1
Indice
Capitolo 1. Introduzione ... 3 Capitolo 2. Stato pro-trombotico nell’ipertensione essenziale
2.1 Generalità sul rischio cardiovascolare ... 24 2.2 Il paradosso pro trombotico... 25 2.3 Disfunzione endoteliale ... 28 2.4 Markers biochimici circolanti di disfunzione endoteliale ... 32 Capitolo 3. Disfunzione piastrinica nell’ipertensione essenziale
3.1 Generalità sulle piastrine: ruolo nella coagulazione e
meccanismi di attivazione ... 37 3.2 Meccanismi che contribuiscono all’ attivazione piastrinica nei pazienti ipertesi ... 40 3.3 Evidenze e markers di attivazione piastrinica
nell’ipertensione essenziale ... 43 Capitolo 4. Effetti del trattamento antipertensivo sull’attivazione piastrinica
4.1 Effetti degli ACE-inibitori e degli antagonisti del recettore AT1 sullo stato pro trombotico e sull’attivazione piastrinica ... 47 4.2 Effetti della terapia con Calcio-antagonisti sull’aggregazione piastrinica ... 54 Capitolo 5. Trattamento antiaggregante piastrinico nell’ipertensione essenziale
5.1 Ruolo dell’aspirina nella prevenzione primaria e secondaria degli eventi cardiovascolari ... 57 5.2 Ruolo del Clopidogrel ... 61 Capitolo 6. Conclusioni ... 64
2
3
Capitolo 1.
Introduzione
L’ipertensione essenziale, primitiva o idiopatica, è definita come una condizione di elevata pressione sanguigna nella quale le cause secondarie, quali malattia renovascolare, insufficienza renale, feocromocitoma, aldosteronismo o altre cause di ipertensione secondaria o forme mendeliane (monogeniche), non sono presenti. Nonostante le continue ricerche, le cause di questa patologia restano ancora parzialmente sconosciute.
Essa rappresenta il 95% di tutti i casi di ipertensione e la sua prevalenza aumenta con l’età. Interessa il 25-30% della popolazione adulta e fino al 60-70% delle persone oltre la settima decade di vita. È un disordine eterogeneo, per il quale sono stati proposti differenti fattori causali, sia genetici che ambientali. Ad oggi, per quanto sia riconosciuto un ruolo importante alla componente genetica nell’eziopatogenesi dell’ipertensione, si conosce molto poco sugli specifici geni coinvolti. Studi familiari hanno suggerito alcuni possibili fenotipi intermedi (tratti genetici) che potrebbero essere correlati con le varianti ereditarie di elevata pressione arteriosa, i.e. alta espressione del contro trasportatore sodio-litio, bassa escrezione urinaria di callicreina, alte concentrazioni di insulina plasmatica a digiuno, alta densità di sottofrazioni LDL.
Jeunemaitre et al. in particolare, hanno individuato un
polimorfismo nel gene dell’angiotensinogeno collegato allo sviluppo di ipertensione essenziale ereditaria. [1-2]
Esistono poi numerosi fattori, collegati all’ambiente e allo stile di vita, che aumentano la pressione sanguigna, tra cui insulino-resistenza, eccessivo introito di alcool, eccessivo consumo di sale, età avanzata e probabilmente uno stile di vita sedentario, lo stress, un basso introito di potassio e di calcio. Infine, sono presenti delle
4
interazioni tra la componente genetica e i fattori ambientali, che influenzano lo sviluppo di fenotipi intermedi, quali l’attività del sistema nervoso simpatico, i sistemi renina-angiotensina-aldosterone (SRAA), il sistema renale callicreina-chinina, e la funzionalità endoteliale, che a loro volta regolano fenomeni come l’escrezione di sodio, la reattività vascolare e la contrattilità cardiaca.[1]
I due maggiori determinanti fisiologici della pressione arteriosa sono la gittata cardiaca e la resistenza vascolare periferica totale. Essi sono costantemente regolati da una combinazione di meccanismi a breve e lungo termine. I primi agiscono su resistenza periferica, capacitanza vascolare, performance cardiaca e sono controllati dal sistema nervoso autonomo ( principalmente dal sistema nervoso simpatico). I meccanismi a lungo termine agiscono invece sulla regolazione del volume sanguigno circolante effettivo, il bilancio del sodio e il volume di fluidi extracellulari, grazie ad una corretta funzionalità renale.
I reni infatti svolgono un ruolo di primo piano nel controllo della pressione arteriosa, avendo a disposizione vari sistemi, quali il meccanismo di pressione- natriuresi (ipotesi sviluppata da Guyton
et al.), il riassorbimento di sodio a livello tubulare ed infine il
sistema renina-angiotensina aldosterone. Inoltre esistono due meccanismi che interagiscono per fornire un’efficiente autoregolazione renale: il feedback tubulo-glomerulare (o meccanismo della macula densa) e il meccanismo miogeno.
Meccanismo pressione-natriuresi e riassorbimento di sodio a livello tubulare:
Per ciascuna condizione data di stato stazionario, esiste una relazione diretta tra la pressione di perfusione renale e la quantità escreta di sodio, così che elevazioni acute della prima provocano
5
natriuresi e diuresi. In condizioni fisiologiche un aumento dell’introito di sale suscita quindi una serie di risposte che innalzano la pendenza della curva di relazione pressione-natriuresi. Il corretto funzionamento del meccanismo dipende ovviamente dall’integrità dei sistemi intrarenali che controllano l’escrezione di sodio, e di quelli recettoriali extrarenali che individuano cambiamenti nel bilancio dello ione, nel volume di sangue circolante, nella pressione arteriosa e lo segnalano ai reni. Di conseguenza, alterazioni in uno o più di questi meccanismi possono ridurre la pendenza della curva di relazione pressione-natriuresi, portando ad un ripristino della pressione arteriosa maggiore rispetto al livello richiesto per mantenere quel dato bilancio di sodio.
Per quanto riguarda il riassorbimento tubulare di sodio, in condizioni normali meno dell’1% del carico di sodio filtrato viene escreto e piccoli cambiamenti percentuali nella frazione di riassorbimento si traducono in ampi cambiamenti nell’escrezione giornaliera di sodio.
Alcuni individui con ipertensione essenziale hanno mostrato una caduta pressoria maggiore a seguito della riduzione dell’introito di sodio rispetto ad altri soggetti con la stessa patologia, e questo ha portato all’idea di suddividere ulteriormente l’ipertensione in “sale-sensibile” e “sale-resistente”.
Sistema renina angiotensina aldosterone
:
La renina è secreta dalle cellule iuxtaglomerulari delle arteriole afferenti a livello renale in risposta ad una ipoperfusione glomerulare o ad un ridotto introito di sodio. Essa genera l’angiotensina I (ATI) dal precursore angiotensinogeno che si forma principalmente nel rene ma anche nel fegato. Il decapeptide ATI
6
viene clivato ad octapeptide angiotensina II (ATII) dall’enzima ACE (enzima di conversione dell’angiotensina), che è presente in notevole quantità a livello del polmone ed inoltre si trova legato alle cellule endoteliali di molti organi, incluso il rene. In questo organo è reperibile anche sulle cellule con orletto a spazzola del tubulo prossimale.
L’angiotensina II è un potente vasocostrittore sistemico, che provoca un aumento nella pressione arteriosa. Essa inoltre stimola il rilascio di aldosterone dalla zona glomerulosa delle ghiandole surrenali, il quale permette l’ulteriore aumento pressorio favorendo il riassorbimento di sodio, e conseguentemente di acqua, e l’escrezione di potassio. L’angiotensina ha due recettori principali , AT1 e AT2, ma la potente azione del sistema RAA si esplica principalmente attraverso il recettore AT1.
Questo ormone esercita multipli effetti intrarenali: provoca una vasocostrizione renale diretta, che risulta in una riduzione del flusso sanguigno a livello renale (RBF, renal blood flow) e, in misura minore, della GFR (glomerular filtration rate), vasocostringendo principalmente l’arteriola efferente, ma anche il segmento pre-glomerulare, fattore che spiega la sua capacità di aumentare la frazione di filtrazione. Inoltre ATII incrementa la quota di riassorbimento tubulare di sodio a livello prossimale e distale, dove agisce stimolando lo scambiatore Na+/H+.
Meccanismi di autoregolazione renale:
Il feedback tubulo-glomerulare risponde a perturbazioni che causano sia cambiamenti nel flusso tubulare distale al di là della macula densa sia cambiamenti di concentrazione dei soluti nel tubulo. Un segnale paracrino è generato dalle cellule della macula densa ed inviato alle arteriole afferenti, cosicché aumenti del flusso oltre la macula portano ad una vasocostrizione, mentre riduzioni
7
dello stesso, portano ad una vasodilatazione. Questo meccanismo aiuta a bilanciare il carico di sodio filtrato, dipendente da fattori emodinamici, con le capacità di riassorbimento del nefrone ed a regolare la quota dello ione che giunge al nefrone distale.
