2.1 “Screening” Genoteca di cDNA
2.1.1 Marcatura di sonde a DNA con “Random priming”
Il metodo dei “Random Priming” si basa sull’uso di un insieme di una miscela di esanucleotidi “Random Hexamers” come “primers” per la sintesi di DNA. In particolare è stato eseguito il protocollo fornito dalla ditta produttrice Armersham Bioscience con il “DNA labelling kit”.
Sono stati utilizzati 25 ng del frammento di DNA da marcare (costituito da DNA linearizzato di nostro interesse) a cui è stato aggiunto 1/10 del volume finale di “Random Primer in 10x reaction buffer”, la miscela è stata poi messa a 100°C per 5 minuti in modo tale da permetterne la denaturazione. Sono stati quindi aggiunti i dNTPs non marcati (mancanti di dCTP), il buffer di reazione 10x, la Klenow (DNA polimerasi specifica), e i dCTPs marcati con l’isotopo radioattivo del fosforo P32, secondo la seguente ricetta: 5microL di buffer 10x, 12microL dNTP(no dCTP), 2microL Klenow, 5microL di dCTP radioattiva e ho portato a 50microL di volume finale con acqua. La miscela è stata fatta incubare per 20 minuti a 37°C in un contenitore piombato per la schermatura dall’emissione radioattiva dell’isotopo di fosforo.
A questo punto la sonda generata è stata purificata usando una colonna cromatografica “Sephadex G-50” in modo da eliminare i dCTPs non incorporati che potrebbero dare origine a segnali aspecifici, e successivamente diluita in 50 mL di Miscela di Ibridazione contenente Salmon Sperm DNA (ssDNA) precedentemente denaturato a 100°C per 5 minuti.
2.1.2 Titolazione genoteca di cDNA
La genoteca utilizzata per lo “Screening” è stata fornita da M. King. Si tratta di una banca di cDNA costruita partendo da mRNA di embrioni st.42 di Xenopus laevis; i frammenti di cDNA sono stati inseriti nel fago λZapII.
La titolazione della banca è stata realizzata mediante diluizioni seriali (1:100; 1:10000; 1:1000000) a partire da un’aliquota di 10microL della banca.
Cellule prelevate da una colonia del ceppo XL1Blue di E.Coli, sono state fatte crescere in 10mL di LB addizionato di 0.2% di maltosio e 10mM di MgSO4 per 8-12 ore. La presenza del maltosio induce l’espressione del recettore per questo zucchero, che è lo stesso recettore riconosciuto dal fago λ.
Con 3microL di ciascuna delle diluizioni della banca di cDNA, si infettano 200microL di cellule a 37°C per 20 minuti. Trascorso questo tempo, si introducono nella miscela 5mL di Top-Agarosio ( LB + 0.7% agarosio autoclavato e raffreddato a 47°C ), si mescola capovolgendo gentilmente il tubo e si versa il tutto in una Piastra Petri piccola contenente Bottom-Agar, preriscaldata a 37°C.
Dopo aver atteso il consolidamento dello strato di Top-Agarosio, la piastra viene messa capovolta a 37°C. Il tempo necessario affinché le placche di lisi diventino ben visibili dipende dal tipo di vettore fagico (minimo 6-7 ore). La crescita deve essere tenuta sotto controllo per evitare che le placche vadano a confluenza ed impediscano un preciso conteggio.
Dal numero di placche di lisi presenti in ciascuna diluizione, è possibile risalire alla quantità di pfu (unità di placche fagiche)/microL.
Soluzioni: Top-Agarosio: 21g/L NZY Broth 0.7g/100mL Agarosio Bottom-Agar (NZY):
20g/L Bottom-Agar in acqua milliQ 21g/L NZY Broth in acqua milliQ
NZY Medium:
10g/L NZamine ( casein hydrolysate enzymatic) 5g/L NaCl 5g/L Bacto-yeast extract 2g/L MgSO4 7H2O LB Medium: 10g/L Bacto-tryptone 5g/L Bacto-yeast extract 10g/L NaCl
Portare a pH 7.0 con NaOH
2.1.3 “Screening” della genoteca di cDNA
Per l’identificazione dei cDNA di nostro interesse, la genoteca, precedentemente titolata, è stata piastrata su Piastre Petri grandi, in modo tale da avere 1.500.000 pfu/piastra; la procedura è analoga a quella adottata per la titolazione, con la differenza che in questo caso si usano 50mL di Top-Agarosio per piastra.
Le placche di lisi vengono arrestate nella crescita al momento opportuno, ponendo le piastre a 4°C. Ciò consente anche l’indurimento del Top-Agarosio che, a sua volta, facilità il trasferimento sul filtro.
Dopo alcune ore si procede al trasferimento su appositi filtri di Nylon delle placche fagiche. Si appoggiano i filtri sullo strato di Top-Agarosio e vi si lasciano per 2 minuti, si sollevano e si mettono per 5 minuti in soluzione denaturante (NaCl 1.5 M + NaOH 0.5 M ). Trascorso questo tempo, si mettono in soluzione neutralizzante per 5 minuti (Tris 0.5M a pH 7.4 + NaCl 1.5M) e poi passo per 2 minuti in SSC 2x. Passo a questo punto all’asciugatura sotto cappa per 2 minuti. Il DNA sui filtri deve essere sempre rivolto verso l’alto. Sulla stessa piastra si pone un secondo filtro, questa volta per 6 minuti. Questa seconda operazione serve per avere una replica che servirà da controllo dei segnali autoradiografici che potrebbero essere falsi positivi e eseguo gli stessi passaggi della prima volta. A questo punto fisso il DNA sui filtri di Nylon sotto gli UV per 3-5 minuti.
I filtri vengono quindi pre-ibridati nella miscela di preibridazione per circa 2 ore e successivamente ibridati a 50°C per tutta la notte con moderata agitazione in una soluzione identica a quella di pre-ibridazione, alla quale ho aggiunto la sonda fatta con “Random Priming”.
Passato il tempo di incubazione, eseguo dei lavaggi di 20 minuti ciascuno: il primo a RT (temperatura ambiente) in SSC 3X addizionato da 0.1% di SDS (sodiododecilsolfato, agente denaturante); il secondo in SSC 3X + 0.1% SDS a 55°C; il terzo in SSC2X + 0.1% SDS a 55°C. Le soluzioni sono precedentemente preriscaldate.
I filtri, asciugati all’aria, sono stati esposti su lastre autoradiografiche (KODAK-X) in cassette con schermi intensificati per 1-2 giorni a -80°C. Le lastre sono state poi sviluppate e fissate usando liquidi opportuni per lo sviluppo e il fissaggio.
Le placche corrispondenti ad un segnale positivo, sia sul primo filtro che sulla replica, sono state recuperate dalle piastre Petri, incubate in 1
mL di SM e 100 microL di cloroformio, lasciate a temperatura ambiente per 6-7 ore e quindi mantenute a 4°C.
