1)
I
NTRODUZIONE
1.1) F
ARMACOGENETICA ED EFFICACIA TERAPEUTICALo studio dei Polimorfismi a Singolo Nucleotide, comunemente detti SNPs, è oggi uno dei campi di maggior rilievo nella genetica molecolare umana, in quanto le varianti puntiformi contribuiscono in maniera determinante alla sostanziale diversità che caratterizza l’unicità di ciascun individuo. In questo ambito si inserisce la farmacogenetica che è un settore che tenta di stabilire un ponte tra la farmacologia e la genetica. Lo scopo è quello di ottimizzare l’approccio terapeutico a patologie complesse, come i tumori, in relazione all’unicità di ciascun individuo. In altre parole, la farmacogenetica mette in relazione i principi dell’approccio terapeutico con la variabilità genetica individuale, cercando di sviluppare conoscenze tali da poter mettere a punto terapie in funzione di specifici genotipi. In questa ottica lo studio delle migliaia di polimorfismi a singolo nucleodite annidati nei nostri geni potrebbe permettere di correlare, ad esempio, l’efficacia di un determinato protocollo terapeutico ad una determinata variante allelica di un particolare gene.
1.2) M
IELOMAM
ULTIPLO:
la patologia
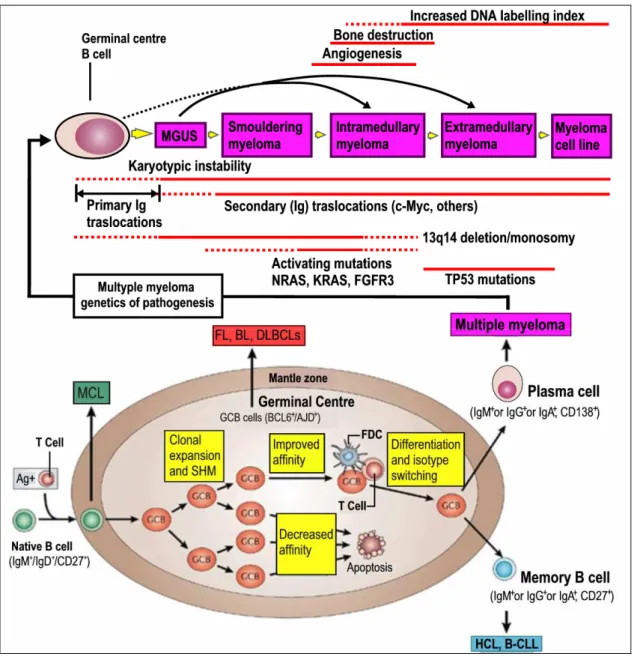
Il Mieloma Multiplo (MM), anche detto Mieloma plasmacellulare o Plasmocitoma, è una patologia tumorale maligna caratterizzata dalla proliferazione e dall’accumulo di un singolo clone di plasmacellule nel midollo osseo.
La definizione “multiplo” associata al mieloma deriva dalla localizzazione dei plasmociti neoplastici in attiva proliferazione, che interessano zone multiple del midollo osseo (plasmociti midollari) ma che
possono essere confinati all’osso o anche interessare i tessuti molli (plasmocitomi extramidollari)[1]. La progressione e lo sviluppo della malattia porta ad una serie di conseguenze fisiopatologiche le quali risultano in un complesso insieme di sintomi e disfunzioni gravi, tra cui si evidenziano dolore osseo associato a lesioni osteolitiche (80% dei casi), anemia, ipercalcemia, la compromissione della produzione di immunoglobuline normali (IgG, IgA, IgD o IgE) con conseguente insufficienza renale (60%) e suscettibilità ad infezioni. In questo quadro clinico, la prognosi dei pazienti è generalmente infausta ed il progressivo declino della qualità della vita è associato ad una aspettativa di sopravvivenza media che oscilla tra i 20 e i 60 mesi.
Il mieloma multiplo è una patologia relativamente rara, rappresentando circa l’1% di tutte le malattie maligne ed in particolare il 10% tra le neoplasie ematologiche[2]. A livello mondiale l’incidenza annua del mieloma multiplo è di circa 3 nuovi casi ogni 100000 individui (superiore negli anziani) ed è maggiore nei maschi, mentre l’età media alla diagnosi si attesta intorno ai 68 anni. I tassi più elevati, inoltre, si osservano tra gli Afro-Americani degli Stati Uniti (33% di tutti i cancri ematologici) mentre i tassi più bassi si registrano tra le popolazioni dell’Asia e dell’Europa Orientale[3].
In Italia, l’incidenza registrata è di circa 2-4 nuovi casi ogni 100.000 abitanti all’anno, con un’età media di insorgenza che va oltre i 50 anni. Anche in Italia, l’incidenza è leggermente maggiore nei maschi rispetto alle femmine. I tassi di mortalità, relativi a tutti i gruppi di età registrati nel 1997, sono pari alla media dell’Unione Europea negli uomini (2,15) e superiori alla media (1,52) nelle donne (1,55).
Le caratteristiche del tumore corrispondono a quelle della plasmacellula mielomatosa, che è interessata da alterazioni genetiche, biochimiche e metaboliche che ne alterano la corretta funzionalità,
conferendole carattere neoplastico. Le plasmacellule patologiche,
generalmente superiori al 20% della cellularità globale, possono arrivare a localizzarsi in sedi specifiche, con una infiltrazione massiva dello stesso spazio midollare e conseguente alterazione della crasi ematica (Fig. 1.1).
Dal punto di vista funzionale, le cellule mielomatose vanno incontro ad un forte aumento dell’attività cellulare, in particolare con una produzione di elevate quantità di una immunoglobulina monoclonale (detta proteina M),
solitamente di tipo IgG o IgA, oppure di un suo componente/frammento, caratteristica della patologia.
Infatti, il mieloma multiplo ha origine dalla trasformazione neoplastica di una cellula della fase differenziativa antigene-dipendente, quali in particolare il plasmoblasto o la cellula B memoria, ovvero elementi cellulari che sono appena passati per la fase di selezione antigenica a livello del centro germinativo del midollo osseo (Fig. 1.2).
Nella fase del ciclo in cui le cellule B immature diventano plasmacellule mature, avviene un processo fisiologico denominato “ricombinazione sito-specifica” dei geni delle immunoglobuline ( V,D e J), che consiste nell’associazione combinatoria di differenti segmenti genici, con la conseguente generazione dell’ampio spettro di anticorpi caratteristico di ciascun individuo. In altre parole, segmenti diversi di geni diversi vengono riarrangiati da un complesso apparato biochimico cellulare, noto come ricombinasi VDJ, a formare un gene che codificherà le componenti necessarie a fomare l’immunoglobulina specifica di ciascun linfocita B.
A causa di questo delicato passaggio di ricombinazione, le cellule B sono caratterizzate da una instabilità genetica intrinseca che è alla base, a sua volta, della maggior parte degli eventi oncogenetici che contribuiscono alla patogenesi del mieloma multiplo. Infatti, nel cariotipo delle plasmacellule mielomatose si osservano numerose alterazioni come delezioni o traslocazioni, localizzate proprio nelle regioni cromosomiche contenenti i geni per le immunoglobuline.
L’alterazione più frequente, che colpisce appunto la regione che contiene i geni codificanti per le catene pesanti delle immunoglobuline, è la traslocazione della banda 14q32, anormale in circa il 75% dei pazienti affetti da mieloma[4]. Il fatto che nella maggior parte delle traslocazioni a carico della banda 14q32 il punto di rottura cada all’interno della regione di “switching” delle catene pesanti fornisce una indicazione indiretta del fatto che l’evento iniziale dello sviluppo della malattia abbia effettivamente luogo nel centro germinativo, con un probabile coinvolgimento di progenitori plasmacellulari precoci.
Figura 1.1: plasmacellule mielomatose nel midollo osseo.
L’80-90% dei pazienti affetti da mieloma multiplo può comunque presentare anomalie cromosomiche numeriche o strutturali (del 13q, del 17q, t(4,14)), anche diverse dalla traslocazione a carico della banda 14q32, indipendentemente dallo stadio della malattia[5].
Dal punto di vista della patogenesi del mieloma, le alterazioni e i riarrangiamenti cromosomici che si osservano nelle plasmacellule sono considerati l’evento primario nell’evoluzione della malattia, in quanto garantiscono l’immortalità della cellula, senza tuttavia essere da soli sufficienti a determinare l’effettiva trasformazione neoplastica della cellula.