L’altro meccanismo di autoregolazione è quello miogeno che risponde ai cambiamenti di tensione nella parete arteriolare concomitanti alla variazione della pressione arteriosa. Esso attiva un sensore che regola il tono delle cellule muscolari lisce del vaso. Le arterie arcuate, interlobulari e le arteriole afferenti, ma non quelle efferenti, esibiscono questa risposta miogena al cambiamento di tensione nella parete vasale. Questo meccanismo permette una risposta autoregolatoria renale residua anche quando il feedback tubulo glomerulare viene bloccato. [3]
Data la complessità dei sistemi coinvolti nella regolazione della pressione arteriosa, i quali sono strettamente intrecciati tra loro anche attraverso la secrezione ormonale sistemica e paracrina (ad. esempio ATII e NO), sono state formulate varie teorie fisiopatologiche che potrebbero spiegare lo sviluppo dell’ipertensione essenziale. Tuttavia i vari ricercatori non sono stati in grado di determinare con certezza se le anomalie riscontrate siano effettivamente primitive oppure secondarie all’instaurarsi della malattia stessa.
L’attenzione si è focalizzata principalmente su: • aumento delle resistenze vascolari periferiche
• eccesso di introito di sodio e suscettibilità individuale ad esso
• alterata fisiologia renale
- difficoltà nell’escrezione di sodio - abnorme attivazione del SRAA
8
- meccanismi acquisiti coinvolti nello sviluppo di ischemia renale, stress ossidativo e infiammazione
• Alterazioni generalizzate a carico delle membrane cellulari • Insulino-resistenza e iperinsulinemia
Aumento delle resistenze vascolari periferiche:
Molti pazienti con ipertensione essenziale mostrano una normale gittata cardiaca insieme con un’aumentata resistenza periferica. Essa è determinata soprattutto dalle piccole arteriole la parete delle quali è costituita da cellule muscolari lisce. Si pensa che una loro contrazione prolungata, probabilmente mediata dall’angiotensina, induca un ispessimento della parete vasale portando così ad una aumento irreversibile delle resistenze periferiche. È stato però ipotizzato che nelle primissime fasi dell’ipertensione, l’elevazione della pressione sia causata da un aumento della gittata cardiaca, collegato ad un’iperattività del sistema simpatico, piuttosto che ad un aumento delle resistenze. [4]
Nei pazienti con ipertensione essenziale i caratteristici risultati includono:
• Riduzione del diametro luminale ( inferiore a 500 µm nei vasi precapillari) ;
• Aumento del rapporto tra diametro dello strato di cellule muscolari lisce del vaso (tunica media) e diametro del lume, denominato rapporto media/lume.
Evidenze crescenti supportano l’ipotesi secondo la quale il cambiamento predominante che avviene nei vasi di resistenza sia il
remodelling, ovvero il riarrangiamento del materiale cellulare già
9
trasversale dello stesso, come avviene invece nel caso della crescita (sia ipertrofia che iperplasia) della componente cellulare. [5]
Eccesso di introito di sodio e suscettibilità individuale ad esso:
Un eccessivo consumo di sale provoca ipertensione aumentando il volume di fluidi circolanti (precarico) e di conseguenza la gittata cardiaca. Inoltre esso può influenzare la reattività vascolare e la funzione renale. Tuttavia solo una parte della popolazione generale è suscettibile agli effetti deleteri dell’alto introito di sodio,
presumibilmente in quanto questi individui hanno un ulteriore difetto nell’escrezione dello ione a livello renale.
Dato che nei paesi occidentali tutti gli individui sono esposti a diete con alto introito salino il fatto che solo la metà della popolazione svilupperà ipertensione, suggerisce una certa variabilità nella suscettibilità della pressione arteriosa al sodio. Weinberger et al. hanno definito la sensibilità al sale come “una riduzione della pressione arteriosa media di 10mmHg o più dal livello misurato dopo 4 ore di infusione di 2l di soluzione fisiologica, rispetto al valore misurato la mattina dopo un intero giorno di dieta contenente 10mmol di sodio, durante il quale vengono somministrate tre dosi di furosemide (alle ore 10, 14 e 18)”. [6] Utilizzando questo criterio, hanno scoperto che il 51% degli ipertesi e solo il 26% dei normotesi sono sodio sensibili. Sono stati proposti molti meccanismi per spiegare questo fenomeno e più precisamente sono stati chiamati in causa:
- difetti nell’escrezione renale di sodio,
- aumento dell’attività dello scambiatore sodio-idrogenioni, - aumento dell’attività del sistema nervoso simpatico,
10
- aumento dell’intake di calcio a livello delle cellule muscolari lisce,
- compromissione della sintesi di NO.
La sensibilità al sale è più frequente nella razza nera ed aumenta con l’età, soprattutto nel sesso femminile.
Alterata fisiologia renale:
Nell’ipertensione essenziale cambiamenti fisiologici e patologici a livello renale spesso anticipano quelli identificabili a carico di altri organi, tuttavia se essi precedano o seguano l’inizio dell’ipertensione stessa non è ancora stato definito chiaramente. L’alterazione più significativa è stata riscontrata a carico della fisiologia vascolare: il GFR è mantenuto, mentre il flusso totale renale è ridotto con aumento della frazione di filtrazione. Questo pattern può essere spiegato dalla presenza di una diffusa vasocostrizione di tutti i nefroni, principalmente a carico dell’a. efferente ma anche di quella afferente, o in alternativa dalla vasocostrizione selettiva a carico di alcuni nefroni, che risulta in una deviazione del flusso sanguigno verso altri, in modo tale da mantenere GFR vicino al valore normale. Questa vasocostrizione renale è reversibile ma può portare ad una riduzione della pressione e del flusso a livello post-glomerulare, fattore che potrebbe predisporre ad un aumento del riassorbimento tubulare di sodio.
Difficoltà nell’escrezione di sodio:
Vari ricercatori hanno proposto diverse ipotesi per spiegare come l’anomala ritenzione di sodio possa essere l’evento che dà inizio all’ipertensione:
11
In condizioni normali la pressione di perfusione è circa 100mmHg e l’escrezione di sodio è 150mEq/die. Questi due meccanismi si trovano in un sostanziale status di bilancio.
Guyton et al. hanno proposto che i diversi processi in grado di
aumentare la pressione arteriosa, come ad esempio l’attivazione del sistema nervoso simpatico, provochino solo un incremento transitorio, mentre un rialzo permanente di essa, richieda invece un adattamento del meccanismo pressione-natriuresi. In più gli stessi autori hanno suggerito che sia l’ipertensione “sale resistente” che quella “sale sensibile”, siano entrambe di derivazione renale. Esse risultano in differenti tipologie di grafico: nel primo caso avremo infatti uno spostamento parallelo verso destra della curva, tale che la gestione del carico di sodio risulterebbe simile a quella osservata negli individui sani, ad eccezione del fatto che si avrebbe per un valore basale più alto di pressione. Al contrario, l’ipertensione “sale-sensibile” sarebbe associata ad eccessivi aumenti o riduzioni della pressione arteriosa a seconda rispettivamente di un aumentato o ridotto introito di sodio. [7] (Fig.1)
12
Fig.1 Effetto del fenotipo dell’ipertensione sulla curva pressione natriuresi.
Ridotto numero di nefroni:
Brenner et al. hanno avanzato l’ipotesi che una bassa dotazione di
nefroni alla nascita sia inversamente correlata al rischio di sviluppare ipertensione durante il corso della vita. La riduzione congenita di questa quota o della superficie di filtrazione glomerulare, limita l’abilità nell’escrezione di sodio e aumenta la pressione arteriosa. Si instaura così un circolo vizioso, per cui tale aumento genera ipertensione glomerulare, che a sua volta incrementa la pressione sistemica.
Questi ricercatori osservano che ben il 40% degli individui under 30 hanno una quota di nefroni inferiore al normale (presumibilmente 600,000 per rene) e “ipotizzano che queste persone, il cui numero di nefroni cade nel basso range, costituiscano il substrato della
13
popolazione che esibisce un’aumentata suscettibilità allo sviluppo di ipertensione essenziale”. [8]
Allo stesso modo una riduzione della superficie di filtrazione, rivelata dalla riduzione del diametro glomerulare o della superficie della membrana basale capillare, potrebbe essere responsabile di un aumentato rischio di sviluppo della patologia ipertensiva anche in presenza di un normale numero di nefroni.