A questo punto è stato effettuato un secondo “screening” piastrando diluizioni opportune delle placche isolate in seguito al primo “screening”, in modo da avere colonie nettamente distinguibili (20-100 placche per piastra).
Per avere la certezza di isolare placche di lisi di un unico fago si è proceduto ad uno “screening” terziario, con procedura identica alle precedenti.
Infine dalle placche singole sono state effettuate “minipreps” di DNA fagico. Soluzione Denaturante: 1.5M NaCl 0.5M NaOH Soluzione Neutralizzante: 0.5M Tris-Cl, pH 7.4 1.5M NaCl
Mix di pre-ibridazione e ibridazione: 5x SSC
5x Denhardt’s 0.1% SDS
0.1mg/mL ssDNA (Salmon SpermaDNA)
Denhardt’s:
1% peso/volume Ficoll 400
2.1.4 Estrazione DNA fagico
Le placche positive allo “screening” terziario sono carotate singolarmente e risospese in un tubo da centrifuga da 1.5 mL contenente 500 microL di “SM Buffer” con aggiunti 20 microL di cloroformio e incubate per tutta la notte (O/N) a 4°C.
Contemporaneamente si fa crescere separatamente O/N a 30°C una coltura di cellule XL1Blue in LB addizionato di 0.2% di Maltosio e 10 mM di MgSO4.
Il giorno seguente, centrifugo un minuto le crescite di cellule a 1000xg. Risospendo entrambe le colture in una quantità di MgSO4 10 mM che mi permetta di avere una densità ottica a 600 nm pari a 1.
A questo punto aggiungo a 200 microL di cellule XL1Blue risospese, 250 microL di Stock di fagi (contiene circa 10 di particelle fagiche) e 1 microL di “ExAssist helper phage”( 10 pfu/microL). Incubo a 37°C per 15 minuti.
Aggiungo 3 mL di LB fresco e incubo in blanda agitazione per 2-3 ore a 37°C.
Eseguo uno shock termico a 65-70°C per 20 minuti e centrifugo il tubo per 15 minuti a 1000xg.
Prelevo il sovranatante e lo metto in un tubo sterile e lo faccio decantare. Nel sovranatante sarà contenuto il plasmide pBluescript con l’inserto libero dalle proteine di impacchettamento del fago.
Prelevo due aliquote da 200 microL di cellule XL1Blue risospese, come descritto precedentemente, e le metto due eppendorf da 1.5 mL. In una aggiungo 100 microL e nell’altra 10 microL di sovranatante contenente il plasmide. Incubo a 37°C per 15 minuti.
Piastro 200 microL di cellule in piastre Petri contenenti LB agar e ampicillina (50 microL/mL) e incubo O/N a 37°C.
Il giorno seguente, eseguo un estrazione di DNA plasmidico da cellule batteriche. ( Vedi dopo).
2.2 Trasformazione di cellule di E. coli ed
amplificazione del DNA plasmidico
2.2.1 Trasformazione
Alcuni dei plasmidi, utilizzati nel presente lavoro di tesi, sono stati ottenuti nel nostro laboratorio attraverso processi di clonazione che prevedono il legame di un gene o di una sua porzione ad un DNA plasmidico, mentre altri provengono da laboratori esterni in forma di DNA diluito in acqua od adsorbito su carta 3 MM. In tutti questi casi è necessario amplificare il plasmide d’interesse per averne una quantità sufficiente e ciò è possibile tramite la trasformazione di cellule batteriche competenti all’acquisizione del plasmide.
Le cellule di E. coli, ceppo DH5α, sono state rese competenti mediante due trattamenti, di 30 minuti ciascuno, a 4°C in CaCl2 50 mM, e
conservate a –80°C fino al momento dell’uso. Per trasformare cellule batteriche con DNA plasmidico è sufficiente aggiungere, ad un’aliquota di 100 μl di cellule competenti, 10 ng di plasmide superavvolto e incubare in ghiaccio per 30 min. Segue un “heat shock” a 42°C, per 90 secondi, ed un’incubazione in ghiaccio per 10 minuti. Si aggiungono, successivamente, 500 μl di LB preriscaldato a 37°C, si incuba in stufa per 60 minuti, infine si piastrano le cellule su terreno solido selettivo (LB con agar e ampicillina 100 μg/ml). Dopo incubazione a 37°C per tutta la notte, sulla piastra Petri compaiono le colonie: l’antibiotico fa sì che crescano solo le cellule che hanno assunto il plasmide poiché esso contiene il gene che conferisce resistenza all’ampicillina.
Ogni volta che si effettua una trasformazione, bisogna aver cura di controllare l’efficacia dell’antibiotico piastrando, in parallelo, 100 μl di
Soluzione:
Luria-Bertani Broth (LB)
NaCl 1%
bacto tryptone 1% bacto yeast extract 0,5%
2.2.2 Estrazione di DNA plasmidico su piccola scala mediante lisi alcalina ("miniprep”)
Tale metodo permette di ottenere ≅2 μg di DNA plasmidico. Da una piastra di cellule di E. coli, recanti il plasmide di interesse, o da uno stock di batteri in glicerolo, viene effettuato in maniera sterile un inoculo di un clone che viene posto in un tubo batteriologico da 10-15 ml contenente 3 ml di brodo di coltura LB con ampicillina (100 μg/ml). Il tubo è incubato per 12-16 ore a 37°C in agitazione, affinché la coltura batterica raggiunga la fase di crescita stazionaria. La coltura viene quindi centrifugata a 12000 rpm per 1-3 min. allo scopo di ottenere un “pellet” di cellule batteriche, che viene risospeso in 400 μl di “soluzione 1”. Alla suddetta sospensione si aggiungono 400 μl di “soluzione 2” e la miscela viene mescolata delicatamente per inversione. La reazione di lisi alcalina non deve procedere per una durata superiore a 5 min., trascorsi i quali si aggiungono 400 μl di “soluzione 3”, necessaria a far precipitare le membrane e le pareti delle cellule lisate, insieme al DNA genomico e all’RNA ad alto peso molecolare, che ad esse sono associati. Dopo una centrifugazione di 15 min a 12000 rpm viene recuperato il sovranatante, contenente il DNA plasmidico, l’RNA a basso peso molecolare, le proteine batteriche. Dopo aver aggiunto 0.7 volumi (V) di isopropanolo, alla fase acquosa contenente il DNA plasmidico, la si lascia 10 min. a temperatura (T) ambiente. In questo modo il DNA plasmidico e l’RNA a basso peso
molecolare precipitano e vengono recuperati mediante centrifugazione (10 min. a 12000 rpm). Seguono il lavaggio del “pellet”, ottenuto in etanolo (EtOH) al 70%, e la sua risospensione, in 20 μl di TE (o H2O ultrapura)
contenente RNasi A ad una concentrazione di 20 μg/ml, allo scopo di eliminare l’RNA a basso peso molecolare.