Per quel che riguarda la traslocazione della banda 14q32, ad esempio, sono stati ad oggi identificati alcuni geni che agiscono come partner primari della traslocazione, quali bcl1, prad1, cicline D1 e D3 ed altri che codificano per proteine coinvolte nelle regolazione del ciclo cellulare, contribuendo nell’evoluzione del carattere neoplastico. Un’altra alterazione comunemente osservata nel cariotipo di pazienti affetti da mieloma multiplo è la delezione a carico del braccio lungo del cromosoma 13 (del 13q), che si verifica precocemente nel corso della malattia ed è stata osservata nel 43% dei casi alla diagnosi e nel 70% dei pazienti con malattia avanzata o evoluta in leucemia plasmacellulare.
Dall’analisi dei pazienti che presentavano riarrangiamenti a livello della banda 14q32 e la delezione 13q sono state recentemente ipotizzate le tappe fondamentali nella patogenesi del mieloma multiplo. L’evento più precoce nello sviluppo della malattia sarebbe la ricombinazione illegittima dei geni codificanti per la catena pesante delle immunoglobuline con la conseguente traslocazione a carico della banda 14q32. Tale ipotesi è confermata dalla correlazione esistente tra i riarrangiamenti a livello della banda 14q32 e il tipo di immunoglobulina prodotta dalle cellule mielomatose. Inoltre, è stato osservato che le traslocazioni della banda 14q32 sono quasi sempre associate (nel 95% dei pazienti) alla delezione del braccio lungo del cromosoma 13. In questi pazienti, pertanto, il riarrangiamento della banda q32 del cromosoma 14 rappresenterebbe l’evento oncogenico primario, mentre la delezione 13q l’evento oncogenico secondario.
Nel 25% dei pazienti, tuttavia, non si evidenziano traslocazioni a carico della banda 14q32, ed in questi soggetti la malattia presenta un
decorso clinico indolente sino al momento in cui non si presenti la delezione 13q[6]. Studi clinici evidenziano che le anomalie a carico del cromosoma 13 sono associate ad una bassa sopravvivenza libera da eventi (EFS) e globale (OS) in seguito alle chemioterapie (convenzionale e ad alte dosi). In particolare, le delezioni, come la delezione 13q, sono associate sia ad una malattia più aggressiva che alla farmaco-resistenza del tumore[7]. Inoltre, come facilmente prevedibile, durante la progressione della malattia aumenta l’instabilità del cariotipo. Questo porta ad un accumulo, nelle cellule mielomatose, di mutazioni ed alterazioni nell’espressione di altri geni importanti per la sopravvivenza ed il controllo cellulare, quali c-myc, Nras, Kras e p53, con un conseguente decorso clinico generalmente sfavorevole (Fig.1.2).
Un’altra importante caratteristica del mieloma multiplo è la completa dipendenza delle plasmacellule neoplastiche dal microambiente del midollo osseo, caratterizzato dalla presenza di proteine della matrice extracellulare, cellule stromali, osteoclasti, osteoblasti ed altre cellule accessorie.
L’interazione delle plasmacellule con questi elementi è alla base dell’attivazione e secrezione di fattori di crescita, di fattori anti-apoptotici e di diverse citochine (in particolare IL-6) che, attraverso una complessa rete di segnali intra- ed extra- cellulari, promuovono la proliferazione, la sopravvivenza, la farmaco-resistenza e la progressione della malattia[8].
L’interleuchina 6 (IL-6) è il principale segnale per la crescita e la sopravvivenza delle plasmacellule neoplastiche che, come le cellule stromali del midollo osseo, la producono e la secernono. In pazienti nei quali la malattia è in fase attiva, i livelli sierici di IL-6 sono aumentati e risultano, senza dubbio, associati ad una prognosi infausta. Le citochine, ed in particolare IL-6 e IL-10, oltre a essere determinanti per la crescita e la proliferazione delle cellule mielomatose, mediano anche i fenomeni di distruzione dell’osso, conseguenza principale del processo di riassorbimento da parte degli osteoclasti attivati.
La comprensione dei meccanismi molecolari della patogenesi e dell’evoluzione del mieloma multiplo è un punto cruciale nella ricerca delle cure efficaci, in quanto permetterebbe di individuare potenziali bersagli
terapeutici per l’identificazione di nuove terapie in grado di superare la farmaco-resistenza e di indurre l’apoptosi delle cellule neoplastiche.
1.3) M
IELOMAM
ULTIPLO: fattori prognostici tradizionali
Sono stati individuati numerosi fattori che, valutati alla diagnosi, hanno un valore prognostico. La componente senza dubbio principale nella valutazione della condizione neoplastica è la presenza nel siero di una immunoglobulina monoclonale, o di un suo frammento, nota come proteina M. In condizioni patologiche, la proteina M è presente nel siero e/o nell’urina in quantità ingenti e viene normalmente identificata, all’elettroforesi proteica, da un “picco” in corrispondenza della regione delle gammaglobuline.
Oltre alla proteina M sono stati caratterizzati numerosi altri fattori prognostici, sierici e non. I fattori prognostici tradizionali, come i livelli di microglobulina β2, della proteina C reattiva (CRP) e di albumina spiegano soltanto il 15-20% dell’eterogeneità della risposta alla terapia. Inoltre, anomalie cariotipiche, presenti in un terzo dei pazienti di nuova diagnosi, sono state associate ad un esito fatale più rapido e meno del 10% dei pazienti che presentano tali anomalie sopravvive oltre i 5 anni. I progressi recenti della citogenetica molecolare hanno identificato nel 40% dei pazienti traslocazioni primarie che coinvolgono il locus della catena pesante delle immunoglobuline, situato a livello della banda 14q32. Mielomi iperdiploidi e positivi per le traslocazioni t(11,14)(q13q32) sono associati con una migliore prognosi, mentre l’assenza di iperdiploidia, spesso associata con traslocazioni diverse dalla t(11,14) e con la delezione di parte del cromosoma
13, predicono una prognosi estremamente peggiore[9].
Un importante limite dei fattori prognostici tradizionali individuati fino a questo momento rimane quello di essere valutabili unicamente alla diagnosi, in quanto sono sensibilmente influenzati dai regimi chemioterapici adottati nella cura della patologia. E’ per questo che si pensa che lo studio dei polimorfismi a singolo nucleotide (SNPs) in geni bersaglio dell’azione dei
farmaci e coinvolti nel loro metabolismo e trasporto possa identificare nuovi fattori prognostici.
1.4) M
IELOMAM
ULTIPLO:
la terapia
Il mieloma multiplo è una malattia progressiva con una prognosi sfavorevole che è sensibilmente migliorata con l’introduzione della chemioterapia. Attualmente la sopravvivenza è di circa 2-3 anni[10], ma si va anche oltre. Approssimativamente il 25% dei pazienti sopravvive più di 5 anni ma circa il 3% sopravvive più di 10 anni.
Lo scopo delle moderne terapie per il trattamento del mieloma multiplo è di ottenere remissione completa (CR) della malattia, sebbene nella sopravvivenza a lungo termine si osservino alcune tracce residue, come la presenza di proteina M nel siero. La definizione di remissione completa (CR), che è stata più volte modificata negli ultimi 20 anni, in genere è identificata dall’assenza della paraproteina monoclonale nel siero e/o nella urina, da un contenuto minore del 5% di plasmacellule nel midollo osseo, dall’assenza di incremento nel numero e nelle dimensioni delle lesioni ossee e nella scomparsa di plasmocitomi nel tessuto molle. La presenza di remissione completa è un parametro importante da valutare, perché è un indice della qualità della vita ed è un fattore prognostico per la sopravvivenza.
Il trattamento del mieloma multiplo è andato via via evolvendo verso terapie sempre più specifiche. Con l’introduzione di una combinazione di melphalan e prednisone (MP), impiegata come chemioterapia specifica per il mieloma multiplo da parte di Alexian e colleghi per la prima volta circa 40 anni fa, furono ottenuti buoni risultati in oltre il 60% dei pazienti trattati. Tuttavia, nessun paziente andava incontro a remissione completa e la sopravvivenza si attestava intorno ai 12-30 mesi. La combinazione MP è stata considerata per lungo tempo il trattamento terapeutico specifico del mieloma multiplo, nonostante presentasse diversi svantaggi tra i quali l’elevata tossicità per le cellule staminali del midollo osseo.
A causa dei limiti evidenziati dalla chemioterapia MP sono stati sviluppati numerosi regimi alternativi di chemioterapia combinatoria (CCT), i quali generalmente includevano infusione continua di vincristina e adriamicina, nel tentativo di migliorare la sopravvivenza e il grado di risposta. Tuttavia, nessuno di questi tentativi ha mostrato una differenza significativa nella riduzione della mortalità rispetto alla chemioterapia MP.
Nel 1998, sono stati riportati alcuni dati relativi alla comparazione della chemioterapia combinatoria CTT con la chemioterapia MP. Secondo questo studio le sopravvivenze risultavano equivalenti (circa 29 mesi), sebbene il grado di risposta fosse significativamente più alto con la CCT (60% vs 53%)[11].