Ormone natriuretico acquisito:
L’ormone endogeno ouabaina è rilasciato dai surreni e probabilmente dal mesencefalo in risposta all’ipossia o all’espansione del volume di fluidi extracellulari indotta dall’aldosterone. Alcune evidenze suggeriscono che, in modo dipendente dalla sua concentrazione, questo ormone si comporti come un modulatore versatile della pompa del sodio espressa in modo ubiquitario. Nelle cellule tubulari renali l’inibizione della pompa Na+/K+ ATPasi da parte dell’ouabaina promuove la
natriuresi.
Inoltre, nel contesto patogenetico dell’ipertensione essenziale questo ormone agisce anche sulle pompe del sodio localizzate a livello dei miociti cardiaci e delle cellule vascolari lisce, fattore che può rallentare lo scambio sarcolemmatico Na+/ Ca2+ e , attraverso
vie calcio dipendenti, stimolare l’accoppiamento eccitazione-contrazione nonché l’espressione di geni legati alla crescita cellulare.
A concentrazioni molto basse, nel range nanomolare, l’ouabaina endogena può aumentare il numero delle pompe del sodio attive nel pool di membrana e, per mezzo di questo ed altri meccanismi ancora sconosciuti, potare ad una conservazione renale del sodio anziché alla sua escrezione.[2]
14
Abnorme attivazione del SRAA:
La renina gioca un ruolo critico nella patogenesi della maggior parte dei casi di ipertensione. Sebbene nell’ipertensione essenziale ci si aspettino bassi livelli di renina, un gran numero di pazienti non ha una soppressione dell’asse renina-angiotensina, ma piuttosto livelli di PRA (Plasma Renin Activity) “inappropriatamente” normali o persino elevati. Tuttavia quando nei pazienti con ipertensione essenziale il profilo reninico viene correttamente eseguito ed indicizzato, circa il 20% dei soggetti mostra alti valori di renina che risultano invece bassi nel 30% dei casi, con la metà rimanente distribuita tra questi due estremi.
Si ha quindi un intervallo più ampio di attività della renina plasmatica nei soggetti ipertesi rispetto ai normotesi. Da qui la necessità di suddividere i pazienti affetti da ipertensione essenziale a bassa renina, da quelli con ipertensione ad alta renina.
Sono stati proposti tre meccanismi per spiegare questa attivazione anomala del SRAA, presente in molti pazienti con ipertensione essenziale:
Eterogeneità dei nefroni:
All’interno dei reni esistono basi strutturali e funzionali per l’instaurarsi di un’anomala secrezione reninica e di un’alterata escrezione di sodio, che sono caratteristiche dello stato ipertensivo. L’ipotesi che sia presente una eterogeneità dei nefroni nei pazienti con ipertensione essenziale è supportata dalle seguenti evidenze:
- Sono presenti nefroni ischemici con una compromessa capacità di espellere sodio frammisti a nefroni che si sono adattati all’iperfiltrazione e all’ipernatriuresi
15
- La secrezione reninica proveniente dai nefroni ischemici è elevata e bassa da quelli iperfiltranti
- I livelli circolanti di renina e angiotensina, che risultano inappropriati, compromettono l’escrezione sodica in quanto nei nefroni con adattamento all’iperfiltrazione aumenta il riassorbimento tubulare di sodio e l’azione del meccanismo a feedback tubulo-glomerulare, mediato dalla vasocostrizione dell’arteriola afferente. Inoltre, in quanto il livello circolante di renina è ridotto a causa della non partecipazione dei nefroni adattati, esso diviene inadeguato per supportare il tono efferente nei nefroni ipoperfusi
- Una perdita del numero di nefroni con l’età e a causa dell’ischemia compromette ulteriormente l’escrezione di sodio.
Non–modulazione:
Questa ipotesi è stata proposta da Williams e Hollenberg per livelli normali e alti di renina, identificati in circa la metà dei pazienti ipertesi e legati ad una difettosa regolazione a feedback del SRAA tra i reni ed i surreni.[9]
Gli individui normali modulano la risposta dei tessuti bersaglio dell’ATII al variare dell’introito salino con la dieta. Con una restrizione sodica la secrezione surrenale di aldosterone è aumentata e la risposta vascolare all’ormone è ridotta, mentre con un carico di sodio la risposta surrenale è soppressa e quella vascolare aumentata. Questo fenomeno è evidente in particolare all’interno della circolazione renale. Con una restrizione di sodio, quindi, RBF (Renal Blood Flow) si riduce, facilitando così la conservazione dello ione, mentre il contrario avviene dopo assunzione di un carico di sodio con la dieta. Questi cambiamenti
16
sono mediati principalmente attraverso variazioni dei livelli di ATII i quali, a loro volta, sono ridotti in caso di carico salino e aumentano dopo una sua restrizione.
Gli individui ipertesi “non modulatori” sono quindi così denominati per l’assenza della modulazione mediata dal sodio nelle risposte dei tessuti target all’ATII.
Questo fenomeno è stato attribuito ad un livello di angiotensina circolante pressoché fisso e regolato in modo anomalo, il quale non va ad aumentare la secrezione di aldosterone nel tessuto surrenale in risposta ad una restrizione sodica e, all’interno della circolazione renale, non permette un aumento del flusso a seguito di un carico salino.
Questa ipotesi è comprovata dalla correzione, dopo soppressione di ATII mediante terapia con ACE- inibitori, sia del difetto renale che di quello surrenale.
Il meccanismo della non–modulazione, a fronte di un alto introito salino che caratterizza la dieta assunta comunemente, può spiegare la patogenesi dell’ipertensione sodio-sensibile.
Una bassa prevalenza di soggetti “non modulatori” è stata riscontrata nelle giovani donne, suggerendo così che gli ormoni sessuali femminili possano conferire una certa protezione verso lo sviluppo fenotipico di questo tipo di ipertensione.
Ipertensione essenziale a bassa renina:
Come precedentemente esposto circa il 30% dei casi di ipertensione essenziale mostra un’attività reninica plasmatica ridotta. I possibili meccanismi per spiegare questo fenomeno riguardano l’espansione di volume circolante, con o senza aumento di mineralcorticoidi. La
17
maggioranza delle analisi più accurate ha tuttavia escluso la presenza delle suddette condizioni.
Studi condotti da Fishar et al. si sono focalizzati sulla risposta surrenale e pressoria all’ATII, in funzione dell’introito di sodio con la dieta, in tre gruppi di pazienti: quelli con ipertensione a bassa renina, ipertensione con livelli normali di renina e controlli. Sono stati individuati vari aspetti similari tra l’ipertensione con renina normale e l’ipertensione essenziale dei soggetti “non-modulatori” con PRA normale, che includono:
• Sensibilità della pressione arteriosa al sale
• Ridotte risposte plasmatiche all’aldosterone all’infusione di ATII e all’assunzione della posizione ortostatica, dopo cinque giorni di dieta con rigida restrizione sodica
• Livelli basali di aldosterone plasmatico relativamente bassi Queste differenze riscontrate rispetto ai controlli e ai soggetti ipertesi “modulatori” scompaiono quando l’introito salino veniva aumentato a 200mEq/die. Questo risulta in accordo con la soppressione dell’attività reninica plasmatica dell’ATII in concomitanza con alte assunzioni di sodio, con una resensitizzazione dei recettori dell’angiotensina e un miglioramento della risposta ad essa.
Questi risultati farebbero ipotizzare che l’attenuazione della risposta all’ATII durante il periodo di restrizione sodica potrebbe riflettere una sottostante condizione di continua generazione dell’ormone con una conseguente dowregulation dei suoi recettori in questi soggetti. [10]
Aumento dell’attività simpatica:
Il sistema nervoso simpatico regola la secrezione di renina in risposta all’ortostatismo, agendo sia direttamente che
18
indirettamente sulle cellule iuxtaglomerulari, nel primo caso tramite un aumento dell’attività dell’adenilciclasi al loro interno, nel secondo agendo mediante una vasocostrizione dell’arteriola afferente.
