31 2. SCOPO DEL LAVORO
In questo lavoro di tesi sono state verificate l’infettività e la capacità di competere con endofiti indigeni di due isolati di Glomus mosseae, IMA1 e AZ225C, introdotti come inoculo in campi sperimentali di M. sativa. La regione di DNA utilizzata per lo studio è stata quella dell’rDNA nucleare comprendente una porzione di SSU, la regione ITS e parte di LSU. La regione SSU è stata utilizzata per l’identificazione della comunità fungina radicale tramite RFLP e successivo sequenziamento. Il sequenziamento della regione ITS2 è stato utilizzato per l’identificazione dei due isolati fungini AM inoculati.
32 3. MATERIALI E METODI
3.1 Materiale fungino e vegetale
3.1.1. Materiale fungino
Gli isolati geografici di funghi AM studiati appartengono alla collezione IMA (International Microbial Archives), mantenuta dalla Prof. Manuela Giovannetti, presso il Dipartimento di Biologia della Piante Agrarie, Università di Pisa.
Glomus mosseae IMA1, Gerdemann and Trappe, origine geografica Kent (U.K.). Deriva dall’isolato Glomus mosseae, originariamente noto come Yellow Vacuolate, raccolto ed identificato da Barbara Mosse e tenuto in coltura presso la Rothamsted Experimental Station, UK. L’isolato è stato ottenuto da inoculo monosporale, mantenuto dal 1981 in coltura a Pisa e depositato presso l’Erbario dell’Orto Botanico (Codice PI-HMZ4).
Glomus mosseae AZ225C, origine geografica Arizona (U.S.A.). Raccolto ed identificato dal Prof. J.C. Stutz e tenuto in coltura presso l’International Culture Collection of VA mycorrhizal fungi (INVAM), Morgantown, W. Va., USA. L’isolato è stato ottenuto da inoculo plurisporale e mantenuto dal 1997 in coltura a Pisa.
3.1.2. Materiale vegetale
Le analisi sono state effettuate su radici di M. sativa cv. Messe, foraggera poliennale, diffusa in tutto il mondo, appartenente alla famiglia delle Leguminosae. Le piante di M. sativa analizzate erano parte di un esperimento avviato a settembre 2004, presso il Dipartimento di Agronomia e Gestione dell’Agroecosistema, in località
33 ‘Rottaia’ su parcelle di 15 m2 (5m x 3m). L’esperimento consisteva in tre trattamenti :
- piante di M. sativa inoculate con l’isolato IMA1;
- piante di M. sativa inoculate con l’isolato AZ225C;
- piante di M. sativa non inoculate (controllo).
L’inoculo era stato distribuito manualmente sul terreno, dopo la vangatura, e adeguatamente interrato. Gli attrezzi adoperati erano stati sterilizzati ad ogni passaggio, ed erano state impiegate protezioni per gli operatori, guanti e calzari in modo da evitare contaminazioni delle parcelle nei passaggi tra i diversi trattamenti. La dose di inoculo distribuita era stata 0.7 Kg/m2.
3.2 Estrazione del DNA da radici
I campioni analizzati erano costituiti da radici di tre piante per singola tesi (non inoculate, inoculate con l’isolato IMA1, inoculate con l’isolato AZ225C). I campioni erano stati prelevati dopo due mesi dalla germinazione.
L’estrazione del DNA dai campioni è stata effettuata con il kit DNeasy Plant Mini QIAGEN secondo il protocollo della ditta produttrice.
I campioni di radici, derivanti da collezione a -80oC, sono stati pesati (Tab. 1) e messi in azoto liquido fino al momento del pestaggio in mortaio. Il DNA estratto è stato eluito in 50 µl di “nuclease-free water”.
E’ stata effettuata un’elettroforesi su gel di agarosio, al fine di valutare la qualità e la quantità dell’estratto. Un’aliquota di 1 µl di DNA genomico è stato visualizzato mediante corsa elettroforetica su gel di agarosio (Biorad, Milano, Italia) all’0,8% in tampone TBE 1X, con Bromuro di Etidio 0.5 µg/ml.