La concentrazione di DNA estratto e purificato viene stimata sottoponendo un’aliquota ad elettroforesi su gel di agarosio, con Bromuro di Etidio (EtBr), insieme ad aliquote di preparazioni a concentrazione nota utilizzate come standard.
Soluzioni: Soluzione 1 Tris-HCl 25 mM pH 8 EDTA 10 mM glucosio 50 mM lisozima 10 mg/ml Soluzione 2 NaOH 0,2 M SDS 1% Soluzione 3 pH 4.8 CH3COOH 5 M KCI 3M TE Tris 10 mM (pH 8.0) EDTA 1 mM (pH 8.0)
2.2.3 Estrazione di DNA plasmidico su media scala mediante colonne NUCLEOBOND
Questa metodica prevede l’utilizzo di colonnine cromatografiche NUCLEOBOND a scambio ionico commercialmente disponibili insieme alle soluzioni necessarie per il loro impiego. Con questa tecnica è possibile estrarre da 45 a 100 μg di DNA altamente purificato. Partendo da una coltura batterica di 50-100 ml, ottenuta dopo incubazione a 37°C in agitazione o/n, se il brodo di crescita è torbido si procede alla centrifugazione della stessa a 4000 rpm per 10 min. Spesso si usano tubi da centrifuga, che hanno una capacità di circa 30 ml, pertanto non tutto il brodo può essere centrifugato contemporaneamente, ma è necessario ripetere il passaggio 2 o 3 volte. Il “pellet” così ottenuto viene risospeso in 4 ml di soluzione E1; quindi si aggiungono 4 ml di E2, si capovolge delicatamente e si lascia a T ambiente, per 5 min. Dopo aver aggiunto 4 ml di soluzione E3 ed aver capovolto delicatamente per cinque volte si centrifuga a 20°C per 10 min. a 15000 x g. Il sovranatante è rimosso velocemente e centrifugato alle stesse precedenti condizioni, al fine di ottenere un lisato meno torbido possibile. Si equilibra una colonnina cromatografica JETSTAR, caricandola con 10 ml di soluzione E4 permettendone lo svuotamento per gravità; si carica quindi la colonnina equilibrata con il lisato, ciò risulta nell’immobilizzazione del DNA da parte della resina. Si libera il DNA dai sali e dall’RNA residuo mediante due lavaggi di 10 ml ciascuno con la soluzione E5. Si procede infine con l’eluizione del DNA dalla colonnina, con 5 ml di soluzione E6.
L’eluato viene precipitato, dopo aggiunta di 0.7 V di isopropanolo, mediante centrifugazione a 4°C per 30 min. a 15000 x g. Il pellet viene lavato con EtOH al 70% e risospeso in TE pH 8.0 (o in H2O mQ).
Per stimare la quantità del DNA estratto, è possibile sottoporre un’aliquota ad elettroforesi su gel di agarosio oppure, se si desidera ottenere una misura più precisa, si può ricorre a tecniche
spettrofotometriche che prevedono la lettura dell’assorbanza alla lunghezza d’onda (λ) di 260 nm di campioni diluiti della preparazione di DNA in esame. Soluzioni: E1 RNasi A 100 μg/ml Tris-HCI 50 mM (pH 8.0) EDTA 10 mM E2 NaOH 200 mM SDS 1 % E3 Potassio acetato/ 3.1 M Acido acetico (pH 5.5) E4 NaCl 600mM Sodio acetato 100 mM TritonX-100 0.15% Acido acetico (pH 5.0) E5 NaCl 800 mM Sodio acetato/ 100 mM Acido acetico (pH 5.0)
E6
Tris/HCl (pH 8.5) 100 mM
NaCl 1250 mM
2.3 Digestione di plasmidi con enzimi di
restrizione
Per ottenere DNA stampo lineari, necessari per effettuare trascrizioni in vitro, si utilizzano enzimi di restrizione che tagliano nel plasmide, ma non nell’inserto, alla fine della regione che si vuole trascrivere. Vengono usati preferibilmente enzimi che lasciano un’estremità al 5’ protrudente o “blunt”; si evita così che l’RNA venga trascritto a partire dal filamento complementare a quello desiderato, cosa che può succedere usando un DNA stampo con estremità 3’ sporgente. Capita talvolta di potere utilizzare solo enzimi che lasciano estremità sporgente al 3’. In questi casi si sfrutta la attività esonucleasica della T4 DNA polimerasi aggiungendo al DNA linearizzato la miscela di trascrizione priva dei nucleotidi e della RNA polimerasi, 5 U/μg di T4 e lasciando agire per 15 minuti, a 22°C. Si procede quindi con la trascrizione, aggiungendo nucleotidi e RNA polimerasi.
Ovviamente l’uso di enzimi di restrizione non è limitato alla produzione di DNA stampo per trascrizione. In questa sede sono stati utilizzati enzimi per clonare un gene, per verificarne l’orientamento in un plasmide e per rimuovere sequenze geniche che interferivano con i nostri scopi.
Le digestioni vengono generalmente effettuate in un volume finale di 20 μl. La miscela di reazione è costituita dal DNA plasmidico, dall’enzima
di restrizione, presente in concentrazione di 1-2 Unità Enzimatiche (UE) per μg di plasmide e dal tampone specifico per il tipo di enzima utilizzato. Occorre che il volume di enzima aggiunto, non superi 1/10 del volume della miscela di reazione, poiché un’eccessiva concentrazione di glicerolo, in cui gli enzimi sono conservati, può interferire con la cinetica di reazione. La reazione di digestione viene fatta procedere per una o più ore, in base alla quantità di DNA che deve essere digerito. In genere sono sufficienti 1-3 ore di incubazione a una temperatura che può variare a seconda del tipo di enzima di restrizione. Al fine di verificare se la digestione è completa si carica su gel di agarosio all’1%, colorato con bromuro di etidio, 1/20 della miscela di reazione accanto ad un’aliquota di plasmide non digerito. Se la digestione, linearizzazione, è completa avremo una banda unica che teoricamente dovrebbe migrare più lentamente rispetto al superavvolto circolare. Se la digestione del DNA ha avuto successo si procede con l’estrazione fenolica.
2.4 Purificazione del DNA
2.4.1 Estrazione fenolica
Per purificare il DNA dalle proteine, si porta la miscela di digestione ad un volume di 200 μl con acqua e si aggiunge un uguale volume di fenolo-cloroformio-alcool isamilico, soluzione 25:24:1 a pH 8. Dopo aver mescolato energicamente, si centrifuga per 3 minuti a 12000 giri. Si ottengono due fasi, una organica contenente le proteine ed una acquosa contenente il DNA. Si prende il sovranatante acquoso e si mette in una provetta pulita.