Meccanismi acquisiti coinvolti nello sviluppo di ischemia renale, stress ossidativo ed infiammazione:
È stato ipotizzato che l’ipertensione rappresenti una forma acquisita di malattia renale che avrebbe l’effetto di limitare l’escrezione di sodio. A seguito di diversi studi su numerosi modelli animali sono state fornite prove evidenti di uno specifico meccanismo patogenetico. In particolare in molti casi l’evento iniziale sembrerebbe essere la vasocostrizione renale, soprattutto a livello dell’arteriola afferente. Modlinger et al. hanno mostrato che la vasocostrizione è mediata da diversi meccanismi tra cui stress ossidativo, trombossani, deficit della produzione di NO, angiotensina II. [11]
Johnson et al. hanno dimostrato che il coinvolgimento di questa via
può risultare da un SNS iperattivo, un’attivazione impropria del SRAA, una disfunzione endoteliale con riduzione del rilascio di NO, da ipokaliemia. Durante questa fase precoce il parenchima renale appare normale eccetto per una lieve infiammazione ed ischemia a livello tubulare, e l’ipertensione è mediata sia dagli effetti dei suddetti sistemi vasocostrittori (vasocostrizione sistemica), sia dai meccanismi collegati all’ischemia renale (rilascio di renina e aumento del riassorbimento di sodio). Fisiologicamente questa è una forma di ipertensione sale resistente, come proposto da
Guyton et al., nella quale l’aumento della pressione sistemica
agirebbe per alleviare l’ischemia renale e permettere alla gestione del carico di sodio di tornare a valori normali a spese appunto di un aumento della pressione sistemica.
19
Tuttavia nel tempo episodi ricorrenti di vasocostrizione renale, in particolare se accompagnati da una stimolazione locale da parte di ATII, possono portare allo sviluppo di malattia arteriolare renale. Finemente associata con quest’ultima è l’infiltrazione di cellule infiammatorie nell’interstizio in particolare cellule T e macrofagi. Il meccanismo primario dell’accumulo di cellule infiammatorie nell’interstizio sembra essere l’ischemia renale, anche se è stato ipotizzato un possibile ulteriore ruolo della proteinuria. È stato dimostrato che le cellule T interstiziali contribuiscono alla sviluppo di ipertensione tramite la generazione in loco di ATII e prodotti ossidanti in loco, i quali sono capaci di ridurre la concentrazione di NO a quel livello. È probabile che il danno intrarenale possa attivare le afferenze simpatiche che giungono a livello del SNC simpatico. Esso può essere inoltre stimolato dall’eccessivo introito di sale con la dieta, producendo come risultato finale un’ulteriore vasocostrizione mediata dalle efferenze simpatiche a livello renale. Tutti questi cambiamenti potrebbero facilitare la ritenzione di sodio sia riducendo la GFR del singolo nefrone, sia stimolando il riassorbimento dello ione stesso. Poiché il danno micro vascolare non è uniforme l’aumento della pressione sistemica risulta nella iperperfusione di alcuni nefroni e nella ipoperfusione di altri. Il risultato finale è che l’ischemia non è totalmente alleviata e cioè porta ad una stimolazione continua dei meccanismi sodio ritentivi, sia renina dipendenti che renina indipendenti. In conseguenza la curva pressione-natriuresi è spostata verso destra e appiattita e l’ipertensione è sale sensibile.
Riassumendo, quindi, possiamo dire che questa ipotesi patogenetica proposta per spiegare lo sviluppo dell’ipertensione vede due fasi:
20
- la prima è condotta da agenti che causano vasocostrizione renale e sistemica, i.e. disfunzione endoteliale, anomala attivazione di SRAA, iperattivazione SNS
- la seconda è dovuta sia all’infiammazione sia al danno microvascolare renale che si viene a produrre nel tempo con il perpetuarsi di questi episodi di attivazione degli agenti vasocostrittori. Questi due fenomeni si vanno a sovrapporre protraendo così la vasocostrizione. [12]
Alterazioni generalizzate a carico delle membrane cellulari:
Nella patogenesi dell’ipertensione sono state implicate anomalie delle proprietà fisiche delle membrane e dei loro molteplici sistemi di trasporto. La maggior parte di esse si riferiscono alle cellule muscolari lisce delle pareti vasali ma poiché tali cellule non sono disponibili per studi negli esseri umani sono stati utilizzati surrogati, quali globuli rossi e globuli bianchi. Negli eritrociti sono presenti sistemi di trasporto, a livello della membrana cellulare, che controllano il movimento di sodio e potassio per mantenere una differenza marcata tra la concentrazione intra- ed extra-cellulare di questi ioni, che fornisce il gradiente elettrochimico necessario per le varie funzioni cellulari.
Esistono prove che lo scambiatore sodio/idrogenioni sia stimolato nei pazienti ipertesi da un aumento della quota di calcio intracellulare o da una sua maggiore entrata dall’esterno. Un aumento dell’attività dello scambiatore Na+/H+ potrebbe giocare un
ruolo significativo nella patogenesi dell’ipertensione, sia stimolando il tono vascolare e la crescita cellulare, sia aumentando il riassorbimento di sodio nelle cellule tubulari prossimali a livello renale.
Inoltre le membrane dei globuli rossi dei pazienti ipertesi mostrano un aumento del rapporto colesterolo/fosfolipidi in associazione
21
con un alto trasporto sodio/litio ed un aumento dei rapporti dei metaboliti degli acidi grassi/precursori, rispetto a quelle di controlli normotesi di età corrispondente. Questi cambiamenti nell’assetto lipidico si riflettono in una maggiore micro viscosità di membrana ed in una riduzione della fluidità, che potrebbero essere responsabili dell’aumentata permeabilità al sodio e di altre alterazioni nel suo trasporto.
Insulino-resistenza ed iperinsulinemia:
Alti livelli di insulina e/o l’insulino-resistenza sono stati indicati come responsabili di un maggiore aumento pressorio in alcuni pazienti affetti da ipertensione. Questo parametro è riconosciuto attualmente come parte della sindrome metabolica assieme con obesità centrale, dislipidemia e alti valori pressori. Mentre appare evidente che una percentuale di pazienti ipertesi presenta iperinsulinemia e resistenza all’insulina, non è del tutto certo che si tratti di un’associazione casuale. Parecchi studi hanno dimostrato che le due suddette condizioni sono presenti anche in pazienti magri, ipertesi, non affetti da diabete mellito, il che suggerisce che questa relazione non sia una coincidenza. Infine, sembra che questi pazienti appartengano in gran parte al fenotipo dei non-modulatori. Esistono vari meccanismi possibili per spiegare questa associazione. L’iperinsulinemia infatti :
- determina aumento del riassorbimento renale di sodio e acqua;
- altera i meccanismi di trasporto degli ioni attraverso la membrana cellulare (i.e. aumenta la quota di sodio intracellulare, riduce l’attività della pompa Na+/K+ ATPasi,
aumenta il trasporto di calcio attraverso la membrana e quindi i suoi livelli citoplasmatici);
22
- stimola la produzione di fattori di crescita in particolare a livello delle cellule muscolari lisce delle pareti vasali;
- stimola l’attività del SNS;
- a livello endoteliale riduce la sintesi di prostaglandine e aumenta la secrezione di endotelina compromettendo la vasodilatazione.
Alla luce di tutto ciò, è importante sottolineare che il ruolo dell’insulina nel controllo della pressione arteriosa ad oggi è stato compreso solo parzialmente e quindi resta da chiarire il peso che essa potrebbe avere nel quadro eziopatogenetico dell’ipertensione. [5]
Diagnosi:
Ogni definizione e classificazione numerica dell’ipertensione sono rese arbitrarie dall’evidenza di una distribuzione unimodale della pressione arteriosa nella popolazione e dalla presenza di una relazione continua tra rischio cardiovascolare e pressione arteriosa sino a valori sisto-diastolici rispettivamente di 115-110mmHg e 75-70mmHg.
Nella pratica clinica, tuttavia, per favorire l’approccio diagnostico e terapeutico, viene utilizzata la classificazione proposta dalle Linee Guida ESH/ESC pubblicate nel 2003, con le seguenti precisazioni:
1. Quando i valori pressori sistolici e diastolici di un paziente si collocano in categorie diverse, si deve considerare la categoria più elevata sia per il calcolo del rischio cardiovascolare totale sia per decidere il trattamento farmacologico e per valutare l’efficacia dello stesso.
2. L’ipertensione sistolica isolata può anch’essa essere suddivisa in gradi (grado 1,2,3) usando gli stessi cut-off impiegati per la definizione dell’ipertensione arteriosa
sisto-23
diastolica. È tuttavia da tener in considerazione che la presenza di valori ridotti (es. 60-70mmHg) può costituire fattore di rischio aggiuntivo.
3. Il valore soglia per definire la presenza di ipertensione e la necessità di iniziare un intervento terapeutico dev’essere flessibile e basarsi sul profilo di rischio cardiovascolare totale.
La diagnosi di ipertensione viene posta quando la media di due o più misurazioni della pressione diastolica in almeno due visite sequenziali è ≥ 90mmHg, o quando la media di più letture della pressione sistolica in due o più visite sequenziali risulta costantemente ≥ 140mmHg. (Tabella 1) [13]
Pressione arteriosa (mmHG) sistolica diastolica
Ottimale < 120 e <80
Normale 120-129 e/o 80-84
Normale-alta 130-139 e/o 85-89 Ipertensione grado 1 140-159 e/o 90-99 Ipertensione grado 2 160-179 e/o 100-109 Ipertensione grado 3 ≥180 e/o ≥110 Ipertensione sistolica isolata ≥140 e <90 Tabella 1: Valori pressori di riferimento
24
Capitolo 2.