34 Tabella 1. Peso in mg dei campioni di radici di M. sativa dai quali verrà effettuata l’estrazione del DNA.
Campioni Peso (mg) CONTROLLO 1 28 CONTROLLO 2 34 CONTROLLO 3 25 IMA1 1 31 IMA1 2 47 IMA1 3 26 AZ225C 1 52 AZ225C 2 46 AZ225C 3 40
Ad 1 µl di DNA sono stati aggiunti 2 µl di tampone di caricamento 6x Mass Loading Dye Solution (Fermentas) (bromofenolo blu 0,09%, glicerolo 60%, EDTA 60 mM).
Come marker di riferimento di concentrazione e peso molecolare è stato usato Mass Ruler DNA Ladder, Low Range (Fermentas), che permette di valutare le dimensioni e la quantità dei frammenti di DNA.
E’ stata poi effettuata una quantificazione del DNA ottenuta attraverso un biofotometro (Eppendorf AG Biophotometer 6131, Amburgo, Germania).
La concentrazione del DNA viene determinata considerando che una densità ottica (OD) pari a 1, misurata a 260 nm, corrisponde a una concentrazione di 50 µg/ml di DNA a doppio filamento. Il biofotometro permette di effettuare anche letture a 230 nm e a 280 nm. Il valore dei rapporti dell’assorbanza alle varie lunghezze d’onda (A260/A280 e A260/A230) permette di valutare la purezza del campione di DNA. Se la qualità del DNA è buona, questi rapporti danno valori vicini a 1,8.
35 3.3 Amplificazione del DNA fungino tramite PCR
3.3.1 Amplificazione con i primers NS31-LSUGlom1
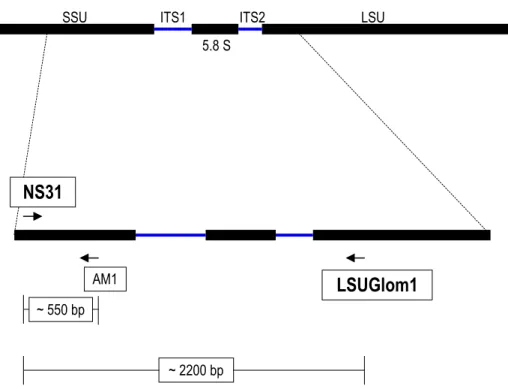
Una porzione della regione del DNA ribosomale di ~2200 bp (Fig. 7) è stata amplificata dagli estratti di DNA radicale, usando la GoTaq DNA polimerasi (Promega) con un primer universale per gli eucarioti, NS31 (5’-TTGGAGGGCAAGTCTGGTGCC-3’) (Simon et al., 1992), e uno
specifico per i funghi AM, LSUGlom1
(5’-CTTCAATCGTTTCCCTTTCA-3’) (Renker et al., 2003). Il primer LSUGlom1 è stato disegnato per essere specifico per i Glomeromycota e per amplificare il terminale 5’.
Figura 7. Posizione schematica dei primers NS31 (Simon et al., 1992) e LSUGlom1 (Renker et al., 2003) all’interno del DNA ribosomale.
SSU 5.8 S LSU ITS1 ITS2
NS31
AM1 ~ 550 bpLSUGlom1
~ 2200 bp36 La reazione di PCR è stata fatta usando un mix di reazione di 25 µl con 0.625 U di GoTaq DNA polimerasi (Promega), 5 µl di buffer di reazione 5X della casa produttrice, MgCl2 2 mM, dNTPs 0.2 mM each,
NS31 e LSUGlom1 0.2 µM ciascuno e 2% V/V di Blotto (De Boer et al., 1995). Un termociclizzatore (Eppendorf Mastercycler® personal, Eppendorf, Milano, Italia) è stato settato con un programma touch down come segue: 95oC per 2 min; 19 cicli a 95oC per 30 sec, da 62oC a 52oC con la diminuzione di 0.5oC per ogni ciclo per 1 min, 72oC per 2 min e 15 sec;19 cicli a 95oC per 30 sec, 52oC per 1 min, 72oC per 2 min e 15 sec; 1 ciclo a 72oC per 10 min.
