è circa 1:1:10, mentre il rapporto di espressione oscilla tra 1:1:8 e 1:1:20 rispettivamente per Vp1, Vp2 e Vp3 (Girod, 2002; Goncalves, 2005; Van Vliet, et al., 2008). Recentemente, è stato identificato un nuovo prodotto genico codificante per una proteina regolatoria non strutturale: AAP “assembly-activating protein” localizzato in una porzione a monte del codone d’inizio che induce la traduzione di Vp3. AAP sembra promuovere il trasporto delle Vps al nucleolo, cioè nel sito cellulare dove poi si formerà il capside (Sonntag et al., 2010).
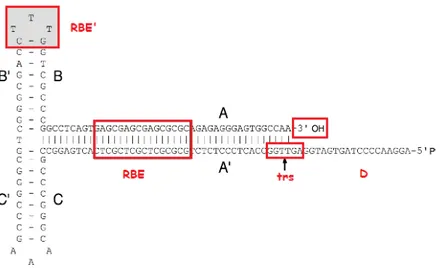
Le ITRs (inverted terminal repeats) comprendono 145 basi ciascuna, ma solo 125
hanno struttura palindromica. La denominazione ITR deriva dalla loro simmetria, che è necessaria affinché il genoma di AAV si possa replicare. Queste sequenze hanno la caratteristica di formare una struttura secondaria a forcina, permettendo la sintesi del secondo filamento di DNA. La struttura a forcina fornisce il gruppo idrossilico al 3’ necessario per dare il via alla replicazione virale e quindi, svolge sia la funzione di origine di replicazione che di primer per la sintesi del secondo filamento da parte delle DNA polimerasi della cellula ospite. Gli ultimi 20 nucleotidi costituiscono la sequenza “D” a singolo filamento e ospitano un sito di legame per la proteina Rep68, detto RBS “Rep binding site”, ed un sito trs (terminal resolution site) dove avverrà il taglio da parte della proteina Rep68 durante la replicazione (Wang et al., 1997; Wang and Srivastava, 1997; Young et al., 2001).
Fig. 9: Organizzazione ITR (Daya and Berns, 2008).
È stato dimostrato che le ITRs contengono in cis tutte le sequenze necessarie per la replicazione, per il packaging e per l’integrazione del DNA di AAV nel genoma della cellula ospite e successivo mantenimento dello stato integrato (Yang et al., 1997).
1.2.3 Regolazione e localizzazione delle componenti virali in
relazione alla formazione delle VLPs
Quando inizia l’assemblaggio del capside, una grande quantità di proteine Rep e di copie del genoma sono già disponibili per il packaging (Gallo-Ramirez et al., 2011). Gli studi riguardanti l’assemblaggio del capside di AAV2 sono stati effettuati prevalentemente in due sistemi cellulari eucariotici evolutivamente lontani: le cellule di mammifero che rappresentano la piattaforma tradizionale per la produzione di AAV, e le cellule d’insetto grazie ad un recente sistema d’espressione sviluppato mediante l’infezione con Baculo virus (Meghrous J. et al., 2005). In entrambi i casi, il processo di assemblaggio delle particelle virali (virus like particles: VLPs) necessita soltanto dell’espressione del gene Cap e di nessun altro gene virale (Ruffing et al., 1992; Wistuba et al., 1997). Tuttavia la produzione di vettori di AAV è un processo complesso che richiede l’interazione di tutte le componenti virali: i capsidi, le proteine Rep e il genoma ricombinante. È necessario quindi che tutte le componenti co-localizzino per il packaging del genoma nei capsidi correttamente assemblati.