Stato pro-trombotico nell’ipertensione
essenziale
2.1
Generalità sul rischio cardiovascolare
La semplice procedura di misurazione della pressione arteriosa consente l’identificazione dei fenotipi a rischio di malattia cardiovascolare. Tuttavia il solo trattamento di tale aumento pressorio non è sufficiente a ridurre in maniera ottimale il rischio cardiovascolare associato ed è quindi raccomandata una stima formale di quest’ultimo. Il calcolo del rischio basato sulla coorte dello studio Framingham usato in USA e UK può dare una sovrastima nella popolazione europea del 7% circa e di una percentuale maggiore in quella asiatica pertanto, l’ European society
of cardiology ha raccomandato l’uso del sistema SCORE.
I sistemi di calcolo del rischio disponibili sono basati su markers convenzionali che possono essere registrati in un ambito clinico di base i.e. pressione arteriosa sistolica, età, sesso, colesterolo sierico, presenza di diabete, tabagismo. Lo studio INTERHEART ha suggerito che più del 90% del rischio attribuibile di popolazione per IMA può essere rappresentato da questi fattori. Elementi aggiuntivi che contribuiscono e amplificano il rischio sono il diabete e la disfunzione renale indicata da una bassa stima di GFR (che tiene conto di età, sesso e BMI) e dalla microalbuminuria o proteinuria. Infine, le anomalie cardiache riscontrate con ECG o ecocardiografia sono correlate con la prognosi.
La soglia per il trattamento attualmente definisce come pazienti ad alto rischio quelli con una probabilità del 20% o più di sviluppare malattia cardiovascolare a 10 anni secondo lo score Framingham.
25
Chiaramente la stima formale del rischio cardiovascolare non è necessaria per pazienti con ipertensione e malattia cardiovascolare, diabete o danno d’organo conclamato. Questi soggetti hanno un rischio sufficiente per beneficiare di un intervento a livello multifattoriale. [14]
2.2
Il paradosso pro trombotico
Evidenze cliniche e di laboratorio sempre maggiori suggeriscono che l’ ipertensione in sé conferisca uno stato pro trombotico o di ipercoagulabilità e che le piastrine e l’endotelio, i quali risultano entrambi attivati nell’ipertensione, abbiano un ruolo cruciale nell’aumentare la tendenza trombotica osservabile in questa condizione clinica. Come è noto, le maggiori complicanze dell’ipertensione sono infarto miocardico acuto ed ictus ed i processi fisiopatologici fondamentali sottostanti ad esse sono la trombogenesi e l’aterogenesi. Infatti, nonostante la parete vasale sia esposta ad alte pressioni, le complicanze dell’ipertensione sono paradossalmente di natura trombotica piuttosto che emorragica: questo è conosciuto come il paradosso pro trombotico dell’ipertensione o “Birmingham paradox”.
Molti componenti della via coagulativa e fibrinolitica sono predittori primitivi e secondari degli eventi cardiovascolari. La stretta associazione di questi markers con l’outcome cardiaco e i comuni fattori di rischio cardiovascolare aumenta la possibilità che questi indici non siano solamente markers o conseguenze della trombosi ma contribuiscano significativamente alla patogenesi del processo aterotrombotico. [15]
Oltre 150 anni fa Virchow postulò che tre caratteristiche predispongono alla formazione del trombo, ovvero: anomalie nel
26
flusso sanguigno, nei costituenti plasmatici e nella parete vasale. Mentre Virchow si stava riferendo alla trombosi venosa il concetto può essere applicato anche a quella arteriosa. Un aggiornamento di questa triade potrebbe essere considerato in riferimento ad anomalie nell’emoreologia e turbolenze a livello delle biforcazioni e delle regioni stenotiche (ovvero anomalie nel flusso sanguigno), anomalie piastriniche e delle vie della coagulazione e fibrinolitica (anomalie dei costituenti plasmatici), ed infine anomalie nell’endotelio (ovvero nella parete vasale). Per conferire all’ipertensione uno stato pro trombotico ciascuna di queste componenti deve essere adeguatamente soddisfatta. Inoltre esse devono essere correlate al danno degli organi bersaglio, prognosi a lungo termine ed alterazioni conseguenti al trattamento.
La principale proprietà reologica del sangue è la sua resistenza al flusso o viscosità. Con un alto shear rate la viscosità è bassa ma se la prima aumenta possono verificarsi, in relazione con l’iperviscosità locale, aterogenesi, disfunzione endoteliale, trombogenesi ed ischemia.
In pazienti ipertesi sono ben note anomalie nel flusso sanguigno come alterazioni della compliance arteriosa, dei fattori emoreologici, e riduzione della riserva di flusso coronarico.
Per quanto riguarda le anomalie nei costituenti plasmatici molti di essi, associati con l’ipertensione e le sue complicanze, sono componenti delle vie coagulatoria e fibrinolitica. Il processo di trombogenesi risulta infatti da un sottile bilancio di queste ultime. Il sistema fibrinolitico è influenzato principalmente dalle interazioni tra attivatori del plasminogeno, come l’attivatore tissutale del plasminogeno (t-PA) che promuove la fibrinolisi, e gli inibitori che modulano la sua attività, come l’inibitore-1 dell’attivatore del plasminogeno (PAI-1).
27
Come riportato dal Framingham Offspring Study [16] i livelli plasmatici degli antigeni PAI-1 e t-PA aumentano in funzione dell’innalzamento pressorio e la loro elevazione è predittrice del rischio cardiovascolare. Anche l’aumento di t-PA riflette in realtà una fibrinolisi compromessa, in quanto la quota misurata è legata per la maggior parte con PAI-1 ed è inattiva. Risulta quindi come livelli elevati di pressione sistolica e diastolica siano in relazione con la compromissione del potenziale fibrinolitico.
La terza componente della triade di Virchow si riferisce alle “anomalie dei vasi sanguigni”. L’ipertensione può anche causare danni all’endotelio come dimostrato da ben documentate anomalie sia nella vasodilatazione arteriosa flusso-mediata che in quella farmacologica. [17]
Esistono infatti due tipi di vasodilatazione denominati endotelio dipendente e non-endotelio dipendente. La dilatazione flusso mediata è un indice che riflette la vasodilatazione endotelio dipendente. Dopo che l’arteria brachiale viene occlusa dalla compressione del bracciale e si creano un’ischemia ed una anossia transitorie. Il rilascio di cellule endoteliali danneggiate in circolo e di fattori di rilassamento endotelio derivati come NO, portano alla vasodilatazione.
La dilatazione nitroglicerina-mediata, dall’altro lato, è un indice riflesso della vasodilatazione non endotelio dipendente. La nitroglicerina e il sodio nitro prussiato hanno effetti diretti sulle cellule vascolari lisce, infatti aumentando la concentrazione di cGMP, causano vasodilatazione.
In circostanze normali i cambiamenti di diametro indotti da questi due tipi di vasodilatazione sono simili. Come riportato nello studio di Yang P. et al., condotto su un totale 81 pazienti con ipertensione essenziale e su un gruppo di controllo costituito da 28 pazienti
28
normotesi, il cambiamento indotto dalla vasodilatazione endotelio dipendente risultava più piccolo rispetto quella non endotelio dipendente, e ciò indica una compromissione della funzione endoteliale nei pazienti ipertesi. Inoltre la dilatazione flusso mediata nel gruppo degli ipertesi risulta significativamente più bassa rispetto al gruppo di controllo. [18]
2.3
Disfunzione endoteliale
L’endotelio è un regolatore chiave dell’omeostasi vascolare. Nonostante sia un semplice strato monocellulare, considerato una barriera permeabile altamente selettiva, è in grado di rispondere a vari segnali chimico-fisici con la produzione di un ampio range di fattori in grado di controllare il tono vascolare, l’adesione cellulare, la tromboresistenza, la proliferazione di cellule muscolari lisce e l’infiammazione a livello della parete vasale.
Tutto ciò viene raggiunto con la produzione ed il rilascio di diverse molecole vasoattive che rilassano o costringono il vaso, così come attraverso la risposta a mediatori circolanti quali bradichinina e trombina.