I prodotti di PCR sono stati visualizzati in un gel di Agarosio (Biorad, Milano, Italia) all’1.5% in TBE 1 X, con Bromuro di Etidio 0.5 µg/ml.
Un marker per DNA 1 Kb plus DNA ladder (Invitrogen) è stato usato per identificare la dimensione delle bande. I profili di DNA sono stati visualizzati e acquisiti con ImageMaster VDS system e le immagini sono state analizzate con il software ImageMaster Elite (Amersham Bioscience).
3.3.2 Amplificazione con i primers AML1-LSUGlom1
Si è verificata, per alcuni estratti, la necessità di utilizzare un’altra coppia di primers, a causa dell’assenza o in molti casi della scarsa quantità di amplificato prodotto ottenuto. Con le stesse condizioni di PCR touch down, è stato utilizzata la coppia di primers AML1 (5’-ATCAACTTTCGATGGTAGGATAGA-3’) (Lee et al., 2008) (Fig. 8)- LSUGlom1 ed è stato ottenuto un prodotto di ~2400 bp.
37 Figura 8. Posizione schematica dei primers AML (Lee et al., 2008) e LSUGlom1 (Renker et al., 2003) all’interno del DNA ribosomale.
3.4 Purificazione dei prodotti di PCR
I prodotti di PCR dotati della sola banda specifica di ~2200 bp, sono stati purificati in colonne Montage® PCR Centrifugal Filter Devices Millipore, come da protocollo della casa produttrice. Se l‘amplificazione produceva anche bande diverse da quella di interesse, si procedeva con l’estrazione da gel con Wizard® SV Gel and PCR Clean-Up System Promega, come da protocollo della casa produttrice.
Entrambi i prodotti di purificazione sono stati utilizzati come inserti per il vettore di clonaggio. Gli inserti sono stati quantificati in termini di concentrazione (ng/µl) al biofotometro.
3.5 Ligazione e clonaggio
La reazione di ligazione è stata fatta in 10 µl contenenti 3 U di T4 DNA ligasi, 5 µl di 2X Rapid ligation buffer, 1 µl di pGEM®-T Easy Vector Promega (50 ng/ µl) (Fig. 9) e 110 ng di inserto.
SSU 5.8 S LSU ITS1 ITS2 NS31
LSUGlom1
AML1
~ 2450 bp38
Figura 9. Schema del vettore di clonaggio pGEM®-T Easy Vector (Promega).
La quantità di inserto è stata calcolata sulla base della seguente relazione, in modo da ottenere un rapporto 3:1 inserto:vettore, in funzione della dimensione in termini di base-pairs dell’inserto:
((50 ng di vettore x 2,2 kb di inserto)/3,0 kb di inserto) x 3/1= 110 ng di inserto
La reazione è stata tenuta in incubazione overnight a 4 oC.
Sono state trasformate cellule di Escherichia coli XL10-Gold® Ultracompetent Cells Stratagene secondo il protocollo di trasformazione fornito dalla casa produttrice, utilizzando i plasmidi ottenuti dalla reazione di ligazione.
Le cellule trasformate sono state messe ad incubare a 37oC su piastre di Petri con substrato Luria Bertani (LB) contenente 15 g/l di Agar, IPTG (Isopropile β-D-1-tiogalattopiranoside) 0,05 mM, 80 µg/ml
39 di X-GAL (5-bromo-4-cloro-3-indolil-Betagalattoside) e 100 µg/ml di ampicillina (Sambrook et al., 1989).
La selezione dei cloni positivi è stata effettuata grazie alla colorazione delle colonie ricombinanti, che appaiono bianche invece che blu.
Il vettore pGEM®-T Easy Vector della serie PUC può esprimere , sotto il controllo del promotore del lattosio, un piccolo peptide, corrispondente alla parte N-terminale della Beta-galattosidasi (il prodotto genico di lacZ – Fig. 9).
Questo peptide é in grado di complementare la funzionalità enzimatica di β-galattosidasi mutanti prive della corrispondente parte N-terminale. Utilizzando, in terreno solido, un substrato cromogenico come l'X-Gal, l'attività enzimatica viene recuperata e le colonie appaiono colorate di blu.