Per cercare di definire i requisiti minimi per la produzione delle VLPs, le tre proteine capsidiche sono state espresse separatamente e in diverse combinazioni, ed è stata investigata la loro capacità di assemblare il capside includendo studi in vivo e in vitro (Steinbach et al., 1997). Questi studi hanno portato a capire che:
Vp1 non è essenziale per la formazione del capside, ma è necessaria per garantire l’infettività virale (Girod., 2002; Goncalves.,2005). Ciò è stato successivamente spiegato grazie alle sequenze di localizzazione nucleare/NLS (detti Basic clusters, BCs) necessarie per il rilascio dall’endosoma al compartimento nucleare e per l’avvio dell’espressione genica, ma anche grazie alla scoperta di un motivo conservato con attività fosfolipasica A2 (PLA2) identificato nella porzione N-terminale di Vp1.
Anche Vp2 come Vp1 presenta due BC elements, sebbene con un ruolo molto ridotto di localizzazione nucleare (Sonntag et al., 2006); infatti uno studio di mutagenesi effettuato su Vp2 identifica una regione specifica localizzata all’N-terminale necessaria al trasferimento nucleare, che non è strutturalmente richiesta per la formazione di VLP indicando che Vp2 è necessaria soltanto per il trasferimento nucleare delle proteine capsidiche (Hoque M. et al., 1999).
Quindi Vp1 o Vp2 possono essere essenziali per l’assemblaggio del capside grazie alla loro abilità di trasportare Vp3 nel nucleolo, la quale costituisce la topologia della superficie di AAV e riesce autonomamente a formare capsidi; tuttavia studi recenti hanno mostrato che Vp3 non è sufficiente per la formazione di un capside corretto, ma è fortemente richiesta l’azione regolatoria della proteina AAP (Sonntag et al., 2010).
Precedenti studi hanno rivelato che tutte e quattro le proteine Rep co-localizzano nel nucleo delle cellule infettate suggerendo che i centri di replicazione per l’AAV sono simili a quelli esistenti per altri virus (Hunter et al., 1992). Inoltre è stato dimostrato che l’assemblaggio dei capsidi di AAV nelle cellule di mammifero avviene nel nucleolo e richiede la traslocazione delle tre proteine strutturali del virus nel nucleo. Un precedente studio ha dimostrato l’esistenza di tre pool distinti nel compartimento nucleare dove si accumulano la maggioranza delle proteine Rep e Cap: uno dove risiedono i monomeri, uno che presenta gli intermedi di assemblaggio e un altro che contiene i capsidi assemblati. I primi due pool contengono prevalentemente le due proteine capsidiche Vp1 e Vp2 mentre l’ultimo pool contiene tutte e tre le Vps in una stechiometria simile a quella delle VLPs purificate. La frazione citoplasmatica non presenta questi tre pool definiti, mostra invece una diffusione continua delle proteine capsidiche (Wistuba et al., 1995).
v Anche in cellule d’insetto come in cellule di mammifero la distribuzione intracellulare delle componenti virali riveste un ruolo chiave nel processo di formazione delle VLPs. La presenza del Baculo virus usato come vettore induce cambiamenti nell’ambiente intracellulare a livello nucleare, modificando così le interazioni spaziali e temporali delle componenti virali. Uno studio effettuato recentemente sulle cellule d’insetto mostra il profilo di localizzazione delle proteine virali di AAV analizzate individualmente (Gallo-Ramirez et al., 2011):
La distribuzione delle Vps non assemblate espresse individualmente dipende dalla propria concentrazione intracellulare, infatti inizialmente si accumulano prevalentemente nel citoplasma e quando la concentrazione aumenta si osserva la presenza delle Vps non assemblate anche nel nucleo. Inoltre le Vps assemblate son state rivelate esclusivamente nei nuclei in cluster ben definiti, indicando che anche per le cellule d’insetto il nucleolo rappresenta la zona di assemblaggio delle Vps.