Questa tecnica si basa sul fatto che le proteine sono più solubili in fase organica che in fase acquosa: fenolo e cloroformio denaturano le proteine, il cloroformio facilita la separazione delle fasi mentre l’alcool isamilico riduce la formazione di schiuma durante l’estrazione.
Procediamo quindi con la precipitazione alcolica.
2.4.2 Precipitazione alcolica
Il DNA viene precipitato, aggiungendo al prodotto dell’estrazione fenolica 2.5V di EtOH assoluto e sali (NaCH3COOH pH 5.2, 0.3M finale).
Dopo aver mescolato, capovolgendo il tubo diverse volte, si mette il tutto a precipitare 60 min. a -80°C oppure 4 ore a -20°C. Quindi si centrifuga per 20 minuti a 12000 rpm e si lava il “pellet” dai sali con EtOH al 70% freddo. Una volta che il “pellet” è asciugato, all’aria o con l’ausilio di una pompa a vuoto, si sospende il DNA in acqua sterile e si conserva a -20°C. Per stimare la concentrazione di linearizzato, si prende un volume di DNA che presumiamo corrispondere a circa 200/300 ng di DNA, supposto un recupero medio del 80% dall’estrazione fenolica, e lo si carica su gel di agarosio per verificarne la quantità reale.
2.5 Elettroforesi su gel di agarosio
Al fine di verificare la purezza del DNA estratto, la purezza dei trascritti, il grado di completezza raggiunto dalla digestione del DNA, nonché di stimare la concentrazione del DNA nelle preparazioni e la lunghezza in paia di basi degli acidi nucleici, si prepara un gel di agarosio allo 0.8-1.5% (peso/volume).
I gel sono preparati sciogliendo l’agarosio in TBE, portato alla temperatura di ebollizione. Prima che il gel polimerizzi si aggiunge EtBr ad una concentrazione finale di 10 μg /ml. Il gel, lasciato un poco a raffreddare viene colato in un lettino da elettroforesi di un apparato orizzontale. Una volta polimerizzato il gel è posto nell’apparato ed immerso in un tampone di corsa, TBE a pH 8.
Frattanto si preparano i campioni che vengono diluiti in acqua e “loading buffer”. La funzione del “loading buffer” è di appesantire il campione, facendolo andare sul fondo del pozzetto, e consentire al contempo di seguire la corsa elettroforetica.
Si caricano i campioni su gel e si applica una differenza di potenziale di 50/120 V per un tempo variabile dai 5 ai 60 minuti. Si visualizzano infine le bande degli acidi nucleici ponendo il gel sotto raggi UV; il bromuro di etidio che si è intercalato alle basi appare in queste condizioni luminescente. Le dimensioni dei frammenti sono stimate in presenza di marcatori con peso molecolare noto. Nel caso si stia lavorando con RNA si prendono le vaschette da usare per colare i gel di agarosio e gli apparati elettroforetici e si rendono “RNasi free” (RF) mediante pretrattamento con NaOH 0.1M per 20 minuti circa.
Soluzioni: TBE pH 8.0 Tris base 0.089 M Acido borico 0.089 M EDTA 0.002 M Loading buffer 6x glicerolo 5% blu di bromofenolo 0.05% xilene cianolo 0.05% Gel di agarosio agarosio 0.8-1.5% (peso/volume) bromuro di etidio 10 μg/ml finale
TBE 1X
2.6 Embrioni di Xenopus laevis
Gli embrioni sono ottenuti mediante la fecondazione in vitro: il maschio viene anestetizzato mediante immersione in una soluzione allo 0,1% di MS222 (metanosulfonato dell’estere etilico dell’acido 3-aminobenzoico), poi l’animale è risciacquato accuratamente sotto acqua di rubinetto, prima di essere operato per la rimozione del testicolo. Il testicolo può essere conservato per qualche giorno a 4°C in 1X MMR, NaCl 20 mM e gentamicina.
Femmine di Xenopus laevis vengono stimolate con 100 UI di Folligon Intervet per uso veterinario, da 4 a 11 giorni prima della deposizione e
con 800/1000 UI di Profasi MP 2000 Serono (gonadotropina corionica) 10-12 ore prima della deposizione. Le gonadotropine vengono somministrate per iniezione nel sacco perilinfatico. Le femmine vengono indotte a deporre esercitando manualmente una pressione sul loro addome. Le uova vengono raccolte in piastre Petri, contenenti MMR 0.1X. Quindi si elimina dalle piastre l’MMR 0.1X e si effettua la fecondazione passando, per circa 2/3 minuti, un frammento di testicolo sopra le uova che vengono poi lasciate a secco per 5 minuti. Si riaggiunge poi MMR 0.1X. La raccolta delle uova può essere fatta ripetutamente ad intervalli di 1-2 ore.
Dopo almeno 25 minuti dalla fecondazione, gli embrioni vengono privati del loro rivestimento gelatinoso, mediante un trattamento con la soluzione degelificante di DTT, seguito da un accurato lavaggio direttamente sotto acqua corrente, infine gli embrioni vengono lasciati sviluppare in MMR 0,1X fino allo stadio opportuno: 2/4/8 cellule nel caso di embrioni da iniettare, stadi più avanzati per esperimenti di ibridazione. Nel caso si debbano raccogliere embrioni da processare per “whole mount” è necessario asportare manualmente anche la membrana vitellina, questo viene effettuato mediante l’ausilio di pinzette Dumont n° 5.
Gli stadi embrionali richiesti sono stati identificati secondo i criteri di Nieuwkoop e Faber (1967).
Gli embrioni da conservare o da sottoporre a ibridazione in situ “whole mount”, vengono fissati in “vials” di vetro da 5 ml, contenenti MEMFA. La fissazione viene fatta durare per 1 ora a T ambiente, trascorsa la quale la soluzione MEMFA viene rimossa e sostituita con EtOH assoluto, che consente di conservare gli embrioni per mesi alla temperatura di -20°C.
Soluzioni: MMR NaCl 0.1 M KCl 2 mM MgSO4 1 mM CaCl2 2 mM HEPES 5 mM pH 7.8 EDTA 0.1 m Soluzione degelificante DTT 3.2 mM Tris-HCl 0.2 M pH 8,8 MEMFA 1X MOPS 0.1 M pH 7.4 EGTA 2 mM MgSO4 1 mM Formaldeide 3.7%
In genere si conservano stocks 10X sterili di una soluzione di sali che si diluiscono e si addizionano con formaldeide al momento dell’uso.