La chemioterapia convenzionale è comunque in grado di indurre vari gradi di risposta parziale (PR) e viene pertanto utilizzata al fine di ottenere una buona risposta nell’arco di 6-8 mesi e soprattutto per raggiungere la cosiddetta fase di plateau della malattia, ovvero un periodo di stabilità di almeno 4-6 mesi dal termine della terapia, senza che si manifestino segni di ripresa.
I limiti della chemioterapia convenzionale (incapacità di eradicazione del clone neoplastico, sviluppo di resistenza ai farmaci e limitata efficacia delle terapie di recupero nei pazienti refrattari) hanno tuttavia suggerito l’opportunità di utilizzare altri protocolli terapeutici. L’introduzione dei fattori di crescita emopoietici e lo sviluppo di nuove tecniche per la raccolta e reinfusione delle cellule staminali hanno reso possibile associare l’autotrapianto alla chemioterapia ad alte dosi e questa strategia innovativa si è rivelata molto migliore della sola chemioterapia convenzionale nel trattamento del mieloma multiplo.
La combinazione di vincristina, doxorubicina e desametasone ad alte dosi (DAV) fu impiegata per la prima volta nel 1984, in pazienti refrattari o in ricaduta e fu osservato un buon miglioramento della prognosi. Inoltre, la chemioterapia DAV induceva remissioni nel 32% dei pazienti resistenti primari e nel 65% dei pazienti recidivanti, sempre che fossero inizialmente trattati con un regime contenente doxorubicina[12]. Negli studi successivi, il protocollo DAV è stato impiegato come terapia di prima linea, ottenendo un
grado di risposta piuttosto alto (55-84%) e rapido, ma senza un incisivo prolungamento della sopravvivenza (36-44 mesi).
Il protocollo DAV si è così affermato come una valida alternativa al MP o alla CCT[13], non tanto in virtù di un miglior risultato globale ma piuttosto perché in grado di produrre una buona risposta (nel 90% dei casi al secondo ciclo), elevate percentuali di risposte globali (60-80%) con un apprezzabile numero di risposte complete (10-25%), senza oltretutto danneggiare le cellule staminali sane del midollo osseo. Tuttavia, anche nei pazienti trattati con DAV, la ricaduta della malattia si manifestava in maniera molto rapida in circa il 90% dei soggetti. Una volta che i pazienti avessero raggiunto una risposta massima con la chemioterapia DAV si rivelava quindi necessaria una strategia aggiuntiva per un buon controllo della malattia.
Oggi, grazie alla sua capacità d’azione, la chemioterapia DAV viene usata come “terapia di induzione”, preliminare ad una chemioterapia ad alte dosi (HDT) a base di melphalan, in quei pazienti candidati ad un approccio trapiantologico, preferenzialmente di tipo autologo (ASCT).
In passato, dopo la somministrazione di mezza dose di melphalan (140mg/m2) e senza alcun recupero di cellule staminali, solo nel 30% dei pazienti si otteneva una remissione completa, che perdurava per circa 3 anni. In seguito, si è visto che si poteva ottenere un risultato migliore attraverso la combinazione della “terapia di induzione” ed il trattamento con 200 mg/m2 di melphalan, poiché le cellule staminali del sangue periferico potevano essere recuperate.
Negli ultimi anni, sono stati condotti una serie di studi da parte di vari gruppi di ricerca per ottenere dati a sostegno dell’efficacia di questo trattamento. In uno studio randomizzato pubblicato dall’Intergroupe Francophone du Myelome (IFM), nel quale sono stati coinvolti 200 pazienti con mieloma non trattati, si è dimostrata chiaramente la superiorità del trapianto autologo di cellule staminali (ASCT) rispetto alla chemioterapia convenzionale. Con la chemioterapia convenzionale la percentuale di risposta osservata era del 57%, di cui il 5% erano risposte complete ed il 12% dei pazienti aveva una sopravvivenza di almeno 5 anni. Con l’ASCT, invece, la percentuale di risposta osservata è stata dell’81%, con il 22% di
risposte complete ed il 52% di pazienti con una sopravvivenza di almeno 5 anni[14].
Se il trapianto di cellule staminali veniva ripetuto in successione (trapianto in tandem), i risultati già buoni miglioravano ulteriormente, soprattutto in termini di maggiori risposte complete (fino al 50%) e di prolungata durata della remissione. La sopravvivenza mediana libera da malattia era di 43 mesi, e la sopravvivenza globale mediana di 68 mesi[15].
Una delle fasi più importanti nell’approccio trapiantologico è rappresentata dalla raccolta delle cellule staminali sane. I primi tentativi di raccolta nei pazienti con mieloma multiplo sono stati effettuati dopo chemioterapia con alte dosi di ciclofosfamide[16] o con regimi contenenti antracicline[17]. Successivamente, grazie all’introduzione dell’utilizzo dei fattori di crescita dopo la chemioterapia, allo scopo di incrementare il numero di cellule staminali mobilizzate nel sangue periferico, divenne uno standard l'associazione della ciclofosfamide ad alte dosi con fattori di crescita diversi[18].
Al di fuori di studi clinici controllati, la personalizzazione della terapia di mobilizzazione tiene conto dello stato di malattia del paziente, dei precedenti trattamenti, dei rischi di tossicità del farmaco e della dose applicata ed, infine, della maggiore probabilità di contaminazione se la scelta ricade sull'utilizzo di un singolo fattore di crescita. Inoltre, il recupero di piastrine dopo una terapia di mobilizzazione con ciclofosfamide è considerato un’indicazione di un breve attecchimento post-trapianto. Infine, è stato segnalato che l'esposizione al melphalan, prima della terapia di mobilizzazione, riduca il numero delle cellule staminali midollari e la loro capacità di espansione, nonché la loro mobilizzazione nel sangue periferico[19].
I fattori che contribuiscono in maniera determinante all'efficacia del trapianto autologo, devono ancora essere identificati. È verosimile che, tra
questi, un fattore importante sia rappresentato dal regime di
condizionamento, considerando che l'effetto anti-mielomatoso dipende principalmente dalla terapia mieloablativa. È evidente che il regime di condizionamento ideale dovrebbe combinare un effetto selettivo anti-mielomatoso ed una bassa tossicità. È stata ampiamente esplorata, nel mieloma multiplo, la relazione che esiste tra la dose di melphalan impiegata
e le risposte cliniche ottenute. Il lavoro pionieristico di McElwain e Powels (1983)[20], che per primi hanno esplorato l'utilizzo del melphalan ad alte dosi (140 mg/m2) in pazienti con mieloma multiplo, resistenti o non trattati, ha dimostrato l'efficacia di tale regime terapeutico ma ha dovuto registrare un 16% di mortalità legata alla marcata mielosoppressione. Successivamente, ed in linea con questi risultati, lo stesso gruppo ed altri autori hanno confermato l'effetto dose-risposta che si traduceva in una percentuale del 30% di risposte complete nei pazienti resistenti. Purtroppo, veniva anche confermata una percentuale di mortalità molto alta, che dal 10% dei casi di mieloma multiplo non trattati[21] passava al 20% per i casi che avevano già effettuato più linee di trattamento[22]. Pertanto, rimaneva, come ultima strada da percorrere, l'uso delle cellule staminali per il recupero del danno midollare indotto dal melphalan. Nel 1986, Barbogie[23] utilizzando come risorsa il midollo osseo autologo, ha ridotto la tossicità ematologica legata al melphalan utilizzato a vari dosaggi. Successivamente, sulla base del profilo di tossicità e sulla tipologia della curva dose-risposta, il melphalan è stato ulteriormente scalato a 200 mg/m2[24]. Sempre L’Intergroupe Francophone du Myelome (IFM) ha pubblicato nel 2002 i risultati di un trial multicentrico randomizzato (di uno studio iniziato nel 1995) ideato per il confronto tra due
dei più usati regimi di condizionamento, il melphalan 140 mg/m2 associato a
TBI 8Gy (regime A ) ed il melphalan 200 mg/m2 (regime B), per il quale furono reclutati pazienti affetti da mieloma multiplo sintomatico alla diagnosi con età inferiore ai 65 anni. Il trattamento ad alte dosi era preceduto da 4 cicli di terapia di induzione con DAV. Non furono evidenziate differenze in termini di sopravvivenza libera da eventi (21% vs 20.5%) mentre la sopravvivenza globale a 45 mesi è risultata più elevata nei pazienti trattati con melphalan 200 mg/m2 (regime B) (65.8% vs 45.5%). Alla luce di questi dati, il gruppo
francese concluse che il melphalan 200 mg/m2 poteva essere indicato come
il regime di condizionamento standard per i pazienti con mieloma multiplo da sottoporre ad un programma di autotrapianto[25].