Anche in questo sistema la localizzazione delle quattro proteine di Rep ha un profilo nucleare, tuttavia la localizzazione esatta all’interno del nucleo dipende finemente dal tempo d’infezione con il Baculovirus (Gallo-Ramirez et al., 2011). È stato osservato che l’espressione e l’accumulo delle Vps nel nucleo non influenza la localizzazione intracellulare delle proteine Rep e che la presenza del genoma ricombinante di AAV non modifica la loro distribuzione, ma ne aumenta la stabilità nel tempo. Anche l’espressione delle Vps aumenta la stabilità delle proteine Rep. Uno Studio di microscopia elettronica in cellule d’insetto ha dimostrato che i capsidi vuoti si accumulano in compartimenti ben definiti nella regione nucleare centrale, mentre quando sono presenti tutte le componenti virali i capsidi si assemblano in piccoli cluster localizzati nella periferia nucleare (Gallo-Ramirez 2011).
1.2.4 Incapsidazione del DNA nei virus Adeno-associati
Capire la dinamica dei meccanismi d’incapsidazione del DNA è fondamentale non solo per comprendere i fattori implicati in questo processo ed avere più chiara la parte finale del ciclo litico, ma anche per riuscire a migliorare la produzione di vettori virali da usare in terapia genica.
Per il processo d’incapsidazione del virus AAV-2 non avviene un’interazione diretta del genoma con il capside come dimostrato per il parvovirus MVM (virus minuto di topi) (Willwand e Hirt, 1993). Il meccanismo di assemblaggio proposto per AAV prevede due fasi diverse (Myers and Carter, 1980). Inizialmente si formano i capsidi vuoti e dopo vengono riempiti col genoma single strand di AAV, generato durante il ciclo di replicazione del virus. È stato osservato che durante le fasi iniziali di un’infezione produttiva, l’assemblaggio del capside e la replicazione del DNA virale sono eventi spazialmente e temporalmente distinti che avvengono in siti specifici all’interno del nucleo della cellula. Successivamente le proteine Rep e il genoma di AAV sembrano essere co-distribuiti in tutto il citoplasma (Wistuba et al., 1997). Le proteine Rep rivestono un ruolo fondamentale per il processo di packaging dei virus adeno-associati. Come già spiegato, mentre le proteine Rep68 e Rep78 coordinano l’inizio della replicazione legando covalentemente i Terminal Resolution Site (trs), posti nelle ITRs del genoma di AAV, le proteine Rep52/40 non sono necessarie per la replicazione del DNA, ma sono coinvolte nella formazione e nell’accumulo del genoma virale a singolo filamento di DNA (Van Vliet et al., 2008). Dubielzig et al. (1999) hanno dimostrato che le quattro proteine Reps interagisco non solo con il capside, ma anche tra loro. Questa osservazione è stata ulteriormente supportata da un altro studio nel quale è stato dimostrato che l’interazione delle proteine Rep68/78 legate al genoma di AAV, con i capsidi vuoti è mediata dal legame con le proteine Rep52 e Rep40 (King JA et al., 2001). In questo studio è stato osservato che i virus prodotti in assenza di proteine Rep52/40 possiedono un titolo infettivo ridotto di 200 volte. Tale risultato non è dovuto ad una diminuzione del processo di formazione del capside, come dimostrato da titolazione dei capsidi assemblati con saggio ELISA, né a differenze nel processo di replicazione del DNA virale, ma è dovuto ad una notevole riduzione (di 20 volte) della quantità di DNA
incapsidato (King JA et al., 2001). Inoltre non è richiesta l’espressione di entrambe le proteine, indicando un certo livello di ridondanza tra la funzione di Rep52 e Rep40. È stato osservato che proteine Rep52/40 con il sito di legame per l’ATP mutato formano complessi con il ssDNA meno efficienti rispetto alle wild type determinando incapsidazione parziale del DNA. Questo avviene perché il genoma è incapsidato fino al punto in cui l’attività elicasica delle proteine Rep52/40 non diventa essenziale per completare il processo d’incapsidazione (King JA et al., 2001).