2.7 Microiniezione in embrioni di
Xenopus laevis
Per gli esperimenti di microiniezione sono stati utilizzati embrioni pigmentati, che permettono di distinguere, allo stadio di 4-8 cellule, il polo
animale da quello vegetativo e i blastomeri dorsali da quelli ventrali. Al momento della microiniezione, si trasferiscono gli embrioni, degelificati, in una piccola piastra Petri alla quale è fissata, sul fondo, una reticella di plastica con maglie di circa 1 mm. La reticella limita gli spostamenti degli embrioni durante la microiniezione. Gli embrioni sono immersi in una soluzione contenente 0.1X MMR e Ficoll 4%; il Ficoll è molto vischioso e permette agli embrioni di mantenere la forma sferica durante la fase di iniezione. Gli embrioni microiniettati sono lasciati sviluppare in 0.1X MMR-4% Ficoll nelle prime ore dopo l’iniezione e poi trasferiti in 0.1X MMR. Quando gli embrioni di controllo, non iniettati, raggiungono lo stadio desiderato, si fissano controlli ed iniettati e si conservano a -20°C.
Le microiniezioni sono state eseguite con un microiniettore Eppendorf FemtoJet, che consente l’iniezione di volumi molto piccoli di liquido (nl).
Gli aghi impiegati sono aghi Eppendorf Femtotips II che hanno un diametro esterno di 0.7 μm e un diametro interno di 0.5 μm. Il caricamento dell’RNA da microiniettare può essere eseguito con una micropipetta Gilson direttamente nell’ago stesso prima che questo venga montato sul microiniettore.
2.7.1 Sintesi in vitro dei trascritti da microiniettare
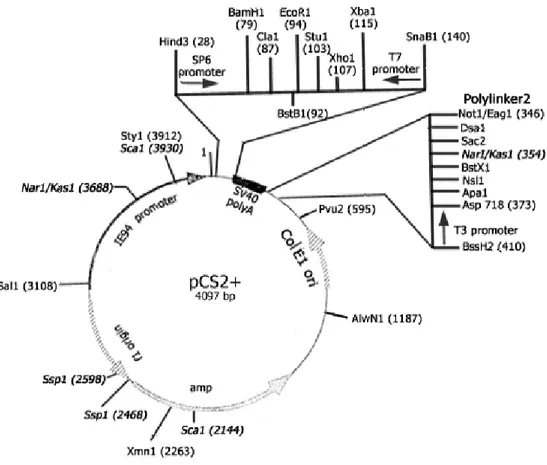
Quando si preparano trascritti da microiniettare, si pone il problema sia della stabilità del trascritto che dell’efficienza della traduzione all’interno della cellula. Per ovviare a questi problemi si inserisce il cDNA dei geni da iniettare all’interno di plasmidi (vedi es. pCS2+) che contengono elementi stabilizzanti l’RNA. Inoltre, per aumentare l’efficienza di traduzione, si aggiunge alla miscela di trascrizione una “terminal CAP
trascritti stabili, in grado di sopravvivere all’interno della cellula. Il “CAP”, tipico di molti RNA cellulari, consiste di una 7-metil-guanosina 5’ trifosfato, che si lega mediante un ponte fosfodiesterico 5’-5’ all’RNA trascritto in vitro, rallentandone la degradazione in ambiente cellulare. Per ottenere trascritti forniti di “CAP” è sufficiente far avvenire la reazione di trascrizione in presenza di una concentrazione di GTP pari a 1/10 di quella del “CAP”, la cui concentrazione eguaglia quella di ATP, UTP e CTP.
I templati vengono di norma preparati, linearizzando per digestione 5-10 μg di DNA plasmidico, usando un sito di restrizione a valle della coda di poli-A o del sito di poliadenilazione virale. La reazione di trascrizione viene generalmente condotta in 20 μl.
Esempio: DNA linearizzato 1-2 μg Mix “CAP” 5X 4 μl RNA-polimerasi 2 μl (50-100 UE) Rnase-lnhibitor 1 μl (20 UE) H2O RF fino a 20 μl
La reazione è condotta a 37°C per 2 ore, trascorse le quali, il DNA stampo viene idrolizzato, mediante incubazione sempre a 37°C per 1 ora con DnasiI RF. Seguono estrazione con TNES e fenolo-clorofornio, precipitazione alcolica, risospensione in TE RF o H2O RF e stima di quantità
su gel di agarosio con l’ausilio di “markers” a pesi molecolari noti.
I trascritti vengono poi diluiti, suddivisi in varie aliquote e conservati a -80°C, questo permette di scongelare la sola quantità necessaria durante la microiniezione.
Fig. 2.1 Plasmide pCS2+
2.7.2 Trascritti microiniettati
• Il costrutto cDNA XPet-1 codifica per un fattore di trascrizione. Quando il messaggero iniettato viene tradotto, viene traslocato nel nucleo dove attiva la trascrizione dei geni “target”. Le dosi iniettate in questo lavoro sono 300-500 pg.
• Per la produzione del trascritto codificante per la GFP (di cui si sono iniettati 100-200 pg in ogni embrione) è stato utilizzato un vettore commerciale.
• La porzione codificante del gene LacZ unita ad un segnale di localizzazione nucleare ed inserita in pCS2+ ci è stata fornita da I.
Dawid. Di questo trascritto sono stati iniettati 100-200 pg per embrione.
2.8 Reazione cromogenica per la
β-galattosidasi
Nella maggior parte degli esperimenti di microiniezione insieme al trascritto di interesse abbiamo iniettato il messaggero per la β-galattosidasi (citoplasmatica o nucleare). Questo metodo ci permette di individuare facilmente il lato iniettato da quello di controllo attraverso una reazione enzimatica il cui prodotto è colorato. Esistono diversi substrati per la β-galattosidasi, ad esempio il Salmon-gal il cui prodotto è di colore rosato, l’X-gal, di colore celeste e il Red-gal, di colore rosso.
Quando gli embrioni hanno raggiunto lo stadio desiderato, vengono fissati in MEMFA per 40 minuti e lavati per 5 minuti in PBS 1X; si aggiunge poi la soluzione con il substrato, “β-gal solution”, e si incuba in stufa a 37°C fino a quando gli embrioni sono sufficientemente colorati. E’ importante controllare spesso la reazione di colorazione poiché la durata di questa fase è variabile (da 15 min. a 1h). Per interrompere la reazione cromogenica, si passano gli embrioni in PBS 1X per qualche minuto, infine, si fissano nuovamente in MEMFA per 20 min. e si disidratano in etanolo (EtOH).
Gli embrioni in EtOH 100% possono essere conservati a -20°C od utilizzati per l’ibridazione in situ “whole mount”.
Soluzioni: PBS 10X (pH 7.3) NaCl 1.37 M KCl 0.027 M Na2HPO4 0.043 M KH2 PO4 0.015 M β-gal solution (10 ml) C6FeK3N6 32.93 mg C6FeK4N6•3H2O 42.24 mg X-gal o Red-gal (20 μg/μl ) 500 μl MgCl2 (1 M) 20 μl PBS fino a vol.
