DIPARTIMENTO DI MEDICINA CLINICA E SPERIMENTALE
Tesi di Specializzazione in Neuropsichiatria Infantile
Caratteristiche neuro-funzionali della sindrome di Down:
plasticità ed effetti dell’arricchimento.
Candidato:
Dr.ssa Margherita Bozza
Relatore:
Chiar.mo Prof. Giovanni Cioni
i
INDICE
Sommario ... 1
1
Introduzione ... 4
2
La sindrome di Down ... 6
2.1
Definizione ed Epidemiologia ... 6
2.2
Eziopatogenesi ... 7
2.3
Diagnosi e Clinica ... 9
2.4
Profilo Neuropsicologico ... 13
2.4.1
Profilo Cognitivo ... 13
2.4.2
Profilo metacognitivo ... 15
2.4.3
Profilo Linguistico ... 16
2.4.4
Profilo adattivo ... 17
2.5
Disturbi psichiatrici ... 20
2.6
Deterioramento Cognitivo ... 23
2.7
Aspetti neuro-anatomici ... 27
2.7.1
Anatomia Macroscopica ... 27
2.7.2
Anatomia Microscopica ... 31
2.8
Trattamento nella SD ... 37
2.8.1
Trattamento riabilitativo ... 37
2.8.2
Arricchimento ambientale ... 37
2.8.3
Trattamento farmacologico ... 40
ii
3.2.1
Disegno dello studio ... 48
3.2.2
Criteri di inclusione/esclusione dallo studio ... 49
3.2.3
Valutazioni ... 49
3.2.4
Tempi delle valutazioni e divisione in gruppi ... 53
3.2.5
Esperienza di arricchimento cognitivo ... 54
3.2.6
Analisi statistica ... 55
3.3
Risultati ... 56
3.3.1
Descrizione del campione ... 56
3.3.2
Risultati di un intervento di arricchimento cognitivo ... 66
3.4
Discussione ... 85
3.5
Conclusioni... 91
1
Sommario
La Sindrome di Down (SD) è la causa più comune di aberrazione cromosomica e la più frequente forma di ritardo mentale geneticamente determinato.
Dalle ultime ricerche emerge che il profilo neuropsicologico degli individui con SD è molto complesso e che alcune abilità sono maggiormente preservate rispetto ad altre. Lo sviluppo cognitivo non avviene semplicemente più lentamente ma assume aspetti qualitativi particolari. Il Quoziente Intellettivo (QI) non si mantiene costante nel corso della vita ma tende a decrescere con l’età. L’acquisizione delle abilità motorie si compie più lentamente e si caratterizza per la presenza di pattern di movimenti atipici. Le abilità linguistiche sono nel complesso deficitarie, la produzione in misura maggiore rispetto alla comprensione lessicale. Un punto di forza sono le abilità visuo-spaziali in particolare nei compiti che implicano il processamento di materiale spaziale, rispetto all’elaborazione del materiale visivo come forme e colori. Le abilità mnestiche sono deficitarie in particolare a livello della Memoria verbale a breve Termine e nella memoria a lungo termine esplicita. Dal punto di vista neuroanatomico, i soggetti con SD presentano un ridotto volume cerebrale in particolare a livello del cervelletto e dei lobi frontali e
2
temporali. Le cause dell’ipotrofia cerebrale nella SD possono essere ricondotte ad un disordine della neurogenesi, aumento dell’Apoptosi cellulare, neurodegenerazione e ipotrofia dendritica. Idealmente, l’identificazione dei meccanismi alla base di queste anomalie può fornire un razionale per ideare terapie che possano portare ad un miglioramento dei difetti di sviluppoRecenti studi con modelli sperimentali sia animali che umani hanno fornito iniziali evidenze scientifiche rispetto al possibile ruolo di un intervento di arricchimento ambientale nella fase preclinica della malattia di Alzheimer per ridurre il rischio di sviluppare la demenza. L’esposizione di topi Ts65Dn ad un ambiente arricchito ha portato ad un incremento della lunghezza dei dendriti ed ad un aumento della densità a livello delle cellule piramidali.