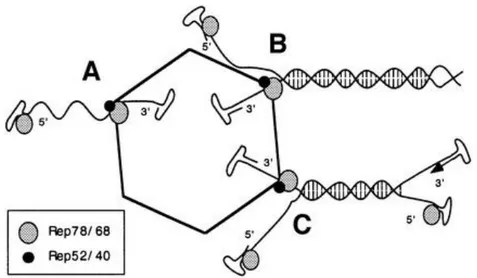
King J. (2001) ha proposto un modello per l’incapsidazione del genoma di AAV guidata dall’attività elicasica di Rep52/40. Come riportato in figura 10 possono manifestarsi tre scenari diversi: nel primo caso (Figura 10-A) il genoma ss libero viene traslocato dal complesso di elicasi. In questo scenario è richiesta soltanto l’attività elicasica per svolgere le ripetizioni terminali. Nel secondo scenario (Figura 10-B), l’attività elicasica di packaging svolge un genoma concatenamerico a doppio filamento sulla superficie del capside e contemporaneamente inserisce l’estremità 3’ di uno dei 2 filamenti singoli nel capside. Nel terzo scenario (figura 10-C): una ITR contentente la parte terminale del monomero a doppio filamento, non coinvolta nella reazione di packaging, rimane aperta consentendo un altro giro di replicazione durante l’incapsidazione.
Fig. 10: Packaging del DNA: I tre possibili scenari (King JA et al., 2001).
superficie del capside non rappresentino la forza motrice per il packaging del DNA, perché se così fosse, la replicazione favorirebbe una direzione di packaging 5’à3’. Anche se è improbabile che questi due eventi siano direttamente collegati, è possibile che il processo di replicazione interferisca con la lunghezza del genoma che viene incapsidato.
Infine è stato visto che Rep78 e Rep68 da sole catalizzano la reazione di packaging fino a 1000 volte meno efficientemente rispetto alla situazione in cui sono presenti anche Rep40 e 52, anche se possiedono le intere regioni di Rep52 e Rep40. È concepibile che il forte legame tra la sequenza di riconoscimento del genoma di AAV e le proteine Rep68/78 impedisca loro di muoversi lungo il genoma in maniera processiva e di consentire l’incapsidazione del DNA.
1.2.5 Produzione di vettori basati sul virus Adeno-associato
Le caratteristiche di AAV lo rendono un ottimo candidato nell’uso come vettore per la terapia genica. Una delle caratteristiche che lo rendono interessante come vettore per la terapia genica è il fatto che il DNA che veicolano dentro la cellula ospite viene mantenuto come episoma ed espresso a lungo termine ed ad alti livelli.
Fig. 11: Gene therapy usando un vettore adeno-associato.
Il primo clonaggio riuscito di AAV, risalente ai primi anni ’80, ha portato a più di due decenni di studi volti a perfezionare le tecnologie ricombinanti per la produzione di vettori basati su AAV (rAAV) ed alla conoscenza di nuovi aspetti ignoti della sua biologia. rAAV hanno mostrato efficacia terapeutica in un ampio range di modelli animali: topi, maiali, cani, pecore etc…
I vettori rAAV si costruiscono sostituendo i geni che compongono il genoma virale, Rep e Cap con il “gene terapeutico” (transgene), mantenendo le ITR che rappresentano gli unici elementi virali in cis indispensabili per la replicazione ed il packaging di rAAV (Fig. 12) (Samulski et al., 1989).
Fig. 12: Differenza tra l’AAV wild type e un vettore derivato da AAV: eAAV.
Tuttavia, per avere replicazione e packaging di rAAV, è necessario fornire le proteine Rep e Cap, e le funzioni helper in trans all’interno di una cellula di packaging (Fig. 13).
Fig. 13: Produzione di vettori AAV nella cellula di packaging tramite tripla trasfezione.