2.9 Ibridazione in situ “whole mount” e “su
sezione”
2.9.1 Preparazione dei “probes” a RNA
Per preparare un “probe” a RNA è necessario digerire il plasmide contenente il gene di interesse con un enzima che taglia una sola volta nel plasmide in un punto posto al 5’ del gene e che non taglia nel gene stesso in modo da ottenere un frammento di DNA lineare. Questo DNA viene purificato mediante estrazione fenolica e poi utilizzato come stampo
promotore posto al 3’ del gene,in modo da ottenere il trascritto “antisenso”.
Fig. 2.2 Plasmide pBluescript SK-
Ad esempio nel caso del plasmide pBluescript SK- per ottenere il "probe" antisenso si può linearizzare con NotI e trascrivere con la T7 RNA polimerasi.
Si effettua una reazione di trascrizione standard (Melton et al., 1985) usando Sp6, T7 o T3 RNA polimerasi, aggiungendo alla miscela di reazione un UTP-ribonucleotide, sostituito in posizione 11, con digossigenina o in posizione 12, con una molecola di fluoresceina. La miscela di reazione viene preparata aggiungendo i reagenti a temperatura ambiente. Solitamente il segnale dato dalla sonda marcata con digossigenina è assai più forte, rispetto al segnale dato dalla stessa sonda floresceinata; l’uso di quest’ultima diventa però necessario nel caso si voglia procedere ad una doppia ibridazione.
Esempio di reazione condotta in 20μl: Tampone di trascrizione 5x 4 μl DTT 100mM 2 μl DNA linearizzato (1 μg/μl) 1 μl Rnase-lnhibitor (20 UE/μl) 1 μl RNA polimerasi 2 μl Miscela di nucleotidi con:
digossigenina-UTP 2.5 mM 2 μl (fluoresceina-UTP 2.5 mM 2 μl)
H2O RF fino a 20 μl
Dopo incubazione a 37°C per 2 ore, si aggiunge 1 μl di DNasiI RF (1mg/ml) e si pone a 37°C, per almeno 15 min. Per effettuare la precipitazione alcolica si aggiungono 2 μl di litio cloruro 5M e 75 μl di EtOH assoluto. Dopo aver mescolato, capovolgendo il tubo diverse volte, si mette il tutto a precipitare a -20°C, per almeno 4 ore, oppure, a -80°C per 1 ora. A questo punto si centrifuga a 12000 rpm a 4°C, per 20 minuti; poi, eliminato il sovranatante, si aggiungono al pellet 150 μl di EtOH 70% pre-raffreddato e si centrifuga a 12000 rpm a 4°C, per 5-10 min. Si libera, infine, il pellet da tutto l’etanolo residuo, lasciandolo evaporare all’aria a temperatura ambiente e si risospende in 20 μl di H2O RF (oppure TE pH
7.5 RF). La stima della quantità del trascritto ottenuto viene effettuata su gel di agarosio, servendosi dell’ausilio di tRNA a concentrazione nota, come “marker” di quantità. I trascritti possono essere conservati in “stock” 10X (10 μg/ml) nella miscela di ibridazione.
2.9.2 Ibridazione in situ “whole mount”
Per saggiare il normale “pattern” di espressione di un gene e per analizzare l’effetto della sovraespressione, o della perdita di funzione, di un gene su un altro gene, putativo “target”, si utilizza la tecnica di ibridazione in situ “whole mount”. Gli esperimenti di ibridazione in situ su embrioni interi di Xenopus laevis sono stati effettuati, fatta eccezione per alcune modifiche, seguendo il protocollo di R. Harland (1991).
L’ibridazione viene realizzata in tubi “vials” di vetro e gli embrioni vengono mantenuti nelle “vials” per tutta la durata dell’esperimento. E’ importante che tutte le soluzioni ed i materiali usati, fino al termine della ibridazione, siano “RNasi free” (RF); la vetreria viene trattata in stufa a 180°C per almeno 4 ore mentre l’acqua distillata e le altre soluzioni vengono sterilizzate in autoclave.
Tutti i passaggi, se non diversamente indicato, sono eseguiti sistemando i “vials” orizzontalmente su un agitatore, aggiungendo e rimuovendo ogni volta 5 ml circa di soluzione. Gli embrioni vengono prima reidratati, effettuando lavaggi di 5 min. ciascuno, in una serie decrescente di alcoli: EtOH 100% EtOH 75% + H2O 25% EtOH 50% + H2O 50% EtOH 25% + PTw 75% PTw 100% per 3 volte
Segue trattamento in una soluzione di 10 μg/ml di proteinasi K in PTw e si incuba, a T ambiente, per 5-20 minuti, senza agitazione; il tempo di incubazione in proteinasi K (PK) va testato in quanto può variare cambiando il “batch” di PK. Si presta molta attenzione a questo passaggio essendo gli embrioni molto sensibili a sfaldamento, ad opera della PK. Si lava quindi 2 volte per 5 min., sempre a T ambiente, in una soluzione di trietanolammina 0.1 M pH 7-8; agli embrioni lavati in trietanolammina si
aggiungono 12.5 μl di anidride acetica, si incuba agitando i tubi per 5 minuti e si aggiungono altri 12.5 μl di anidride acetica. Seguono 2 lavaggi di 5 min. ciascuno in PTw. Gli embrioni vengono quindi rifissati in 4% paraformaldeide ( 4 ml di PTw + 1 ml di 20% paraformaldeide) a T ambiente sull’agitatore. Seguono 3 lavaggi in PTw, di 5 min. ciascuno.
Si rimuove quindi il PTw da ogni tubo e lo si sostituisce con 600 μl di PTw e 250 μl di miscela di ibridazione, disponendo i tubi sull’agitatore verticalmente per circa 10 minuti: questo passaggio è necessario per abituare gli embrioni alla nuova densità della miscela di ibridazione. Il passaggio successivo prevede il trattamento degli embrioni con la sola miscela di ibridazione per 4-6 ore a 62 °C .
Si sostituisce, infine, con 0.5 ml di tampone di ibridazione addizionato con la sonda marcata alla concentrazione finale di 0.5-1 μg/ml e si ibrida a 62°C per almeno 12 ore.
Una volta rimosso, il tampone di ibridazione contenente la/le sonda/e può essere conservato e riutilizzato fino a due/tre volte. A questo punto è necessario lavare l’eccesso di sonda non ibridata o ibridata in maniera aspecifica; si fanno a tale scopo lavaggi in soluzioni a forza ionica decrescente, per aumentarne la stringenza, tenendo presente che è importante aggiungere le soluzioni preriscaldate alla temperatura del lavaggio corrispondente.
Si sostituisce con 1 ml di tampone di ibridazione fresco e si incuba a 62°C per 10 min.
Seguono 3 lavaggi in 2x SSC a 62°C di 20 min. ciascuno.
Il lavaggio successivo si effettua in 2x SSC addizionato di RNasi A, a concentrazione finale di 20 μg/ml, a 37°C per 30 minuti. Per rimuovere l’RNasi, si lava in 2x SSC a T ambiente per 10 min e due volte in 0.2x SSC a 62°C, per 30 minuti. Prima di iniziare la procedura di incubazione con anticorpo, si lava due volte in MAB a T ambiente per 10-15 minuti.