Tuttavia, fu evidenziato che l'esposizione al melphalan, prima della terapia di mobilizzazione riduceva il numero delle cellule staminali midollari e la loro capacità di espansione, nonché la loro mobilizzazione nel sangue periferico[19].
Con questo nuovo approccio terapeutico, quindi, si è ottenuto un sostanziale miglioramento sia nella velocità di risposta che nella sopravvivenza dei pazienti, anche se si è ben lontani dal poterlo considerare curativo, in quanto circa il 90% dei pazienti presenta una ricaduta. Oggi, infatti, questi dosaggi del farmaco sono utilizzati come terapia di condizionamento in quanto, dal punto di vista della tossicità, sono ben tollerati.
In pratica, oggi, il trattamento standard, a cui sono sottoposti i pazienti più giovani di 70 anni, è la terapia di induzione DAV, seguita da melphalan ad alte dosi (200mg/m2) e da un successivo trapianto di cellule staminali (autotrapianto singolo o in tandem in cui il primo trattamento ad alte dosi è seguita da un altro nell’arco di 2-4 mesi). La mortalità relativa al trattamento con un autotrapianto è minore del 3% e oggi questa procedura è stata estesa anche ai pazienti più anziani di 70 anni, con opportuna riduzione della dose
di melphalan (100mg/m2). D’altra parte, procedure trapiantologiche
allogeniche, seppur teoricamente più indicate all’eradicazione, sono da valutare con attenzione visto il maggior rischio di effetti collaterali.
Riassumendo le fasi del trattamento sono:
1) CHEMIOTERAPIA DI INDUZIONE: chemioterapia DAV che, grazie all’attività citolitica dei farmaci (vincristina, doxorubicina e desametasone), riduce le cellule mielomatose risparmiando il compartimento delle cellule staminali ed, inoltre, produce elevate percentuali di risposte globali (60-80%), un apprezzabile numero di risposte complete (10-25%) e determina risposte precoci (nel 90% dei casi al secondo ciclo).
2) MOBILIZZAZIONE: fase necessaria a mettere in circolo le cellule staminali nel sangue periferico.
3) LEUCOAFERESI: prelievo delle cellule staminali dal sangue periferico.
4) CONDIZIONAMENTO: chemioterapia pre-trapianto con melphalan ad alte dosi che ha l’obiettivo di eradicare completamente la malattia, distruggendo tutte le cellule mielomatose.
5) RE-INFUSIONE: somministrazione delle cellule staminali precedentemente prelevate nella fase di leucoaferesi. Dopo la chemioterapia ad alte dosi, le cellule staminali vengono scongelate e attraverso il catetere venoso centrale o una vena periferica vengono restituite al paziente.
1.4.1) Nuove prospettive nella terapia del Mieloma Multiplo
Il mieloma multiplo, tuttavia, rimane una patologia il cui trattamento ha raggiunto incoraggianti risultati unicamente nel prolungamento della speranza di vita media e nel miglioramento della qualità della vita.
Recentemente, le terapie convenzionali sono state riviste attraverso l’introduzione di nuovi agenti terapeutici quali il bortezomib, la talidomide e la lenalidomide.
Il bortezomib è un inibitore del proteasoma ed il suo utilizzo nel trattamento di pazienti pre-trattati, da solo o in combinazione con altri chemioterapici, ha dimostrato una notevole efficacia. Infatti, è stata osservata una massimizzazione dell’efficacia terapeutica, in termini di elevati tassi di risposta, con un consistente tasso di remissioni complete e di una miglior durata della risposta stessa, combinate con il mantenimento di un livello accettabile di tossicità[26].
In questi anni si stanno sviluppando strategie che associano l’azione del bortezomib a quella dei chemioterapici convenzionali, quali l’adriamicina e la vinscristina, unitamente a modulatori dell’immunità quali la talidomide e la lenalidomide.
La talidomide è stato introdotta nella terapia di soggetti affetti da mieloma multiplo refrattari alla chemioterapia. Studi successivi hanno dimostrato l’efficacia di questo farmaco nell’indurre una buona risposta e un’alta percentuale di remissione completa in pazienti pre-trattati con chemioterapia ad alte dosi. Ancora, è stato dimostrato che l’efficacia della
talidomide aumenta se è associata al trattamento con il desametasone[27] con
con desametasone è approvato anche come terapia di prima linea e nei pazienti in ricaduta.
Anche l’uso della lenalidomide, un analogo della talidamide, è stato recentemente approvato, in combinazione col desametasone, nel trattamento di pazienti già trattati con altri regimi terapeutici[29]. Comunque, anche nei pazienti non trattati, differenti combinazioni di desametasone, bortezomib, talidomide e lenalidomide hanno prodotto tassi di risposta globale tra l’80% e il 90%, con percentuali di risposte complete del 10-32%. L’intensificazione del trattamento con chemioterapia ad alte dosi in preparazione del trapianto autologo di cellule staminali (ASCT) ha permesso ai pazienti più giovani di raggiungere risposte parziali e complete con un notevole prolungamento della sopravvivenza[30].
1.4.2) Meccanismo d’azione dei farmaci
La maggior parte dei farmaci antitumorali attualmente in uso agisce a livello del DNA, alterando irreversibilmente il processo di divisione cellulare ed inducendo apoptosi. Per tale motivo, i farmaci antitumorali sono detti farmaci citotossici. Gli agenti antitumorali che agiscono a livello di una specifica fase del ciclo cellulare sono detti agenti “fase-specifici”, mentre i farmaci antitumorali che agiscono su cellule in replicazione ma non colpiscono una specifica fase sono detti agenti “non fase-specifici”. Infine, esistono farmaci indipendenti dallo stato di replicazione della cellula, che colpiscono anche cellule in fase G0, e sono detti agenti “non ciclo-specifici”.
Un limite degli agenti chemioterapici è l’incapacità di distinguere tra il DNA di una cellula sana e quello di una cellula neoplastica. La selezione delle cellule neoplastiche, quindi, avviene fondamentalmente in base all’intensa attività proliferativa di queste ultime, che determina una maggiore probabilità che i farmaci interferiscano con la loro attività piuttosto che con quella delle cellule sane.
Il protocollo DAV, o VAD-like, prende il nome dagli agenti terapeutici impiegati, che sono il desametasone, l’adriamicina (o doxorubicina) e la
vincristina. L’azione antitumorale è svolta dall’adriamicina e dalla vincristina, mentre il desametasone è un derivato cortisonico utilizzato per fronteggiare la risposta infiammatoria nei tessuti sani.
L’adriamicina (Fig. 1.3) è un antibiotico naturale appartenente alla famiglia delle antracicline citotossiche, isolata da colture di Streptomyces peucetius var. caesius, che agisce interagendo in maniera non covalente con il DNA. Questo farmaco è considerato “non fase-specifico”, anche se il suo massimo effetto è evidente nella fase S del ciclo cellulare (Fig. 1.3). La sua azione citotossica si esplica sulle cellule, tumorali e non, in modalità differenti: si può comportare da agente intercalante o da inibitore della topoisomerasi II. Il nucleo planare dell’adriamicina, infatti, è in grado di frapporsi perpendicolarmente all’asse maggiore della doppia elica del DNA, determinandone così un parziale srotolamento, che impedisce la replicazione e la trascrizione e, conseguentemente, la sintesi proteica, indirizzando la cellula verso la morte cellulare programmata o apoptosi. L’estrema compattezza del DNA organizzato in cromatina fornisce alle cellule protezione contro questo effetto del farmaco e perciò la fase S è la più sensibile.
Tuttavia, l’adriamicina forma complessi anche con alcuni enzimi nucleari, tra i quali la Topoisomerasi II, inibendone l’azione[31]. La Topoisomerasi II è un enzima responsabile del rilassamento delle regioni eterocromatiniche di DNA, evento necessario per i processi di replicazione e di trascrizione. L’adriamicina intrappola la Topoisomerasi II sul DNA per mezzo di un legame covalente, formando un complesso ternario stabile Adriamicina-DNA-Topoisomerasi II, detto “cleavable complex”, che ostacola la progressione della Topoisomerasi II nello svolgimento della doppia elica. In questo modo, sono inibite ulteriormente la replicazione e la trascrizione RNA, con la conseguente induzione di apoptosi (Fig 1.4).
E’ stato anche dimostrato che il doxorubicinolo, un metabolita dell’adriamicina, è responsabile di un effetto cardiotossico che aumenta con la dose di adriamicina somministrata[32]. Inoltre la produzione di radicali liberi contribuisce alla cardiotossicità, ma anche a provocare rotture del DNA, e altri eventi tali da mandare la cellula in apoptosi[33].
Figura 1.4: Inibizione della topoisomerasi II da parte della doxorubicina.