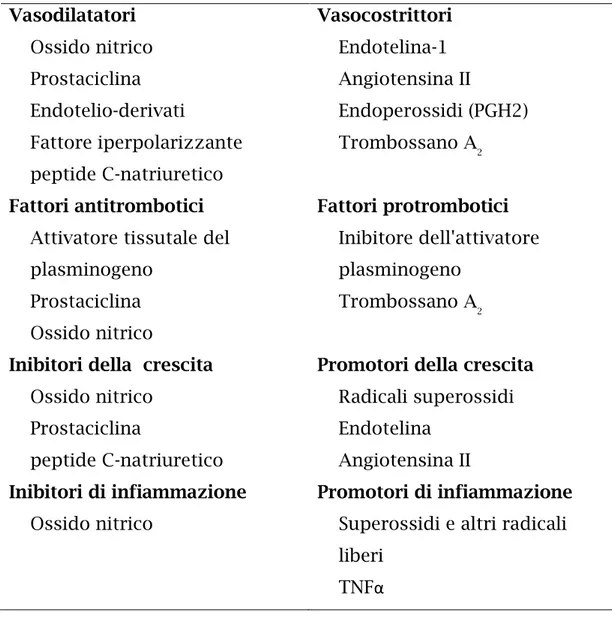
Lo shear stress è il principale fattore fisiologico che stimola l’endotelio a produrre tutte le sostanze suddette riassunte in Tabella 2.
Il protagonista principale nel mantenimento di questo equilibrio omeostatico è l’ossido nitrico (NO) prodotto dalle cellule endoteliali a partire da L-arginina, attraverso l’azione di una sintasi (e-NOS), in presenza del cofattore tetraidrobiopterina. Esso gioca un ruolo essenziale nel mantenere la parete vascolare in uno stato di quiescenza con inibizione dell’infiammazione, della proliferazione cellulare e trombosi. Le proteine target di NO includono, infatti,
29
anche quelle coinvolte nella generazione del Fattore Tissutale (TF), una glicoproteina situata a livello della matrice sub endoteliale la cui esposizione, a seguito di lesione, dà inizio alla cascata emocoagulativa. Inoltre NO inibisce l’adesione piastrinica.
Vasodilatatori Vasocostrittori
Ossido nitrico Endotelina-1 Prostaciclina Angiotensina II
Endotelio-derivati Endoperossidi (PGH2) Fattore iperpolarizzante Trombossano A2 peptide C-natriuretico
Fattori antitrombotici Fattori protrombotici
Attivatore tissutale del plasminogeno
Inibitore dell'attivatore plasminogeno
Prostaciclina Trombossano A2 Ossido nitrico
Inibitori della crescita Promotori della crescita
Ossido nitrico Radicali superossidi
Prostaciclina Endotelina
peptide C-natriuretico Angiotensina II
Inibitori di infiammazione Promotori di infiammazione
Ossido nitrico Superossidi e altri radicali liberi
TNFα
Tabella 2: Fattori endoteliali con proprietà vasoattive, emostatiche, modulatrici della crescita o infiammatorie.
L’altro mediatore vasodilatatorio è la Prostaciclina la quale, tuttavia, sembra avere un ruolo più limitato nel mantenimento del tono vascolare.
30
L’endotelio modula la vasomotilità producendo anche sostanze vasocostrittrici, quali l’Endotelina-1, o convertendo sulla sua superficie angiotensina I in angiotensina II.
La disfunzione endoteliale è caratterizzata principalmente da una compromissione della vasodilatazione mediata dall’endotelio. Più appropriatamente dovrebbe essere considerata come una
attivazione endoteliale, in quanto rappresenta un passaggio da un
fenotipo quiescente, dominato dall’azione di NO, verso uno che coinvolge una risposta di difesa dell’ospite. Molti fattori di rischio cardiovascolare attivano l’apparato molecolare dell’endotelio che risulta nell’espressione di chemochine, citochine e molecole di adesione per piastrine e leucociti. Quindi essa potrebbe essere la conseguenza di uno squilibrio tra fattori che rilassano o contraggono la parete vasale, agenti pro- ed anticoagulanti, o che promuovono/ inibiscono la crescita cellulare, a seguito di fattori dannosi di origine meccanica o biochimica, o di stimolazione del monostrato cellulare verso una secrezione fisio-patologica inappropriata o anomala. [19,20] Ad esempio uno shear stress protratto, cronico, può portare ad un rimodellamento dell’endotelio vascolare, trasformandolo da una superficie anticoagulante in una pro coagulante [14]
Una riduzione nella produzione di NO potrebbe derivare da: • Cambiamenti nella disponibilità intracellulare del substrato • Riduzione dell’attività di e-NOS (presenza di inibitori, deficit
di cofattore)
• Riduzione dell’espressione di e-NOS a causa di fattori quali ipossia, TNFα, o riduzioni nel flusso e shear stress
• Alterazioni del rilascio di NO, recettore-mediato, in risposta allo stress laminare
31
Un aumento della degradazione del mediatore deriva, invece, dalla sua interazione con i radicali liberi dell’ossigeno, la produzione dei quali, a livello delle cellule endoteliali e/o delle cellule muscolari lisce vasali, risulta aumentata nell’ipertensione essenziale come dimostrato da vari studi clinici.
Esistono diversi gradi e forme di disfunzione endoteliale: 1. Danno della subunità alfa delle proteine G
2. Riduzione del rilascio di NO, prostaciclina e fattore iperpolarizzante endotelio derivato (EDHF)
3. Aumento rilascio di endoperossidasi 4. Aumento specie reattive dell’ossigeno 5. Aumento produzione di endotelina-1
6. Riduzione della sensibilità delle cellule muscolari lisce a No, prostaciclina e/o EDHF
Disfunzione endoteliale ed ipertensione:
La rilevanza dell’endotelio vascolare per lo sviluppo dell’ipertensione arteriosa, collegata con il danno alla parete vasale, è ad oggi ampiamente accettata così come il fatto che l’ipertensione tende a peggiorare la funzione vasodilatatrice dell’endotelio. La patologia ipertensiva si verifica in associazione con cambiamenti morfologici e funzionali dell’endotelio. Essi sono espressi in parte dall’accumulo sottoendoteliale di fibrina e dall’infiltrazione delle cellule endoteliali e in parte attraverso modifiche nei processi mediati da NO, endotelina e prodotti di COX che influenzano il tono vascolare endotelio-dipendente.
I pazienti ipertesi mostrano infatti una compromissione della vasodilatazione endotelio dipendente associata con un funzionamento anomalo del sistema endoteliale di NO. In accordo
32
con recenti scoperte anche i radicali liberi dell’ossigeno (ROS) giocano un ruolo rilevante in questo processo.
Lo squilibrio tra attività di NO e ATII osservato nella disfunzione endoteliale e la presenza di altri fattori di rischio di malattia cardiovascolare, causano stress ossidativo che deriva dell’eccessiva produzione di radicali liberi e conseguente aumento del catabolismo di NO.
L’alta generazione di ROS si oppone quindi agli effetti vasodilatatori dell’ossido nitrico, stimola l’attività delle molecole di adesione, favorendo l’attacco dei leucociti all’endotelio, lo sviluppo di una risposta infiammatoria acuta, la proliferazione di cellule muscolari lisce e l’ ulteriore sintesi di matrice extracellulare, contribuendo quindi, nel complesso, allo sviluppo di malattia cardiovascolare.
Le conseguenza dell’ipertensione sull’endotelio non si limitano ad influenzare la regolazione che esso esercita sulla vasomotilità. In particolare è ben noto che l’endotelio possiede potenti proprietà antiinfiammatorie. Infatti NO produce una riduzione dell’adesione leucocitaria e piastrinica all’endotelio, limitando l’espressione di molecole di adesione a livello delle cellule, quali ICAM-1, VCAM-1, selectine, MCP-1.
La disfunzione endoteliale nella malattia ipertensiva potrebbe quindi rappresentare un probabile innesco per lo sviluppo di un’aumentata risposta infiammatoria nella parete vascolare. [21,22]
2.4
Markers biochimici circolanti di disfunzione endoteliale
Una più ampia valutazione delle funzioni dell’endotelio può essere ottenuta studiando i livelli di molecole di derivazione endoteliale presenti nel sangue circolante.
33
Questi markers includono prodotti di derivazione diretta dalle cellule endoteliali, che cambiano a seconda dello stato di attivazione dell’endotelio quali citochine infiammatore , molecole di adesione e fattori regolatori della trombosi, così come markers di danno e riparazione endoteliale.
La misurazione di molti di essi è costosa e difficile e viene effettuata solo nell’ambito della ricerca clinica. Come risultato della disponibilità e variabilità biologica dei test, ad oggi questi fattori hanno un ruolo molto limitato nella valutazione del singolo paziente. [20]
La disfunzione endoteliale è stata associata con l’aumento dell’espressione di glicoproteine del gruppo delle selectine (E-selectina e P-(E-selectina) e del gruppo delle immunoglobuline (ICAM-1, VCAM-1). Queste molecole di adesione permettono ai leucociti di aderire alla superficie delle cellule endoteliali e conseguentemente di migrare all’interno della parte vasale.