Poiché il polilinker di pGEM®-T Easy Vector é situato nel gene che codifica l' α-peptide, l'inserzione di un frammento di DNA, l’inserto, ne interrompe l'integrità funzionale e la capacità di dare α -complementazione (Sambrook et al., 1989). Le colonie corrispondenti appaiono bianche e segnalano la presenza di un clone positivo.
3.6 Selezione mediante PCR delle colonie ricombinanti (colony PCR)
Le colonie ricombinanti sono state amplificate con la coppia di primers specifica del vettore, SP6 (5’-ATTTAGGTGACACTATAGAAA-3’) e T7 (5’-TAATACGACTCACTATAGGG-3’) (Fig. 9) per verificare l’effettiva presenza dell’inserto.
Una parte della colonia bianca è stata sospesa in un tubo eppendorf e utilizzata come stampo in una reazione di PCR contenente i primers SP6 e T7 e contemporaneamente è stata strisciata su una piastra di Petri contenente LB e ampicillina (100
40 µg/ml) per essere replicata. La reazione di PCR è stata fatta usando un mix di reazione di 12.5 µl con 0.3125 U di GoTaq DNA polimerasi (Promega), 2.5 µl di buffer di reazione 5X della casa produttrice, MgCl2
2 mM, dNTPs 0.2 mM ciascun nucleotide e SP6 e T7 0.4 µM ciascuno. Il termociclizzatore (Eppendorf Mastercycler® personal) è stato così programmato: 95oC per 2 min; 29 cicli a 95oC per 30 sec, 50oC per 30 sec, 72oC per 2 min e 30 sec; 1 ciclo a 72oC per 10 min.
3.7 Nested PCR dei prodotti positivi della “colony PCR”
Una nested PCR è stata effettuata diluendo (1/10-100) il prodotto della reazione con i primers SP6/T7 (colony PCR), e usando 1 µl della diluizione come stampo per una seconda reazione di amplificazione in un volume di 25 µl. La reazione è avvenuta a partire dalla coppia di primers NS31 e AM1 (5’-GTTTCCCGTAAGGCGCCGAA-3’) (Fig. 7) (0.2 µM ciascuno). Le concentrazioni di GoTaq DNA polimerasi, dNTPs e MgCl2 sono le stesse descritte nella colony PCR. Il
termociclizzatore è stato settato con il seguente programma: 95oC per 2 min; 31 cicli a 95oC per 30 sec, 58oC per 1 min, 72oC per 1 min ; 1 ciclo a 72oC per 10 min.
3.8 Analisi RFLPs (Restriction Fragment Lenght Polymorphisms)
Gli ampliconi dei cloni ottenuti a seguito della nested PCR sono stati digeriti con gli enzimi di restrizione HinfI e Hsp92II (Promega) (Tab. 2). Il livello di risoluzione del metodo RFLP è incrementato usando più di un enzima di restrizione per generare polimorfismi. Almeno 20 cloni positivi NS31/AM1 per ogni campione sono stati analizzati tramite RFLPs. Le reazioni, separate per ognuno dei due enzimi, sono state effettuate in un volume di 20 µl, contenente 2 µl di tampone di reazione 10X della casa produttrice, BSA acetilata 0.1 µg/µl, 1 U di
41 enzima di restrizione e 4 µl del prodotto della nested PCR. La reazione è stata lasciata ad incubare tutta la notte a 37oC.
Tabella 2. Enzimi utilizzati nelle analisi di restrizione con le rispettive sequenze di ricoscimento.
Enzima Sito di riconoscimento
HinfI G ▼ANTC CTNA▲G Hsp92II CATG ▼ ▲GTAC
Il DNA digerito è stato visualizzato in un gel di agarosio (Biorad) al 2% in TBE 1 X, con Bromuro di Etidio 0.5 µg/ml, e il marker utilizzato è stato 100 bp DNA ladder (Promega). I profili di DNA sono stati visualizzati e acquisiti con ImageMaster VDS system e le immagini sono state analizzate con il software ImageMaster Elite (Amersham Bioscience). Sono stati analizzati 350 cloni positivi all’amplificazione NS31/AM1 su un totale di 954 cloni analizzati mediante “colony PCR”.