La vincristina (Fig. 1.3) è un alcaloide vegetale che deriva dalla pianta Pervinca Rosea (Catharanthus roseus). Appartiene al gruppo degli “alcaloidi della Vinca”, un’importante classe di farmaci che agiscono inibendo la fase M del ciclo cellulare, per cui sono considerati farmaci “fase specifici” (Fig. 1.3)[34]. Agiscono perturbando l’equilibrio dinamico tra i microtubuli assemblati e i dimeri di tubulina necessari alla polimerizzazione dei microtubuli stessi a livello dell’estremità di crescita dei microtubuli, legandosi ad uno specifico sito della proteina[35]. In altre parole, il complesso farmaco-tubulina riduce la concentrazione dei dimeri disponibili per la polimerizzazione, spostando l’equilibrio verso la depolimerizzazione (Fig. 1.5). In questa maniera, l’azione degli alcaloidi della vinca impedisce la corretta segregazione dei cromosomi, spingendo la cellula verso l’apoptosi[36].
Sia il melphalan che la ciclofosfamide agiscono come agenti alchilanti, che vengono trasformati in composti ionici carichi positivamente altamente reattivi e possono formare legami covalenti con strutture ricche di elettroni come molte biomolecole, tra le quali gli acidi nucleici, le proteine e gli amminoacidi. In condizioni fisiologiche, gli agenti alchilanti sono responsabili del processo di alchilazione che consiste nella sostituzione di un atomo di idrogeno con un gruppo alchilico in un composto organico.
L’alchilazione delle basi è responsabile della attività antitumorale e citotossica di questi farmaci in quanto può portare alla lettura errata o all’appaiamento anomalo durante la replicazione del DNA. Il principale evento citotossico è la produzione di un legame crociato “cross-link” interfilamento, generalmente tra due guanine, che è responsabile di rotture nella doppia elica di DNA. In questo modo, gli agenti alchilanti inducono un danno che non può essere riparato e di conseguenza la cellula va incontro ad apoptosi. Inoltre, il cross-linking può impedire la despiralizzazione della catena di DNA bloccando il processo di replicazione e di trascrizione.
Gli agenti alchilanti possono legarsi al DNA durante qualsiasi fase del ciclo cellulare, ma il loro effetto citotossico si esplica quando la cellula entra in divisione, per cui sono detti agenti S-dipendenti. Appartengono a questo gruppo di farmaci le mostarde azotate, tra cui il melphalan e la
ciclofosfamide che sono utilizzati anche nel trattamento del mieloma multiplo, prima del trapianto (Fig 1.3).
.La ciclofosfamide (CPA) è un pro-farmaco antitumorale che deve essere attivato metabolicamente e il suo meccanismo d’azione e la sua tossicità sono stati associati all’attivazione epatica da parte del sistema Citocromo P450, attraverso due vie principali che sono rispettivamente reazioni di attivazione e di inattivazione del farmaco. Dalla prima via si formano metaboliti attivi coinvolti nel cross-link del DNA, mentre la via alternativa forma metaboliti inattivi ed è secondaria, interessando meno del 10% della dose[37].
L’efficacia di questi farmaci è spesso limitata dalla comparsa della resistenza acquisita (ADR), in cui dopo dosi ripetute del farmaco si osserva una citotossicità sempre più ridotta e quindi una minore efficacia terapeutica.
1.5) L
A FARMACO RESISTENZAUna discreta percentuale di pazienti affetti da mieloma multiplo possono risultare refrattari al trattamento chemioterapico. La mancata risposta è essenzialmente determinata da un fenomeno noto come “resistenza”, che rende la cellula tumorale insensibile all’azione di un ampio spettro di farmaci. Durante il trattamento le cellule farmaco-resistenti diventano la componente principale nella popolazione tumorale.
Almeno due tipi di alterazioni genetiche sono capaci di conferire questa caratteristica alle cellule tumorali; le mutazioni puntiformi (SNPs) e i fenomeni di amplificazione genica, che possono alterare l’espressione e/o l’attività di proteine coinvolte nel meccanismo di azione o di biotrasformazione del farmaco. Queste caratteristiche possono essere intrinseche oppure acquisite durante il processo patogenetico o per effetto del trattamento chemioterapico.
Cellule tumorali diverse possono essere dotate di una resistenza semplice, quando interessa un singolo agente terapeutico, oppure multipla, quando si rivolge verso più agenti chemioterapici. La resistenza multipla, osservata principalmente in vivo, è il risultato dell’interazione di diversi meccanismi e viene complessivamente indicata con il termine di Multidrug Resistance (MDR)[38].
Tra le cause più studiate che influenzano la resistenza ai farmaci ci sono le alterazioni dei sistemi di trasporto attraverso la membrana cellulare, responsabili delle variazioni di concentrazione del farmaco nella cellula. Anomalie del sistema di trasporto attraverso le membrane, infatti, possono impedire l’accumulo del farmaco nel versante intracellulare, che non raggiunge una concentrazione sufficiente a produrre effetti citotossici. In generale, l’aumento dell’efflusso extracellulare di un determinato farmaco è dovuto ad una maggiore attività di particolari trasportatori di membrana, quali la P-Glicoproteina 1 (Pgp1) e la MRP (Multidrug Resistance associated Protein).
Già nel 1983, il fenomeno MDR fu messo in relazione all’attività di una proteina di membrana espressa in cellule resistenti all’azione di farmaci[39,40]. Bouhris et al. (1990)[41] associarono la resistenza multipla all’azione di una pompa ATP-asica di membrana nota come P-Glicoproteina 1 (Pgp1), codificata dal gene MDR1 (o ABCB1), la cui espressione aumentava drasticamente nelle cellule tumorali. Oggi sono stati descritti anche altri meccanismi di redistribuzione dei farmaci nei compartimenti organici (intra- ed extracellulare), che coinvolgono altre proteine come MRP o LRP (Lung Resistance Protein). La Pgp1 è coinvolta sia nella farmaco-resistenza intrinseca che in quella acquisita.
1.6) I
L GENE E LA PROTEINA:
MDR1
e
P-Glicoproteina 1
Il gene MDR1 è situato sul cromosoma 7q21.1 (Fig. 1.6), è lungo 209 kb ed è
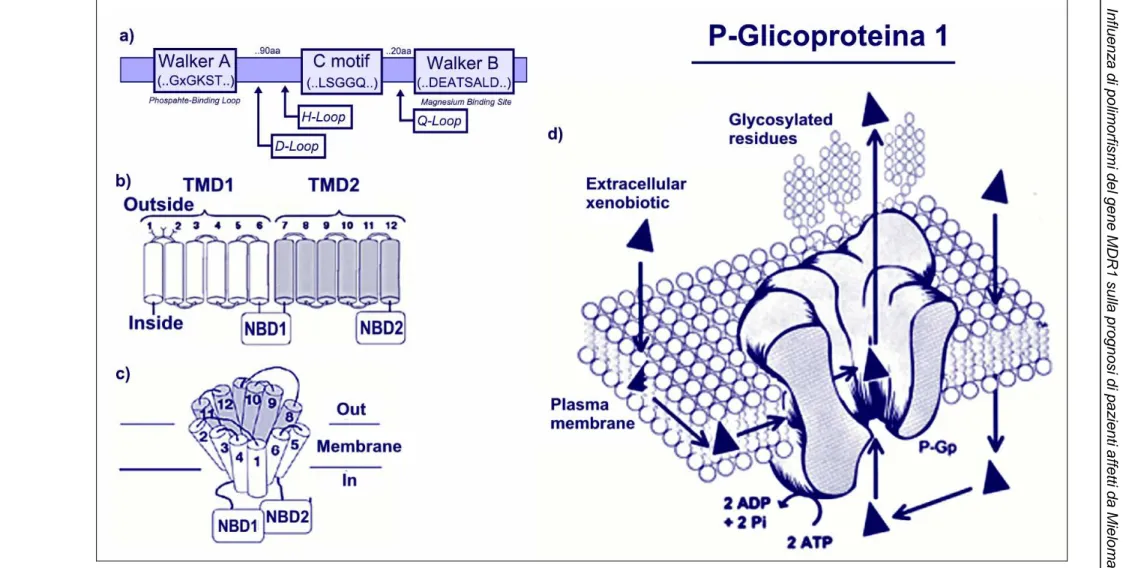
state descritte varie forme di splicing alternativo[42]. La sequenza codificante costituisce circa il 2,5% del gene, con un RNA messaggero lungo circa 4,7kb[43]. Il gene MDR1, o ABCB1, codifica per la P-Glicoproteina 1, una proteina integrale di membrana di circa 170 KDa, costituita da 1280 amminoacidi, appartenente alla superfamiglia dei trasportatori ABC (ATP-Binding Cassette).