Fonte: www.vectorbiolabs.com
La produzione di AAV inizialmente era effettuata trasfettando le cellule umane HEK 293 (cellule renali embrionali) con il vettore contenente il transgene fiancheggiato dalle ITRs e infettando con l’adenovirus. Questa metodica è stata abbandonata in quanto era difficile ottenere preparati di rAAV non contaminati da adenovirus,
patogene per l’uomo (Grimm et al., 1998; Salvetti et al., 1998; Xiao et al., 1998). Per ovviare a questo problema, si effettua una trasfezione nelle cellule HEK 293 con un vettore che contiene il genoma ricombinante rAAV, un plasmide che fornisce le proteine Rep e Cap in trans ed un plasmide contenente i geni adenovirali codificanti per E1, E2a, E4 VAI (Huang et al., 2004; Durocher et al., 2007).
I vettori virali rAAV contengono solo 300 nucleotidi di AAV e perciò il rischio di ricombinazione con il virus wild-type è minimo, inoltre essendo privati dei geni virali non sono capaci di integrarsi nel genoma della cellula ospite in maniera sito specifica. Il processamento del genoma di rAAV può metterlo in condizione di assumere una forma episomica circolare a doppio filamento, sia attraverso la sintesi del secondo filamento, sia attraverso l’appaiamento di filamenti complementari (Russell, 1999; Thomas, 2004). Questi episomi possono essere ulteriormente convertiti in concatenameri di dsDNA virale ad alto peso molecolare che si formano grazie alla ricombinazione dei genomi monomerici (Clark et al., 1996). Si pensa che questi concatemeri siano la principale sorgente d’espressione a lungo termine del transgene, ancor più nelle cellule non in divisione (Duan, 1999). Il meccanismo di produzione dei vettori virali di AAV appena descritto elimina il rischio di mutagenesi inserzionale o silenziamento, tipico dei vettori retrovirali.
Trasduzione di rAAV
È stato dimostrato che numerose influenze ambientali migliorano la trasduzione degli AAV ricombinanti (Sanlioglu et al., 1999) e ciò è alla base dello sviluppo di nuove ed innovative strategie per migliorare l’efficienza di AAV come vettore della terapia genica. Molti di questi fattori includono lo stress genotossico, i raggi UV e i raggi X. Sembra che questi stimoli siano coinvolti nella modificazione di alcuni processi fondamentali per la trasduzione degli AAV, tra i quali: endocitosi, diffusione del virus nel nucleo, conversione del genoma virale da ssDNA a dsDNA. È stato dimostrato che due intermedi molecolari della replicazione del genoma di AAV, in forma replicativa di monomeri, dimeri e intermedi circolari, possono influenzare sia l’espressione del transgene che la persistenza di AAV nell’ospite. L’aumento di trasduzione di AAV mediante irradiazione UV e mediante la proteina adenovirale E4orf6 sono correlate rispettivamente con un aumento della forma circolare o degli
ad un aumento di intermedi di replicazione senza alcuna interazione nell’abbondanza degli intermedi circolari. Questi risultati dimostrano che la trasduzione di AAV può avvenire attraverso due percorsi molecolarmente indipendenti che convertono il singolo filamento di AAV in forme di DNA esprimibili.
Un metodo di produzione dei vettori virali sviluppato recentemente è quello basato sull’uso di Baculovirus e di sospensioni cellulari d’insetto (Virag et al., 2009). Come riportato in letteratura (Smith et al., 1983), il virus ricombinante Autographa californica (AcMNPV), che è un baculovirus, e le linee cellulari derivate da larve di Spodoptera frugiperda (Sf9 e Sf21) possono essere una nuova ed economica piattaforma per la produzione di proteine ricombinanti. È stato osservato che le cellule Sf9 sono idonee per la replicazione del DNA di AAV e per la formazione di particelle di AAV, notando che tali particelle virali sono indistinguibili fisicamente, biochimicamente e biologicamente da quelle della linea cellulare di mammifero (Urabe et al., 2002). Tuttavia l’uso del sistema cellule d’insetto-baculovirus per la produzione di rAAV presenta degli svantaggi come l’instabilità genomica dei vettori basati su baculovirus e la possibilità di contaminazione del preparato di rAAV con baculovirus (Galibert e Merten, 2011).