Tutte le molecole suddette possono essere misurate in circolo con immunodosaggi commerciali e, tra di esse, E-selectina è probabilmente la più specifica per indicare attivazione delle cellule endoteliali. [20,23]
Diversi studi hanno mostrato un aumento dei livelli plasmatici di E-selectina e delle altre glicoproteine di adesione nei pazienti con ipertensione essenziale rispetto ai controlli normotesi.[24,25]
Inoltre, mentre studi precedenti avevano dimostrato l’aumento di tali molecole in pazienti con ipertensione di grado severo e concomitante presenza di altri fattori di rischio di aterosclerosi, fino alla malattia aterosclerotica conclamata, lo studio di Hlubocká
et al, dimostra come tale aumento sia presente anche in pazienti
34
come elevati livelli di molecole di adesione riflettano direttamente l’attivazione endoteliale in presenza di un’aumentata pressione arteriosa anche nei primi stadi della malattia. [23]
Una molecola proposta già circa una decina di anni fa, come marker di disfunzione/danno endoteliale, è il fattore di von Willebrand. Si tratta di una glicoproteina multimerica prodotta dalle cellule endoteliali e dai megacariociti. La maggioranza del vWF in circolo sembra essere di derivazione dalle cellule dell’endotelio, infatti ampie quantità di questo fattore vengono mobilizzate a seguito della loro attivazione . [26]
Diversi studi hanno dimostrato non solo un aumento di questo marker nei pazienti con ipertensione essenziale ma anche come esso sia significativamente correlato con il danno agli organi bersaglio, e con gli score dello studio Framingham . Il riscontro di livelli elevati correla infatti, sia con una maggior incidenza di cardiopatia ischemica sia con un aumento della mortalità dovuta ad eventi cardiovascolari. [27,28]
Il fattore di von Willebrand può essere dosato in modo relativamente facile, quindi potrebbe avere un certo valore clinico, in quanto la sua misurazione nei pazienti ipertesi sarebbe un metodo non invasivo che assiste il medico nel porre diagnosi e soprattutto potrebbe essere utilizzato come indicatore di progressione e prognosi della malattia.
Esso ha un ruolo cruciale nel mediare l’adesione piastrinica alla parete vasale lesionata, quindi il riscontro di alte concentrazioni di tale fattore potrebbe essere un indicatore indiretto di aterosclerosi e/o trombosi. [26]
L’osservazione che la funzione endoteliale riflette il bilancio netto tra danno e riparazione, ha portato allo sviluppo di test per
35
quantificare il distacco di cellule endoteliali mature e delle microparticelle derivate, volti a rappresentare il grado di danno. [20]
Le microparticelle endoteliali (EMP) sono piccole vescicole formate dalla membrana delle cellule dell’endotelio che, dalla loro superficie, vengono riversate in circolo a seguito di attivazione, danno e/o apoptosi.
Il rilascio può essere causato da diverse citochine, come IL-1 e TNFα, e da una shear pressure elevata.
Come mostrato dallo studio di Preston et al. i valori di EMP sono risultati elevati nel gruppo di pazienti con ipertensione severa e ad alto rischio per danno vascolare acuto, rispetto a quello dei pazienti con ipertensione lieve e ai controlli. Inoltre esiste una stretta correlazione positiva tra i livelli di EMP e quelli di pressione sistolica/diastolica. Suddetta connessione persiste anche in presenza di una coesistenza di più fattori di rischio per danno/attivazione endoteliale suggerendo che EMP potrebbero essere di per sé un marker degli effetti della pressione arteriosa sull’endotelio.
Questi dati sono più coerenti con l’ipotesi che il rilascio di EMP e di altri markers di attivazione endoteliale ampiamente utilizzati, quali sICAM-1, sVCAM-1 e vWF, siano modulati in modo diverso dai vari fattori di rischio che portano alla disfunzione endoteliale. Le EMP possono essere quindi markers più specifici degli effetti indotti sull’endotelio da un’elevata pressione arteriosa rispetto agli altri fin qui considerati. Fattore importante è che il riscontro di elevate EMP può permettere di distinguere meglio i pazienti a rischio di imminente lesione vascolare di derivazione dallo stato ipertensivo.
36
Infatti è noto che le microparticelle endoteliali possiedono l’attività pro-coagulante del fattore piastrinico 3 (PF3). Essa è legata all’esposizione sulla superficie delle micro vescicole di fosfolipidi anionici come la fosfatidil serina, che supportano l’assemblaggio dei fattori della coagulazione. Le EMP hanno anche dimostrato di esprimere il fattore tissutale (TF) che può dare inizio alla cascata emocoagulativa. Le micro particelle circolanti sono in grado, infine, di compromettere la funzione endoteliale vasodilatatoria.
Alla luce di tutto ciò, le EMP possono quindi essere più che dei semplici markers di disfunzione/attivazione endoteliale, ma piuttosto contribuire effettivamente alla patogenesi del danno vascolare osservato nei pazienti con ipertensione grave non controllata.[29]
37
Capitolo 3.
Disfunzione piastrinica nell’ipertensione
essenziale
3.1
Generalità sulle piastrine: ruolo nella coagulazione e
meccanismi di attivazione
L’adesione piastrinica alla matrice extracellulare è lo step iniziale nel meccanismo dell’emostasi primaria. Le piastrine, a seguito del rolling sulla parete vasale, aderiscono e ricoprono la matrice collagene, formando un monostrato attivato. [30] La loro adesione è mediata da due diverse interazioni: la prima avviene tra il complesso recettoriale glicoproteina (GP) Ib/V/IX , sito sulla superficie piastrinica, e il fattore di von Willebrand (vWF) , mentre la seconda tra le glicoproteine GPVI e GPIa, ed il collagene presente a livello della parete vascolare danneggiata.
L’interazione tra vWF e la GP Ib/V/IX è cruciale per dare inizio all’adesione piastrinica al sub endotelio in condizioni di alto shear rate, come quello presente a livello di piccole arterie, arteriole, e siti di stenosi.
In condizioni normali infatti, il vWF non è sottoposto a interazioni significative con il suo recettore a livello piastrinico, ma quando esposto a livello del collagene nel sito danneggiato diviene un forte substrato adesivo.
Il reclutamento e l’attivazione piastrinica sono stimolati dai prodotti di secrezione generati dalle piastrine stesse dopo il loro legame, e da fattori locali pro trombotici (i.e. fattore tissutale) che portano alla generazione del coagulo emostatico. Diverse vie permettono l’attivazione piastrinica, tra cui quelle stimolate dal collagene, ADP, trombossano A2, epinefrina, serotonina e trombina. [30-33]
38
L’azione complessiva di questi attivatori risulta nel reclutamento di ulteriori piastrine dalla circolazione, che conduce, a sua volta, a distinte manifestazioni di attivazione piastrinica. Queste includono il cambiamento della conformazione, l’espressione di molecole pro-infiammatorie, come P selectina e ligando solubile CD40 (sCD40L), l’espressione di attività piastrinica pro-coagulante, e la conversione di GP IIb/IIIa (αII b/ß3-integrina) nella sua forma attiva, che consente l’aggregazione piastrinica e quindi lo sviluppo del potenziale per la trombosi patologica.
L’accumulo locale di questi agonisti recluta piastrine circolanti nel coagulo in formazione, stabilizzandolo ulteriormente. [30]
La glicoproteina GPIIb/IIIa è il recettore che riveste un ruolo centrale nel mediare l’aggregazione piastrinica . Dopo la sua attivazione, promuove l’adesione delle piastrine, l’aggregazione e la diffusione sulla matrice extracellulare esposta a livello del sito danneggiato sulla parete vasale. Inoltre, dopo il legame con il fibrinogeno sulla superficie piastrine attivate, permette la formazione di ponti intercellulari e quindi la stabilizzazione del trombo.
I coaguli ricchi di fibrina sono generati dall’azione della trombina, che viene prodotta inzialmente attraverso la via del TF, a livello ad esempio, delle placche aterosclerotiche erose o in via di rottura. Mediante l’attivazione piastrinica, che viene raggiunta attraverso molteplici pathways, un coagulo può, in ultima analisi, trasformarsi da semplice meccanismo difensivo, in un trombo occlusivo ricco di piastrine.
Vie di attivazione piastrinica:
Molteplici vie contribuiscono all’attivazione piastrinica. I principali agonisti che agiscono in questo processo sono:
39 - ADP - Trombossano A2 - Serotonina - Epinefrina - Collagene - Trombina
L’adenosina difosfato (ADP) è immagazzinata ad alte concentrazioni nei granuli densi e rilasciata dalle piastrine aggregate, durante la loro attivazione.