Per questo gene sono stati descritti numerosi polimorfismi a singolo nucleotide disseminati lungo l’intera sequenza, sia nella regione codificante che non codificante (Fig. 1.6).
e n z a d i p o lim o rfi s m i d e l g e n e M D R 1 s u lla p ro g n o s i d i p a z ie n ti a ffe tti d a M ie lo m a M u lti p lo IN T R O D U Z IO N E 2 9
Figura 1.6: Dal gene alla proteina. Il gene MDR1 è localizzato nella sottobanda 21.1 del braccio lungo del cromosoma 7. E’ formato da 29 esoni, dalla cui trascrizione e processing
1.6.1) P-GLICOPROTEINA 1: struttura e meccanismo d’azione
La P-Glicoproteina 1 (Pgp1) è una potente pompa di efflusso cellulare ATP-dipendente, in grado di regolare l’uscita dalla cellula di una vasta gamma di substrati lipofilici differenti (xenobiotici), tra cui molti farmaci antitumorali, ormoni steroidei ed antibiotici.
La proteina è codificata sotto forma di un unico polipetide, il quale è costituito da due regioni amminoacidiche ripetute, risultato di eventi di
duplicazione genica, separate da una regione idrofilica di collegamento (Fig.
1.7b). Ognuna di queste due regioni è costituita da sei segmenti amminoacidici che attraversano la membrana cellulare, formando complessivamente un dominio transmembrana (TBD – Transmembrane Binding Domain). Nella parte terminale di ciascun dominio transmembrana si trova una regione amminoacidica idrofilica che costituisce il sito di legame per l’ATP (NBD – Nucleotide Binding Domain)[44].
La P-Glicoproteina 1, che è stata isolata per la prima volta in cellule di criceto resistenti alla colchicina[45], è stata descritta come un largo alloggiamento sul lato extracellulare, che assume una forma ad imbuto sul versante citoplasmatico. Recentemente si sono registrati progressi nella comprensione della struttura e del meccanismo di azione di questa proteina. Le due metà della molecola si dispongono specularmene attorno ad un asse centrale, delimitando un canale centrale la cui forma è quella di un tronco di cono rovesciato. Dal punto di vista della sua sequenza amminoacidica primaria, la P-Glicoproteina 1, come tutti i membri della famiglia dei trasportatori ABC, è caratterizzata da domini conservati. In particolare, si ritrovano i motivi Walker A e B, presenti in moltissimi domini di legame per l’ATP, ed il motivo C, anche detto “signature”, che è peculiare dei trasportatori ABC. Altri motivi proteici, come i loop H, D e Q sono importanti nella formazione del dominio di legame per il nucleotide (ATP) (Fig. 1.7a). La struttura dei NBDs mostra che i due domini formano un “nucleotide-sandwitch dimer”, nel quale l’ATP si lega all’interfaccia dei due domini, circondato dai domini Walker A e B di una subunità e dal motivo C e dal loop
D dell’altra. L’anello dell’Adenina interagisce con il loop Q, e, quando possibile, con il loop H[46].
La struttura della P-Glicoproteina 1 è stata determinata mediante microscopia elettronica. E’ stato ricostruito il primo modello tridimensionale della proteina, nel quale si evidenziano le relazioni tra le α-eliche dei domini transmembrana e i due domini di legame per l’ATP. Con questi studi, si sono ottenute importanti indicazioni circa le modalità di azione della proteina. Nella sua conformazione nativa la regione TMD della proteina ricorda un cilindro di 5/6 nm di diametro e circa 5 nm di lunghezza. Questo cilindro circonda un poro centrale che sembra essere aperto verso il lato extracellulare e chiuso alla base [47,48,49]. Tale struttura evidenzia la disposizione speculare delle due metà della proteina a determinare il canale centrale, il cui diametro varia da un minimo di 9 Å ad un massimo di 50 Å[46] (Fig. 1.7d).
Il meccanismo di azione di questa pompa ATP-asica non è stato ancora determinato con precisione. Studi recenti di mutagenesi sito-specifica hanno evidenziato che il sito di legame del substrato è collocato all’interfaccia di contatto tra i due domini transmembrana[44] e che il legame tra substrato e trasportatore è indipendente dall’azione ATP-asica della proteina. Sono stati proposti differenti meccanismi per l’azione della pompa.
L’idrolisi dell’ATP sembra, invece, in grado di determinare cambiamenti conformazionali nei segmenti transmembrana che costituiscono il dominio di legame per il substrato, portando a profondi sconvolgimenti nella struttura della proteina, con la rotazione di una α-elica (TMD 6) e l’apertura di lacune laterali che mettono in contatto il canale centrale con l’ambiente della
membrana cellulare[48]. Questa osservazione confermerebbe l’ipotesi
secondo la quale la P-Glicoproteina 1 interagisce anche con quei substrati ancora sospesi nella fase lipidica della membrana, escludendoli prima ancora che entrino nella cellula. L’azione dell’ATP risulta cruciale nella regolazione dei cambiamenti conformazionali. Il legame della molecola è infatti l’evento che fornisce l’energia necessaria per il riarrangiamento della struttura della proteina. Tali cambiamenti permangono fintanto che l’ADP resta legato alla proteina, mentre il suo rilascio porta la struttura a ritornare alla configurazione nativa.
e n z a d i p o lim o rfi s m i d e l g e n e M D R 1 s u lla p ro g n o s i d i p a z ie n ti a ffe tti d a M ie lo m a M u lti p lo IN T R O D U Z IO N E 3 2
Figura 1.7: a) Strutture e organizzazione della P-Glicoproteina 1. a) Nella sua sequenza primaria si ritrovano i motivi amminoacidici che caratterizzano i membri della superfamiglia
ABC. b) La proteina è costituita da un’unica sequenza amminoacidica organizzata in due domini transmembrana (TMD1, TMD2), e presenta due domini per il legame di nucleotidi (NBD1, NBD2). c) Organizzazione spaziale dei segmenti transmembrana dei due domini e formazione di interfacce funzionali tra domini differenti. d) Rappresentazione della P-Glicoproteina 1 nella membrana cellulare e suo funzionamento.
Uno dei meccanismi di azione proposti per l’azione della proteina è riportato nello schema in figura 1.8.
Figura 1.8: Possibile meccanismo di azione della Pgp1.
Secondo questa ipotesi, il legame del substrato sarebbe indipendente dal legame dell’ATP, mentre quest’ultimo determinerebbe i cambiamenti conformazionali necessari all’efflusso del substrato. Il rilascio dell’ADP determinerebbe il ripristino della configurazione nativa[48].
Sebbene permangano ancora molte lacune sul meccanismo di azione della P-Glicoproteina 1 e non esistano ancora dati precisi circa la localizzazione e la struttura del sito di legame del substrato, è ben dimostrato che la P-Glicoproteina 1 ha un’attività estremamente aspecifica, per cui è in grado di modulare il trasporto di una vastissima gamma si sostanze differenti. Studi di mutagenesi sito-specifica hanno chiarito che il substrato interagisce con alcuni residui amminoacidici situati all’interfaccia delle α
-eliche 4,5,6 del TMD1 e 9,10,11 del TMD2[44], mentre i cambiamenti
conformazionali indotti dal legame dell’ATP interessano in particolare l’α -elica 6 del TMD1[48]. L’ipotesi del “substrate-induced fit” prevede che il sito di legame del substrato sia variabile, ed in particolare che dipenda dal substrato. Ciascuna molecola che interagisca con la P-Glicoproteina 1, all’interno dell’alloggiamento proteico, stabilirebbe relazioni con residui amminoacidici differenti delle sei α-eliche coinvolte, secondo la sua natura
chimica. In questo modo, substrati diversi interagirebbero con residui diversi e soprattutto con forza differente[44].
I farmaci utilizzati nel protocollo terapeutico DAV, ovvero adriamicina[50], vincristina[51], e desametasone[52], sono compresi tra i substrati della P-Glicoproteina 1.
1.6.2) P-GLICOPROTEINA 1: localizzazione ed espressione
Per molti anni si è pensato che la P-Glicoproteina 1 fosse espressa unicamente nelle cellule tumorali, dove si riteneva che fosse in larga misura responsabile del fenomeno della farmaco-resistenza multipla. Tuttavia, è stato ampiamente dimostrato come questa proteina sia comunemente espressa in diversi organi e tessuti, dove è presumibilmente associata ad altre funzioni fisiologiche.
Infatti, Pgp1 è espressa a livello del fegato, dei reni, del sistema nervoso centrale (plessi corioidei e barriera emato-encefalica), della placenta, delle ovaie e dei testicoli. Ancora, la P-Glicoproteina 1 è stata individuata nelle cellule staminali ematopoietiche, nelle cellule PBM (Peripheral Blood Mononuclear Cells), nei macrofagi, nelle cellule APC (Antigen-Presenting Cells) e nei linfociti T e B (Fig. 1.22). Sia la funzione che la localizzazione, prevalentemente in zone di scambio, suggeriscono che la P-Glicoproteina 1 abbia un ruolo determinante come “barriera” in grado di eliminare tossine e molecole dannose, provvedendo alla loro escrezione nella bile, nelle urine e nel lume intestinale[53].