1.3 Il lievito Saccharomyces cerevisiae
Fig. 14: Cellule di lievito viste al microscopio elettronico a scansione, si può
osservare gli eventi di gemmazione e le cicatrici lasciate da questi fenomeni.
Il Lievito Saccharomyces cerevisiae è utilizzato in numerosi studi di citologia e in genetica come organismo modello. Appartiene al Regno dei funghi, Phylum Ascomiceti che contiene funghi chiamati ascomiceti, comunemente noti come funghi a sacco. Un lievito si definisce come un fungo unicellulare contenente un singolo nucleo e avente tallo globoso, ovoidale o sferico, talvolta allungato o irregolre. Ad oggi sono state catalogate più di mille specie di lieviti. La maggior parte appartengono proprio al gruppo degli Ascomiceti. S.cerevisiae è noto fin dall’antichità per la panificazione e la produzione di birra e vino. È un organismo anaerobio facoltativo: in presenza di ossigeno ha un metabolismo di tipo respiratorio, in condizioni anossiche fermenta il glucosio a etanolo e anidride carbonica.
S.cerevisiae, di forma ellissoidale, esiste in tre forme cellulari differenti, due aploidi (a
e α) e una diploide. La cellula diploide oltre ad avere una forma più ellissoidale presenta un volume quasi doppio rispetto alla cellula aploide. La riproduzione può essere asessuata (gemmazione) o sessuata. La gemmazione avviene in entrambe le
ploidie, il tempo di gemmazione del lievito è di circa 90 minuti. Da una stessa cellula madre si possono avere dalle 30 alle 40 divisioni, ciascuna divisione lascia sulla cellula madre una cicatrice ed il numero di cicatrici indica l’età della cellula. La conversione tra lo stato aploide e quello diploide avviene attraverso un processo di accoppiamento detto coniugazione, le cellule aploidi possono essere di due tipi coniugativi (mating type): a ed α. Cellule aploidi di tipo a si possono fondere solo con cellule aploidi di tipo α per generare cellule diploidi di tipo a/α.
Il genoma di S.cerevisiae è stato il primo ad essere sequenziato, nel 1996. Le dimensioni sono di circa 12 Mbp, più un cluster di 9137 bp fatto da circa cento segmenti ripetuti codificanti rRNA. È costituito da 16 cromosomi (n) e circa 6000 geni. Il genoma mitocondriale è circolare e contiene intorno alle 7 Kbp, anche se sono presenti dei ceppi che non lo possiedono, essendo il lievito un aerobio facoltativo. La cellula eucariotica ha il citoplasma organizzato in granuli (apparato di Golgi, citoscheletro, reticolo endoplasmatico, mitocondri, nucleo) ed il lievito ha una parete esterna alla membrana, spessa 100-200 nm che rappresenta il 15-20% della massa secca, costituita dall’80-90% in polisaccaridi e per il 2-4% in chitina, la parte superficiale è ricoperta invece da mannoproteine.
S.cerevisiae offre diversi vantaggi per lo studio della biologia molecolare e cellulare:
o È un organismo eucariote, dotato di un nucleo ed organuli, e può quindi essere usato come modello per lo studio di cellule eucariotiche;
o Ha una rapida velocità di crescita, ed è facilmente e velocemente coltivabile; o Ha un genoma semplice e completamente sequenziato;
o Non è patogeno (è considerato un organismo GRAS, “generally regarded as safe”);
o Ad ogni stadio del ciclo cellulare corrisponde un particolare stato morfologico; o Si divide per gemmazione;
o Possono essere generati facilmente mutanti;
o Può vivere nelle due forme: aploide (n) e diploide (2n);
o Può essere facilmente trasformato, ed inoltre la ricombinazione omologa avviene con alta efficacia, consentendo una manipolazione mirata del genoma.