I dati ottenuti mediante RFLP sono stati usati per determinare sia la ricchezza di ribotipi (presenza/assenza degli RFLPs) sia l’abbondanza relativa dei ribotipi (frequenza degli RFLPs). Per cui, il numero totale di RFLPs è stato usato come misura della diversità fungina (Ricchezza delle specie, SR). Inoltre è stata stimata la diversità genetica per ciascun campione radicale mediante la formula: Ho =1 - ∑pi ln pi (dove pi è la frequenza di un dato RFLP).
3.9 Estrazione del DNA plasmidico dai cloni con profili di RFLPs rappresentativi
Un sottocampione rappresentativo di ciascuno profilo RFLP è stato scelto per l’estrazione del DNA pasmidico e il successivo sequenziamento. Dai cloni selezionati sulla base dei risultati
42 dell’analisi con gli enzimi di restrizione, sono stati quindi estratti 288 plasmidi attraverso il kit Wizard® Plus SV Minipreps DNA Purification System della Promega, seguendo il protocollo della ditta produttrice.
3.10 Sequenziamento
I plasmidi estratti sono stati sequenziati in un ABI Prism® 377 sequencer (Applied Biosystem, Forster City, CA, USA) presso la University of Washington, High-Throughput Genomics Unit (Seattle Wa, USA), usando SP6 e T7 come primers per il sequenziamento. In totale, 144 frammenti clonati sono stati sequenziati (96 e 48, per il primo mese e il secondo anno rispettivamente).
3.11 Analisi statistiche per comparare la diversità e struttura AMF ottenuta mediante RFLPs
Sui parametri RS e Ho è stata fatta un’analisi ANOVA ad una via, per
dividere le variazioni nella variabile di risposta utilizzando il fattore trattamento fungino come variabile indipendente. L’ipotesi nulla testata era: nessun effetto del trattamento fungino sulla comunità AM. Le differenze tra le medie sono state determinate mediante il test di Tukey.
Sulle percentuali di ciascun RFLP presente nelle radici, dopo opportuna trasformazione angolare, è stata fatta un’analisi non parametrica a coppie (AZ vs Controllo, IMA1 vs Controllo, AZ225C vs IMA1) mediante Mann-Whitney Test , dato che l’omogeneità delle varianze valutata tramite il Test di Levene aveva rilevato valori significativi.
Il software SPSS 15.0 (SPSS Inc., Chicago, IL, USA) è stato usato per entrambe le precedenti analisi.
43 Il disegno sperimentale ha permesso di valutare la composizione fungina AM entro le radici di piante inoculate con i diversi isolati e il controllo. I dati (0/1 e le abbondanze relative) sono stati inoltre analizzati, dopo valutazione del gradiente (>4), mediante Canonical Correspondence Analysis (CCA) per i dati di RFLP e (<4) mediante Redundancy Analysis (RDA) per i dati relativi ai tipi di sequenze in CANOCO per Windows (ter Braak e Smilauer, 2002), e utilizzando il test di Montecarlo per determinare la significatività del trattamento fungino (Lepš e Šmilauer, 2003; Anderson e Willis, 2003; Anderson, 2006). L’RDA e la CCA sono tecniche di ordinazione “constrained” che si basano sull’analisi diretta del gradiente (Lepš e Šmilauer, 2003). Il test di permutazione di Montecarlo è stato condotto usando 999 permutazioni random.
3.12 Analisi delle sequenze
Le sequenze SSU e ITS ottenute sono state allineate usando il programma BioEdit con sequenze di Glomeromycota presenti in GenBank. . L’analisi è stata effettuata mediante il metodo Neighbour-Joining (NJ) utilizzando il programma TREECON per il software Windows. Le distanze per l’albero NJ sono state calcolate con il modello Kimura dove il valore del rapporto transizioni/transversioni è stato settato a 2 (van de Peer e de Wachter, 1994). La confidenza dei singoli rami dei dendrogramma è stata valutata usando un “boostrap” di 1000 ricampionamenti. Geosyphon pyriforme è stato usato come outgroup nell’ albero SSU e Glomus geosporum come outgroup nell’albero ITS. Gli alberi sono stati disegnati mediante TREECON.