1.6.3) MDR1: polimorfismi a singolo nucleotide (SNPs)
Il primo lavoro sui polimorfismi del gene MDR1 risale al 1989, quando Kioka et al.[54] individuarono due sostituzioni amminoacidiche, Gly185Val e Ala893Ser, nella sequenza della proteina, rivelando l’esistenza di due probabili polimorfismi genetici. Nel 1998, 11 anni dopo, Mickley et al.[55] individuarono due polimorfismi genetici, gli SNPs G2677T, situato nell’esone 21, e G2995A, situato nell’esone 24. Il primo screening sistematico del locus MDR1 fu effettuato nel 2000 da Hoffmeyer e colleghi[56], i quali analizzarono i 29 esoni del gene MDR1 in un campione di 21 volontari caucasici sani, individuando 15 SNPs. Un altro importante screening fu effettuato da Cascorbi et al. (2001)[57] su 461 volontari tedeschi e portò alla luce nuovi polimorfismi del gene MDR1. Ad oggi sono stati identificati più di 40 SNPs, di cui 29 ampiamente caratterizzati e descritti, 19 nelle sequenze esoniche e di cui solo 11 causano una sostituzione amminoacidica[43] (Tab. 1.1).
SNP POSITION EFFECT SNP POSITION EFFECT
Ala/-41G Intron noncoding C12/44T Intron
C-145G Exon 1a noncoding C1474T Exon 13 Arg492Cys T-129C Exon 1b noncoding T17/-76A Intron
C-4T Exon 2 noncoding A17/137G Intron
G-1A Exon 2 noncoding C2650T Exon 21 Silent A61G Exon 2 Ans21Asp
G5/-25T Intron
G2677T/A Exon21 Ala893Ser Ala893Thr G5/-35C Intron A2956G Exon 24 Met986Val T307C Exon Phe103Leu G2995A Exon 24 Ala999Thr C6/139T Intron A3320C Exon 26 Gln1107Pro C6/145T Intron C3396T Exon 26 Silent A548G Exon7 Asn183Ser T3421A Exon 26 Ser1141Thr G1199A Exon 11 Ser400Asn C3435T Exon 26 Silent C1236T Exon 12 Silent G4030C Exon 28 Silent A4036G Exon 28 silent
Tabella 1.1: SNPs individuati e caratterizzati entro la sequenza del gene MDR1. Di questi (29), 19 sono situati
su sequenze esoniche, e ben 11 determinano sostituzione amminoacidica.
Il ruolo della Pgp1 sull’azione dei farmaci ha reso i polimorfismi di questo gene oggetto di un gran numero di ricerche volte a stabilire una eventuale correlazione tra l’espressione o l’attività della P-Glicoproteina 1 ed una specifica configurazione allelica. Nel loro lavoro pionieristico, Hoffmeyer e colleghi (2000)[56] osservarono una alterazione dell’espressione della P-Glicoproteina 1 in relazione al polimorfismo C3435T. In particolare, la
quantità della proteina, rilevata mediante metodi immunoistochimici, decresce in funzione del genotipo ed è maggiore negli omozigoti C/C, intermedia negli eterozigoti C/T e ridotta (fino a due volte inferiore) negli omozigoti T/T[56]. L’impatto di questa osservazione è stato notevole, tanto che le ricerche in ambito clinico degli ultimi 5 anni si sono focalizzate principalmente proprio sul polimorfismo silente C3435T. Altri ricercatori, a distanza di poco tempo, hanno confermato le osservazioni di Hoffmeyer. Misurando sia i livelli di mRNA che l’efflusso del substrato rodamina in 31 individui caucasici, Hitzl e colleghi (2000)[58] hanno osservato come l’espressione di Pgp1 nelle cellule natural killer CD56+ variasse in maniera genotipo-dipendente (CC>CT>TT). Questi risultati sono stati ancora confermati in uno studio su 59 pazienti affetti da HIV, in cui l’mRNA di MDR1 e i livelli di proteina nelle cellule PBM sono più alti per il genotipo C/C, decrescono negli eterozigoti C/T, diminuendo ulteriormente negli omozigoti T/T[59].
Tuttavia, con l’aumentare delle osservazioni sono emersi anche risultati in controtendenza. Infatti, in uno studio condotto su 13 volontari giapponesi Nakamura e colleghi (2002)[60] hanno osservato una maggiore espressione della P-Glicoproteina 1 in relazione al genotipo T/T, anziché al genotipo C/C. In particolare, la maggiore espressione della P-Glicoproteina 1 a livello del duodeno, quantificata attraverso l’analisi dell’mRNA, risultava associata al genotipo omozigote T/T, mentre è intermedia per gli individui C/T e minore per gli individui C/C[60]. Un risultato simile è stato messo in evidenza da Illmer e colleghi (2002)[61], i quali hanno evidenziato come i livelli di mRNA per MDR1 sono minori in pazienti affetti da leucemia mieloide acuta di genotipo CC[61].
Ancora oggi non esiste una visione comunemente accettata circa gli effetti del polimorfismo C3435T sull’espressione o sulla funzionalità della proteina. Recentemente, tuttavia, alcuni lavori hanno avanzato nuove ipotesi ed aggiunto interessanti spunti al dibattito. Uno degli elementi che contribuiscono ad alimentare la controversia deriva dalla natura silente del polimorfismo C3435T, la quale non consente spiegazioni intuitive delle modalità attraverso cui si possano determinare cambiamenti nella funzionalità e/o nell’espressione della proteina. Due lavori differenti, apparsi
in letteratura in tempi recenti, hanno aggiunto al dibattito nuovi punti di riflessione circa le modalità secondo cui il polimorfismo C3435T potrebbe influenzare l’espressione della P-Glicoproteina 1.
Un primo spunto interessante deriva dal lavoro di Wang e colleghi (2005, 2006)[62,63] che in due pubblicazioni successive hanno evidenziato come la stabilità dell’mRNA di MDR1 sia influenzata dal polimorfismo C3435T. In cellule eterozigoti (C/T), Wang e colleghi hanno osservato che l’mRNA codificato dall’allele T risultava presente in concentrazioni nettamente inferiori. Studi in silico sul folding dell’mRNA hanno suggerito che l’allele 3435T potrebbe determinare un drastico cambiamento nella conformazione dell’mRNA, spingendo gli autori a concludere che proprio questo cambiamento conformazionale potrebbe essere alla base della minore stabilità dell’mRNA codificato dall’allele T[62,63] (Fig. 1.9).
Tuttavia, in tempi ancora più recenti è stato proposto un ulteriore meccanismo attraverso il quale il polimorfismo silente C3435T potrebbe influenzare l’espressione della P-Glicoproteina 1. Sebbene non comporti sostituzione amminoacidica, il polimorfismo determina il cambiamento del codone, da ATC a ATT, sempre specifico per l’isoleucina. Questa sostituzione determina un calo nella frequenza del “codon usage” che passa dal 47% (ATC) al 35% (ATT). L’utilizzo di codoni rari potrebbe alterare il tasso di traduzione, e quindi l’espressione della proteina. Di conseguenza, la presenza dell’allele 3435T potrebbe determinare una diminuzione del tasso di traduzione per la presenza di un codone più “raro”. Se combinato in aplotipi con altri SNPs, come il G2677T/A e il C1236T, l’effetto sull’espressione della proteina può essere molto più forte[64].
La caratterizzazione del gene MDR1, comunque, ha riguardato anche altri polimorfismi. Dopo il C3435T, il polimorfismo triallelico G2677A/T, localizzato sull’esone 21, che comporta ben tre possibili sostituzioni amminoacidiche (893Ala, 893Thr e 893Ser), è stato oggetto del maggior numero di osservazioni. Le ragioni sono da ricercare prevalentemente nel fatto che si tratta di un polimorfismo non sinonimo.
Figura 1.9: Cambiamenti conformazionali nella struttura secondaria dell’mRNA di MDR1 determinati dagli alleli
Una delle ipotesi più accreditate nella comunità scientifica è infatti quella per cui le alterazioni dell’espressione attribuite allo SNP C3435T siano in realtà dovute al suo linkage con un’altra mutazione, che sarebbe la vera responsabile dell’effetto fenotipico. In particolare, il polimorfismo G2677T/A è stato a lungo sospettato di essere la vera causa delle alterazioni osservate anche se, ad oggi, lo studio di Schaefer et al. (2006)[65] è l’unico che dimostri un effetto di questo polimorfismo sull’attività della proteina. In questo studio, effettuato in vitro su sistemi di espressione ricombinanti, è stato osservato che le tre varianti alleliche erano associate ad un’alterata funzionalità della proteina, misurata come efficienza di trasporto della vincristina. In particolare, l’allele 2677A (893Thr) determina un aumento di circa 4 volte della funzionalità della proteina rispetto all’allele wild type (2677G – 893Ala), mentre l’allele 2677T (893Ser) determina un aumento di due volte rispetto al wild type. Nessuna delle tre varianti influenzerebbe però l’espressione della proteina[65].