Si incuba per 4 ore a temperatura ambiente o per tutta la notte a 4°C con anticorpo opportuno, diluito in una soluzione di MAB + reagente bloccante 2% + siero di pecora 15% + omogenato di embrioni 5%.
Per rimuovere l’eccesso di anticorpo dagli embrioni si compiono almeno 5 lavaggi con MAB a T ambiente di 60 minuti ciascuno, uno dei lavaggi è preferibile farlo o/n a 4°C.
L’ultimo lavaggio viene sostituito con il tampone per la fosfatasi alcalina (APB), 2 volte per 5 minuti. Nell’ultimo lavaggio con l’APB si aggiunge una sostanza in grado di inibire le fosfatasi endogene detta Levamisole.
Si aggiunge infine la soluzione di rivelazione più opportuna. In questo lavoro il substrato della fosfatasi alcalina utilizzato è il “BM purple” che si trova in forma liquida pronta per l’utilizzo e porta ad una colorazione blu intenso.
In questa fase la fosfatasi alcalina coniugata all’anticorpo scinde il substrato cromogenico (BM purple) generando un prodotto colorato che rende possibile evidenziare le zone in cui la sonda si è ibridata e dove il gene in esame è espresso. Si usano 0.5-1 ml per “vial” a T ambiente fino a quando la colorazione è soddisfacente (da 1 ora a 7 giorni). La reazione deve avvenire al buio così i tubi possono essere ricoperti da un foglio di alluminio. Per osservare la colorazione, gli embrioni possono essere posti in una piastra Petri contenente il tampone per la fosfatasi alcalina. Se il segnale è insoddisfacente si ripongono gli embrioni nella soluzione di colorazione, altrimenti si procede bloccando la reazione cromogenica rimuovendo il tampone per la fosfatasi alcalina e fissando o/n con MEMFA per stabilizzare la colorazione. Gli embrioni così fissati possono essere conservati in EtOH a -20°C.
Soluzioni: PTw
PBS 1X Tween-20 0.1%
Soluzione di trietanolammina
Si diluisce la trietanolammina con acqua distillata in modo da ottenere una concentrazione finale di 0.1 M. Si porta a pH 7-8 con HCl.
Soluzione di paraformaldeide
Si può preparare uno “stock” di paraformaldeide al 20%, che si conserva in frigo a 4°C per diversi mesi. Si scioglie la paraformaldeide in acqua RF ad una temperatura di 60°C e si chiarifica aggiungendo 10μl di NaOH 10N in 100 ml di paraformaldeide. Quando la soluzione è diventata limpida si lascia raffreddare, si porta a volume con acqua distillata e si filtra con carta 3MM.
Al momento dell’uso si diluisce la paraformaldeide così preparata con PTw. Tampone di ibridazione Formammide 50% SSC 5x Torula RNA 1mg/ml Eparina 100 μg/ml Denhart’s 1x Tween-20 0.1% CHAPS 0.1%
MAB (Maleic Acid Buffer) pH 7.5
Acido Maleico 100 mM
NaCl 150 mM
Tampone di reazione cromogenica della fosfatasi alcalina (APB)
Tris 100 mM pH 9.5
MgCl2 50 mM
NaCl 100 mM
2.9.3 Depigmentazione ("bleaching”)
Nell’ibridazione in situ “whole mount” abbiamo processato embrioni pigmentati che derivavano da esperimenti di microiniezione, in questi casi è possibile eliminare il pigmento, in poco tempo, con perdita minima di segnale. A questo scopo è necessario lavare gli embrioni diverse volte in EtOH al 70%, questo serve a rimuovere eventuali reagenti che potrebbero interferire con la reazione di “bleaching”. Si passano, successivamente, per qualche minuto in una soluzione contenente 0.5X SSC ed EtOH 50% che si sostituisce con 0.5x SSC, 5% formammide, 2% H2O2. Gli embrioni
vengono posti su un agitatore, sotto una lampada a luce fluorescente, per un periodo variabile di 1-2 ore. Quando gli embrioni si sono chiarificati a sufficienza, si rimuove la soluzione contenente acqua ossigenata, per passare gli embrioni in EtOH assoluto e conservarli a –20 °C.
2.9.4 Taglio dei preparati da processare su sezione
Gli embrioni sono fissati o/n in 4% paraformaldeide (PFA) / 1x PBS a 4°C. Successivamente vengono effettuati tre lavaggi da 5 minuti in PBS per rimuovere la PFA. A questo punto i preparati sono equilibrati in 30% saccarosio / 1x PBS a 4°C. Quando è avvenuta l’equilibratura i preparati sono messi nel mezzo di inclusione, “Tissue Tek”, e congelati a –80°C. Successivamente vengono tagliati al criostato. I vetrini porta-oggetto con le sezioni sono conservati a –20°C. Alternativamente, gli embrioni o gli encefali estratti possono essere deidratati in una serie crescente di alcool metilico in PBS fino all’equilibratura in 100% metanolo (MeOH). I preparati così trattati possono essere conservati a –20°C per alcuni mesi ed impiegati in esperimenti di immunoistochimica ed ibridazione in situ “whole mount”.
2.9.5 Ibridazione in situ su sezione
I vetrini vengono lasciati scongelare a RT. Si effettuano lavaggi successivi in PBS per rimuovere l’eccesso di “Tissue Teck”. Su ogni vetrino vengono aggiunti 200 μl di “Hybridisation buffer” addizionato con la sonda marcata alla concentrazione finale di 0.5-1 μg/ml e si ibrida a 65°C o/n. Si effettuano 4 lavaggi con “washing solution” per 30 minuti ciascuno a 65°C. Successivamente si fanno 2 lavaggi con MAB per 30 minuti ciascuno a RT. Segue una preincubazione di 1 ora a RT in MAB + 2% reagente bloccante + 20%. siero di pecora. Si incuba per tutta la notte a 4°C con anticorpo antidigossigenina (1:2000), diluito in una soluzione di MAB + reagente bloccante 2% reagente bloccante + 20%. siero di pecora. Per rimuovere l’eccesso di anticorpo dalle sezioni si compiono lavaggi con MAB
tampone per la fosfatasi alcalina (APB), 2 volte per 10 minuti. Nell’ultimo lavaggio con l’APB si aggiunge Levamisole (0.0005 gr/ml), una sostanza in grado di inibire le fosfatasi endogene .
Si aggiunge infine la soluzione di rivelazione composta da NBT e BCIP (3.5 μl/ml di ciascun substrato) in APB. Se il segnale è soddisfacente si procede bloccando la reazione cromogenica rimuovendo il tampone per la fosfatasi alcalina e lavando i vetrini in PBT. A questo punto si effettua un passaggio veloce in acqua per rimuovere l’eccesso di sali dalle sezioni che successivamente vengono lasciate asciugare all’aria e poi coperte con vetrino copri oggetto ed un montante tipo “Eukitt”.