L’ADP contribuisce all’attivazione piastrinica, sia nel caso in cui essa avvenga come meccanismo di emostasi protettiva (i.e. formazione del monostrato di piastrine iniziale) , sia durante la formazione di un trombo occlusivo ricco di piastrine.
Il rilascio di Trombossano A2 dalle piastrine adese, aumenta il reclutamento e l’aggregazione al coagulo primitivo, e attiva le piastrine in entrambe le condizioni suddette.
Il collagene è un forte substrato trombogenico. In condizioni di alto shear stress, l’adesione piastrinica è mediata dal legame del VWF, immobilizzato sul collagene stesso o sulla superficie delle piastrine attivate, alla GPIb. Questa interazione porta all’attivazione di GPIIb/IIIa ( la quale non è in grado di legare ligandi solubili nel suo stato inattivo) e alla stabilità degli aggregati piastrinici mediata da vWF.
Tuttavia il legame tra GPIb e vWF non è sufficiente a produrre un’adesione stabile. È richiesta infatti la partecipazione di GP VI, ovvero il principale recettore per il collagene che media l’attivazione piastrinica, che è necessario per l’adesione, l’aggregazione, la degranulazione e l’attività coagulante a livello della matrice sub endoteliale. [32]
40
La trombina è l’attivatore piastrinico più potente. Essa è in grado di attivare le piastrine a concentrazioni bassissime, nel range nano molare, inferiori a quelle richieste per l’attivazione della cascata emocoagulativa. [34,35] Dopo il legame al suo recettore PAR-1, sito sulla superficie piastrinica, avviene il clivaggio e conseguentemente l’esposizione di un ligando ancorato al recettore stesso che è in grado di attivarlo. [36-40]
Le piastrine in condizioni di riposo, non sono in grado di rimanere a contatto l’una con l’altra in modo stabile , ma sviluppano questa capacità una volta attivate. Queste manifestazioni avvengono tramite un processo di “outside-in signalling”, che si ha a seguito dell’attivazione delle integrine (αIIb/ß3) dopo che esse hanno preso contatto con il ligando (fibrinogeno). [41]
L’attivazione piastrinica innesca così, una complessa serie di vie di segnalazione intracellulare che portano a cambiamenti della conformazione cellulare e al rilascio dei vari componenti immagazzinati nei granuli. Tutti questi eventi dipendono da un aumento della concentrazione citosolica di Calcio. [42]
La maggior parte degli agonisti attiva, infatti, l’enzima fosfolipasi C (PLC), un secondo messaggero che catalizza la formazione di inositolo trifosfato (IP3) e diacilglicerolo (DAG), all’interno delle piastrine. Quest’ultimo stimola il rilascio di Ca2+ dai depositi
intracellulari e promuove l’entrata dello ione dall’esterno attraverso la membrana cellulare. [43,44]
3.2
Meccanismi che contribuiscono all’ attivazione
piastrinica nei pazienti ipertesi
L’attivazione piastrinica contribuisce in maniera importante all’aumento del rischio trombotico nell’ipertensione essenziale.
41
Alcuni studi hanno riportato che nei pazienti ipertesi esiste un’ iperattivazione piastrinica, oltre ad una disfunzione endoteliale, ed è presente una relazione lineare positiva tra pressione arteriosa e predisposizione all’aggregazione piastrinica. [45,46]
Nei pazienti ipertesi sono stati dimostrati un aumento in vitro dell’aggregabilità piastrinica ad ADP, epinefrina e collagene, ed un maggior rilascio in vivo di ß-tromboglobulina (ß-TG) e trombossano A2. [47]
Il fenomeno dell’attivazione piastrinica nel contesto dell’ipertensione coinvolge le shear forces, l’attivazione del SRA, la disfunzione endoteliale, l’aumento dei livelli di catecolamine circolanti.
L’incremento della pressione arteriosa si associa ad elevate shear forces, in particolare nelle vicinanze della superficie endoteliale, e ciò può portare a degranulazione piastrinica e conseguente attivazione. [48]
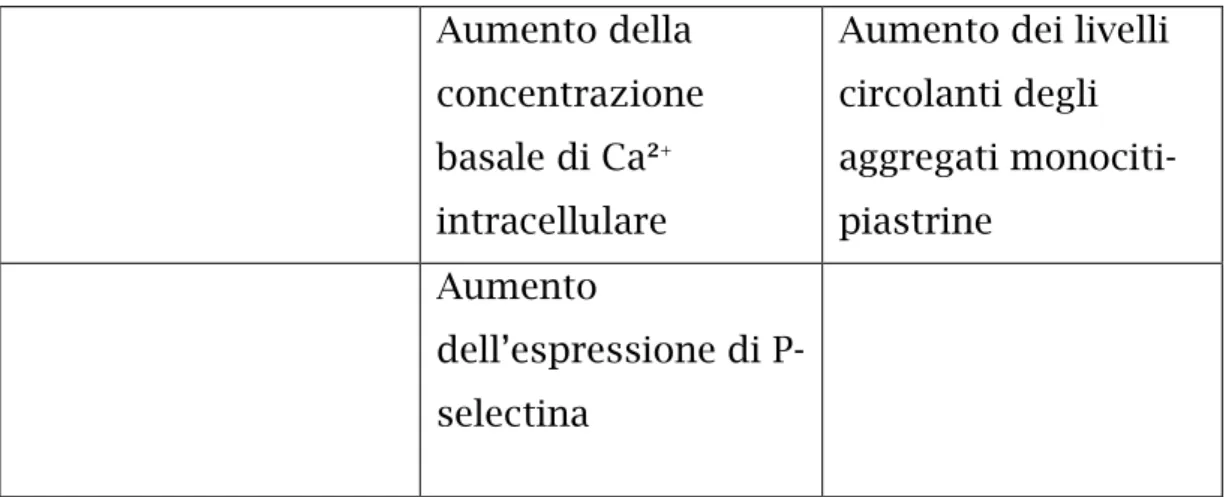
Il sistema renina angiotensina gioca un ruolo di primo piano nella patogenesi dell’ipertensione essenziale in alcuni soggetti. Esso influenza anche l’attivazione piastrinica tramite il suo principale mediatore, l’Angiotensina II. Quest’ultima aumenta i livelli di calcio intracellulare nelle piastrine, che si associano ad una maggiore aggregazione.[49] Dopo l’infusione di questo ormone in volontari sani, è stato dimostrato un aumento dei livelli circolanti di P-selectina solubile e ß-tromboglobulina, ed una maggior capacità da parte delle piastrine di legare il fibrinogeno. Tutti questi effetti sembrano essere mediati dall’interazione con i recettori AT1 presenti sulla superficie piastrinica e dalla conseguente attivazione della fosfolipasi C. [50]
42
Nella disfunzione endoteliale viene generata una quota ridotta di agenti antiaggreganti quali NO in particolare, e prostaciclina, mentre si ha un’eccessiva produzione di endotelina-1 che favorisce l’aggregazione piastrinica. [51]
L’aumento del tono simpatico attiva le piastrine contribuendo allo stato di ipercoagulabilità osservato nell’ipertensione. Inoltre, l’ up regulation degli α2-adrenorecettori piastrinici nei pazienti con ipertensione essenziale, porta ad un aumento dell’effetto pro-aggregante delle catecolamine. [52]
Infine è stato indagato anche il ruolo del NO prodotto dalle piastrine nel provocare un aumento della loro aggregazione. L’ossido nitrico sopprime l’aggregazione piastrinica, riduce l’adesione alle cellule endoteliali ed inibisce la formazione di aggregati piastrine-moniciti (MPAs). [53] Interferisce inoltre con la mobilizzazione del calcio a livello piastrinico, il principale ione coinvolto nel processo di attivazione. NO riduce sia il flusso di Ca²+
attraverso la membrana plasmatica, sia il suo rilascio dai depositi intracellulari. [54]
Le piastrine esprimono la NO sintasi di tipo 3 e un certo numero di studi suggerisce come la sua funzione sia alterata nei pazienti con ipertensione essenziale. Ad esempio, durante la loro aggregazione, le piastrine dei soggetti ipertesi mostrano una risposta ridotta alla N-monometil-L-arginina, un inibitore della NO sintasi, rispetto ai controlli normotesi. [55]
Inoltre, nell’ipertensione, la NDP(H) ossidasi piastrinica produce una quota maggiore di anioni superossido che riducono la biodisponibilità di NO ed aumentano la funzionalità piastrinica. [56] Tutto ciò anche grazie agli alti livelli di Angiotensina II osservati nei pazienti ipertesi, la quale attiva la NADP(H) via recettore AT1.