Grazie a tutte queste caratteristiche il lievito rappresenta un organismo modello utilizzato per numerosi studi di svariato genere, infatti S.cerevisiae è utilizzato per la produzione di proteine umane: viene trasfettato con costrutti plasmidici contenenti cDNA per ottenere proteine sotto il controllo di promotori specifici per il lievito. Il vantaggio di usare il lievito anziché i batteri è da ricercare nell’origine eucariotica di questo organismo modello; questa caratteristica consente infatti alle proteine di subire un appropriato folding e tutta quella serie di modifiche post-traduzionali (fosforilazioni, glicosilazioni etc..) necessarie alla funzionalità delle proteine. Vista l’importanza e la variabilità nell’utilizzo di S.cerevisiae come organismo modello, sono nati dei database aggiornati, dai quali è possibile ricavare informazioni sui geni e sulle proteine di lievito; si ha inoltre anche la possibilità di ordinare ceppi con mutazioni desiderata.
1.3.1 Il lievito S.cerevisiae in campo virologico
S.cerevisiae è capace di sostenere la replicazione di molti virus sia a RNA che a
DNA (Raghavan et al., 2004; Galao et al., 2007) che hanno i loro ospiti naturali sia nelle piante che negli animali, compreso l’uomo. I virus a RNA che riescono a replicarsi nel lievito includono tre virus di piante come Brome mosaic virus (BMW), Carnation Italian ringspot virus (CIRV) e Tomato bushy stunt virus (TBSV), e due virus degli animali come Flock House virus (FHV) e Nodamura virus (NoV). Mentre tra i virus a DNA con genoma circolare sono inclusi: Papilloma virus umano (HPV), Papilloma virus bovino (BPV) e virus Mung bean yellow mosaic Indian (MYMIV) (Alver-Rodriguez et al., 2006; Raghavan et al., 2004), un virus con DNA a singolo filamento circolare. Agli inizi degli anni ’80 S.cerevisiae è stato usato per la produzione del vaccino contro il virus dell’epatite B (antigene di superficie del virus dell’Epatite B), contro il papilloma virus (proteina strutturale L1) (Angeletti et al., 2002) e per studiare le funzioni di proteine virali di virus patogeni umani tra i quali HIV, il virus dell’epatite C e il virus di Epstein-Barr (Alves-Rodriguez et al., 2006; Hughes, 2002).
Data la sua larga applicazione nella produzione di vaccini, risulta essere un ottimo modello per la ricerca biomolecolare e virologica; in particolare riveste un ruolo molto
sono particelle virali simili ai Virus wild type ma non infettive perchè non contengono il genoma virale. Molte VLPs dispongono soltanto dell’esatta conformazione delle proteine strutturali che formano il capside virale, questo permette di ottenere vaccini virali perché è possibile l’auto-assemblaggio delle particelle VLPs senza andare incontro ai rischi associati alla replicazione virale. Proprio per queste caratteristiche VLPs di diversi virus sono già stati prodotti in lievito (Hale et al., 2002; Lowin et al., 2005; Rodriguez-limas, 2011).
È stato recentemente dimostrato nel nostro laboratorio, che il lievito S. cerevisiae è in grado di sostenere la formazione del ssDNA di AAV direttamente da un plasmide a doppio filamento contenente le sequenze in cis necessarie per la replicazione (ITR), qualora venga espressa la proteina regolatoria di AAV Rep68/78 (Cervelli et al., 2011) e la formazione di VLPs di AAV da un plasmine contenente le proteine Vp1, 2 e 3 sotto il controllo di un promotore inducibilie di lievito (Backovivc et al 2012).
2.0 Scopo della tesi
Il progetto da cui nasce questa tesi ha l’obiettivo di valutare se il lievito Saccharomyces cerevisiae possa essere utilizzato come sistema genetico per lo studio della biologia e per la produzione di vettori derivati da AAV. Recenti studi hanno dimostrato che tale organismo modello è in grado di sostenere la formazione di VLPs di AAV da un plasmide contenente le proteine Vp1, 2 e 3 sotto il controllo di un promotore inducibile di lievito.