La lista dei polimorfismi di MDR1 indagati a fondo ne comprende molti altri, riportati in tabella 1.1, tra cui in particolar modo gli SNP C1236T e T-129C. Una delle evidenze che sta emergendo con maggior forza dai dati è quello della presenza di blocchi di linkage disequilibrium più o meno estesi all’interno del gene MDR1. Sono ormai molte le evidenze a supporto di un
linkage disequilibrium tra gli SNPs C3435T e G2677T/A[43,66,67,68], ma anche
di blocchi di linkage disequilibrium più ampi, comprendenti anche gli SNP
C1236T e T-129C[67,68]. Per questo motivo, oggi, uno degli ambiti che viene
esplorato con maggiore curiosità è quello che riguarda le combinazioni aplotipiche dei polimorfismi di MDR1.
Lo studio dei polimorfismi di MDR1 è quindi un campo di investigazione vasto ed importante per la ricerca degli effetti sulla farmaco-resistenza mediata da questo gene.
1.6.4) MDR1: SNPs e frequenze alleliche
La superfamiglia dei trasportatori ABC è forse la famiglia genica più ampia e antica studiata fino ad oggi. Nell’uomo sono stati individuati 48 geni codificanti per altrettanti trasportatori, classificati in 7 famiglie differenti, attraverso l’osservazione di omologia di sequenze amminoacidiche o nucleotidiche. I trasportatori ABC, infatti, sono caratterizzati dalla presenza di un dominio di legame dell’ATP (ATP Binding Domain) estremamente conservato, le cui tracce hanno permesso di individuare e ricostruire la storia evolutiva di questa superfamiglia genica[69].
La sottofamiglia ABCB, di cui ABCB1 (o MDR1) è forse il membro meglio caratterizzato, comprende 11 geni. L’ampia caratterizzazione che ha riguardato ABCB1 ha portato alla luce alcuni aspetti collaterali di grandissimo interesse nella comprensione delle dinamiche evolutive sia del gene in questione che della superfamiglia ABC. ABCB1 si pensa infatti che sia uno dei primi geni ABC che si sia formato nel corso dell’evoluzione di questa famiglia, in seguito alla duplicazione di un gene ancestrale che ha portato all’attuale organizzazione strutturale del gene e della proteina[70].
Le origini evolutive remote di MDR1 giustificano la presenza nella popolazione mondiale di numerose varianti alleliche, la maggior parte delle quali derivano da mutazioni puntiformi. La gran mole di dati disponibili inoltre ha permesso una valutazione delle frequenze alleliche e genotipiche, almeno per i polimorfismi più studiati, nell’intera popolazione mondiale.
Tra i tanti polimorfismi, la sostituzione silente C3435T è sicuramente la più studiata, a causa della sua frequente associazione con alterazioni dell’espressione e/o della funzionalità della proteina. Un dato interessante, diverso da quello funzionale, emerso in numerosi studi è quello di una distribuzione delle frequenze alleliche, e quindi genotipiche, variabile tra le popolazioni.
Uno studio di Ameyaw et al. (2001)[71], condotto su un campione di 1280 soggetti appartenenti a 10 popolazioni differenti ha messo in luce differenze nella frequenza dell’allele 3435C (Tab. 1.2), che è assai più frequente nelle popolazioni africane (Ganesi, Keniani, Afro-americani) che nelle popolazioni europee e asiatiche. Al contrario, gli individui omozigoti T/T
sono pressoché assenti nelle popolazioni di origine africana. Sebbene l’effettivo ruolo di questo SNP nell’alterazione della funzionalità proteica sia piuttosto controverso, alcuni autori ipotizzano che una maggiore frequenza dell’allele C possa aver rappresentato un vantaggio evolutivo nella difesa da fattori ambientali o presenti nella dieta nelle popolazioni africane. Anche se un fenomeno selettivo di questo tipo potrebbe plausibilmente spiegare la variazione delle frequenze alleliche, le informazioni attualmente disponibili non permettono di giungere ad una conclusione ovvia.
POPULATION N ALLELE FREQUENCY GENOTYPE FREQUENCY
3435 C 3435 T C/C C/T T/T Ghanian 206 0,83 0,17 0,67 0,34 0,00 Kenyan 80 0,83 0,17 0,70 0,26 0,04 African American 88 0,84 0,16 0,68 0,31 0,01 Sudanese 51 0,73 0,27 0,52 0,43 0,06 Caucasian, UK 190 0,48 0,52 0,24 0,48 0,28 Caucasian, Ger 188 0,52 0,48 0,28 0,48 0,24 Portuguese 100 0,43 0,57 0,22 0,42 0,36 South-west Asians 89 0,34 0,66 0,15 0,38 0,47 Chinese 132 0,53 0,47 0,32 0,42 0,26 Filipino 60 0,59 0,41 0,38 0,42 0,20 Saudi 96 0,55 0,45 0,37 0,38 0,26
Tabella 1.2. Frequenze alleliche e genotipiche al sito C3435T in 1280 individui di 10 popolazioni.
Con l’aumentare di dati relativi a molti polimorfismi, l’analisi delle frequenze alleliche all’interno di popolazioni differenti ha potuto farsi più intensa ed esaustiva. Tuttavia, piuttosto che soffermarsi all’analisi delle variazioni nelle frequenze dei singoli polimorfismi, oggi la maggior parte degli studi si è incentrata sulla loro combinazione in aplotipi.
A causa della loro rilevanza funzionale e dell’ormai accertata presenza di linkage disequilibrium, lo studio degli aplotipi ha spesso riguardato gli SNPs C3435T, G2677T/A, C1236T e T-129C. Uno degli studi più completi è quello di Tang e colleghi (2002)[67], il quale fornisce un quadro delle frequenze alleliche relative a ciascuno dei quattro polimorfismi (Tab. 1.3). Osservando i dati riportati in tabella, non può sfuggire come anche per gli altri tre SNP (G2677A/T, C1236T e T-129C) le frequenze alleliche variano in maniera considerevole a seconda del gruppo etnico, variazioni piuttosto significative che disegnano un quadro di profonda diversità a livello della popolazione mondiale.
SNP Population N Allele frequency (%) T C Tedeschi1 85 94,1 5,9 Giapponesi2 100 94 6 Malesi9 80 95,7 4,3 Cinesi9 92 98,4 1,6 Exon 1 -129T>C Indiani9 61 98,4 1,6 T C Afro-Americani3 23 15 85 Tedeschi1 188 37,8 62,2 Tedeschi4 461 41 59 Americani Europei3 37 42 58 Indiani9 62 60,5 39,5 Giapponesi2 100 65,5 34,5 Giapponesi5 48 61,5 38,5 Cinesi9 92 68,6 31,4 Exon 12 1236C>T Malesi9 85 69,4 30,6 G T A Indiani9 68 33,8 61,8 4,4 Giapponesi2 100 43 39 18 Cinesi9 103 50,5 43,7 5,8 Malesi9 93 57,5 36 6,5 Americani Europei3 37 54 46 0 Tedeschi4 461 56,5 41,6 1,9 Exon 21 2677G>T/A Afro-Americani3 23 93,5 6,5 0 C T South-west Asian6 89 34 66 Indiani9 68 39,7 60,3 Portoghesi6 100 43 57 Americani Europei3 37 46 54 Tedeschi4 461 46,1 53,9 Britannici6 190 48 52 Caucasici7 537 50 50 Tedeschi1 188 52 48 Cinesi6 132 53 47 Sauditi6 96 55 45 Giapponesi7 50 57 43 Giapponesi2 100 58 42 Giapponesi8 114 61,4 38,6 Filippini6 60 59 41 Cinesi9 104 59,6 40,4 Malesi9 92 63 37 Sudanesi6 51 73 27 Afro-Americani3 23 74 26 Afro-Amricani7 41 79 21 Ganesi7 172 90 10 Ganesi6 206 83 17 Keniani6 80 83 17 Exon 26 3435C>T Afro-Amricani6 88 84 16
Tabella 1.3. Frequenze alleliche in diverse popolazioni di 4 tra i polimorfismi più comuni del gene MDR1.
Nella tabella 1.4 sono state riassunte le frequenze medie per ciascun polimorfismo raggruppando i dati precedenti in tre macroclassi: popolazioni di origine europea (Caucasici, Tedeschi, Portoghesi, Britannici), africana (Sudanesi, Afro-Amricani, Ganesi, Keniani) e asiatica (Indiani, Cinesi, Malesi,