Soluzioni: Hybridisation buffer: 10 x salt 1 x Formammide 50% Solfato di destrano 10% rRNA 1 mg/ml 100x Denhardt’s 1x H2O a volume Washing solution: Formammide 50% SSC 1x Tween 20 0.1%
2.9.6 Immagini
I preparati sono stati analizzati con un microscopio convenzionale Nikon ed uno stereomicroscopio Nikon. Le immagini sono state acquisite con fotocamera “CoolSnap” Photometric. Le tavole delle figure sono state ottenute impiegando software di grafica tipo Photoshop e programmi dedicati per la sovrapposizione delle immagini in esperimenti di co-localizzazione.
2.10 Immunoistochimica“whole mount” e su
sezione
2.10.1 Immunoistochimica “whole mount”
Gli embrioni vengono fissati in un “vial” per un’ora in paraformaldeide 4% o “Memfa 1x”, poi risciacquati in “PBS 1x”. A questo punto mettiamo gli embrioni in “PBS1x” a cui aggiungiamo la proteinasi K, concentrata 10 microL/ mL, e incubiamo otto minuti a 37°C in modo tale da intaccare i tessuti per facilitare l’ingresso delle varie soluzioni anche agli strati tissutali più interni . Finito il tempo di incubazione, risciacquo in “PBS1x” per eliminare la proteinasi K, e metto ad incubare gli embrioni in PBS 1x addizionato con acqua ossigenata allo 0.3% per trenta minuti,in questo modo vado a rompere le perossidasi endogene dei tessuti, procedimento utile poiché la rivelazione verrà fatta con un anticorpo secondario che porta legata la perossidasi. Dopodichè risciacquo in PTw fino a che gli embrioni non precipitano nuovamente sul fondo visto che il
depositati sul fondo li metto in Blocking ( PBS1x + 0.1% Triton + 20% Siero +2% Blocking) per circa un’ora. Alla fine aggiungo un anticorpo primario anti-5HT di coniglio(1:1000) e incubo a 4°C “overnight”. Il giorno successivo lavo l’eccesso di anticorpo, con passaggi da circa un’ora, in PTw per almeno tre ore. Aggiungo un anticorpo secondario che riconosce il frammento Fc degli anticorpi di coniglio e che porta legata l’attività enzimatica di una perossidasi GARPO ( Goat AntiRabbit Perossidase) (rapporto 1:500). Incubiamo “overnight” a 4°C. Il giorno seguente Lavo nuovamente in PBT e rivelo con la DAB,un substrato idoneo per la per ossidasi legata all’anticorpo secondario.
2.10.2 Immunoistochimica su sezione
Sono state fatte sezioni al criostato sia di embrioni che di cervello adulto dello spessore di 15 micron. E si conservano a -80°C. Prima di cominciare l’esperimento si fanno asciugare a temperatura ambiente e poi si esegue qualche passaggio in PBS1x in modo da allontanare il “Tessue Teck” rimanente. Eseguo poi, un passaggio in PBS1x + 0.5% di Acqua Ossigenata da circa trenta minuti, per la stesse motivazione del Protocollo “Whole Mount”. Sciacquo in PBS1x e metto in Blocking (PTw + 2% Blocking Solution + 20% Siero) per circa un’ora. Passato, questo tempo di incubazione, aggiungo un anticorpo primario anti-5HT (1:1000) e incubo “overnight” a 4°C. Il giorno seguente, elimino l’eccesso di primario con passaggi in PTw e metto anticorpo secondario GARPO (Goat Anti-Rabbit PerOssidase)(1:500) e rimetto in incubazione “overnight” a 4°C. Il terzo giorno, lavo l’eccesso di secondario, in modo tale da eliminare segnali di fondo PTw, e metto in rivelazione con la DAB. Ottenuto il segnale blocco la reazione in PBS1x e prima del montaggio eseguo una reazione con una soluzione di PBs1x e Hoecst (1:1000), per cinque minuti, visto che tale sostanza è fluorescente ed in grado di mettere in risalto la localizzazione
del nucleo delle cellule grazie al legame che va ad instaurare con le componenti nucleari. Monto il vetrino con Acqua Polimount.
2.10.3 Immagini
I preparati sono stati analizzati con un microscopio Nikon ed uno stereomicroscopio. Le immagini sono state acquisite con fotocamera “CoolSnap” Photometric. Le tavole delle figure sono state ottenute impiegando software di grafica tipo Photoshop e programmi dedicati per la sovrapposizione delle immagini in esperimenti di co-localizzazione.
2.11 Taglio degli embrioni processati per “in
situ” e immunoistochimica “whole mount”
Gli embrioni processati per “whole mount” vengono poi inclusi in paraffina per poter essere tagliati e analizzati nel dettaglio. Procediamo ad un ulteriore fissaggio per circa due ore a RT o O/N a 4°C in PFA. Sciacquiamo in PBS 1X e procediamo alla disidratazione graduale, su una basculla, da PBS 1X a Etanolo (EtOH), utile per passaggio in Xilene.
EtOH 25% + PBS 1X 75% 10 minuti EtOH 50% + PBS 1X 50% 2 da 10 minuti EtOH 75% + PBS 1X 25% 15 minuti EtOH 100% 2 da 10 minuti
A questo punto passiamo in Xilene gradualmente utile per rendere l’embrione permeabile alla paraffina, infatti lo Xilene verrà rimpiazzato nei tessuti dalla paraffina durante i vari passaggi. Nel frattempo metto a filtrare mentre scioglie la Paraffina in un beker in una stufa a 65°C.
Eseguo passaggi graduali da EtOH a Xilene, togliendo dalla “vial” 1 mL di EtOH (nei passaggi successivi di EtOH+Xilene) sostituendolo con 1 mL di Xilene nuovo. I passaggi sono da 5 minuti l’uno fino ad arrivare a EtOH50%+Xilene50%. Tenendo in agitazione su una basculla.
Successivamente si tolgono 4mL della soluzione dalla “vial” e ne aggiungo 4mL di Xilene fresco in modo da ottenere Xilene75%+EtOH25% e faccio agitare per 15 minuti.
Passiamo adesso alla sostituzione con la paraffina. Xilene50%+Paraffina50% 30 minuti a 65°C Paraffina100% 45 minuti a 65°C Paraffina100% 15 minuti a 65°C Paraffina100% O/N a 65°C
Il giorno dopo metto a scaldare nella stufa a 65°C delle piastrine Petri in cui colerò gli embrioni inclusi, in modo da darmi il tempo di orientarli nella direzione che preferisco.
Faccio freddare prima a RT e poi a 4°C per dare consistenza e procedo con il taglio al Microtomo.