Lo scopo di questa tesi è pertanto quello di caratterizzare ulteriormente la formazione dei capsidi sia andando a studiare la localizzazione delle proteine del capside Vp1, Vp2 e Vp3, sia sviluppando una nuova metodica d’investigazione (Immuno blot) che riveli con più facilità la produzione di vettori virali in S.cerevisiae.
Un altro aspetto approfondito durante il lavoro di tesi è lo studio sul genoma virale di AAV. Era già stato dimostrato che tale organismo modello è in grado di sostenere la formazione del ssDNA di AAV direttamente da un plasmide a doppio filamento contenente le sequenze in cis necessarie per la replicazione (ITR), qualora venga espressa la proteina regolatoria di AAV Rep68/78. In questo lavoro di tesi abbiamo valutato se nel lievito S. cerevisiae avviene l’incapsidazione del ssDNA genomico all’interno dei capsidi.
3.0 Materiali e metodi
3.1 Ceppi di Saccharomices Cerevisiae
3.1.1 Ceppo RSY12-Rep68
Uno dei ceppi di S.cerevisiae utilizzati per questo lavoro di tesi è il ceppo aploide RSY12-Rep68 che deriva dal ceppo RSY12 WT con il seguente genotipo MATa leu2-3, 112 his3-11,15 URA3::HIS3 (Schiestl, 1991). Il ceppo ha integrato nel genoma il plasmide pG.Rep68. Le sue caratteristiche sono:
Ø La presenza di una doppia mutazione nel gene LEU2 che codifica per l’enzima idropropilmalato deidrogenasi implicato nella biosintesi della leucina;
Ø La delezione completa del gene URA3, che codifica l’enzima ortodontina-5’-fosfatodecarbossilasi, coinvolto nella biosintesi dell’uracile, mediante sostituzione con il gene HIS3 coinvolto nella biosintesi dell’istidina;
Ø Contiene il gene Rep68 di AAV, sotto il controllo del promotorecostitutivo di lievito ADH;
Ø Contiene il gene LEU2 come marcatore di selezione.
Tale ceppo è stato modificato durante questo lavoro di tesi sostituendo il gene LEU2 con il gene per la resistenza all’igromicina.
3.1.2 Ceppo BY4743
Un altro ceppo sul quale sono stati effettuati studi durante questo lavoro è il ceppo diploide BY4743 le cui caratteristiche sono: MATa; his3Δ1/his3Δ1; leu2Δ0/leu2Δ0; met15Δ0/MET15; LYS2/lys2Δ0; ura3Δ0/ura3Δ0/ MATα; his3Δ1/his3Δ1; leu2Δ0/leu2Δ0; met15Δ0/MET15; LYS2/lys2Δ0; ura3Δ0/ura3Δ0.
3.2 Terreni di coltura per il lievito
I terreni usati per la crescita e selezione dei ceppi di lievito sono stati: i terreni completi YAPD e YAPG e i terreni selettivi, cioè mezzi di coltura in cui si conosce l’esatta formulazione chimica di ogni ingrediente, che vengono utilizzati per la selezione di particolari ceppi. Tutti i tipi di terreni possono essere sotto forma liquida o solida con l’aggiunta di agar.
Terreni completi: Terreno YAPD (1 L) • 20 g Bactopeptone • 10 g Yeast exstrct • 20 g Glucosio • 0,2 g Solfunato di adenina • 900 ml acqua distillata
• Eventuale aggiunta di 20 g di AGAR nel caso della preparazione del terreno
solido Terreno YAPG (1 L) • 20 g Bactopeptone • 10 g Yeast exctract • 50 g Galattosio • 0,2 g Solfunato di adenina • 900 ml acqua distillata
• Eventuale aggiunta di 10 g di AGAR nel caso della preparazione del terreno