L’obiettivo principale del progetto è di determinare gli effetti di un intervento di arricchimento ambientale, basato su un training cognitivo, nel prevenire il deterioramento nei soggetti adulti con SD. Il training è stato finalizzato ad una stimolazione della memoria, delle capacità di attenzione e di concentrazione, e delle capacità di ragionamento logico.
Il campione di studio è costituito dai 40 soggetti (21 maschi e 19 femmine), con età media di 33,7 anni. Questo è stato suddiviso in modo randomizzato in due gruppi; di cui uno ha eseguito il training (gruppo Tr), l’altro ha proseguito le proprie abituali attività costituendo il gruppo di controllo (gruppo Cont). Tutti i partecipanti sono stati valutati al baseline (T0), dopo 3 mesi (T1) e dopo 6 mesi da T0 (T2). Nel corso di ogni valutazione sono stati eseguiti: protocollo psicologico, psicopatologico e linguistico.
Al termine del trattamento abbiamo osservato una riduzione statisticamente significativa dei sintomi psichiatrici (Iperattività, Letargia, Stereotipie e Irritabilità) nel gruppo Tr rispetto al gruppo Cont.
Nell’ambito del profilo cognitivo valutato con test OLC, non vi sono state modificazioni statisticamente significative ma si osserva una tendenza al miglioramento nelle prove di Seriazione e di Numerazione. Sempre una tendenza alla significatività si osservava nelle prove di memoria a breve termine (SPAN parole e DIGITspan) nel gruppo Tr con ritardo mentale lieve. Dalla somministrazione della WISC-III si osservava una modificazione statisticamente significativa nell’item delle Informazioni ed una tendenza alla significatività nella prova di Ricostruzione degli Oggetti. Nella scala di sviluppo della integrazione
3
visuo-motoria si otteneva un miglioramento statisticamente significativo sia considerando i punteggi grezzi sia l’età equivalente. Per quanto riguarda le capacità adattive, pur non avendo avuto delle differenze statisticamente significative si osservava una tendenza alla significatività nella Età Equivalente media della scala Totale e di alcune sottoscale (Abilità Quotidiane, Gioco, Socializzazione).L’esperienza condotta ha evidenziato in tutti i soggetti una certa sensibilità verso un trattamento, portando delle fondamentali modificazioni qualitative come l’ incremento dell’iniziativa comunicativa e relazionale, l’aumento dei tempi di attenzione, la maggiore tolleranza alla frustrazione.
Tali modificazioni, seppur dopo un trattamento relativamente breve, supportano l’idea di una possibile modificabilità anche nei soggetti adulti con SD.
4
1 Introduzione
Fino a qualche anno fa, la struttura e l’organizzazione cerebrale adulta erano considerate come relativamente stabili. Recenti studi, invece, dimostrano che anche dopo lo sviluppo, il cervello continua a modificarsi in risposta alle nostre esigenze. L’ambito di ricerche che va sotto il nome di plasticità del sistema nervoso, è dunque lo studio delle variazioni funzionali e strutturali del cervello durante lo sviluppo e la vita adulta.
Nell’età evolutiva, anche se la formazione iniziale delle sinapsi è determinata geneticamente, l’esperienza ha un ruolo fondamentale nella maturazione e nel mantenimento delle connessioni.
L’allargamento del concetto di plasticità cerebrale fornisce un importante contributo anche alla ricerca neuropsicologica e in particolare alla possibilità di dimostrare come modificazioni riorganizzative siano presenti anche nell’età adulta e non solo durante lo sviluppo. La condizione di arricchimento guida le modificazioni strutturali e comportamentali del sistema nervoso.
Attraverso l’utilizzo del modello murino, ed in particolare del modello murino della sindrome di Down (Ts65Dn), è stato dimostrato come un’esperienza di arricchimento ambientale abbia degli effetti positivi dal punto di vista comportamentale dimostrando una efficace sia sull’apprendimento che sulla
5
memoria. Riguardo ai cambiamenti anatomici del sistema nervoso centrale, l’esposizione degli animali ad un ambiente arricchito causa un incremento del peso cerebrale globale, del peso corticale, e della densità neuronale della corteccia cerebrale; la densità dei neuroni ippocampali risulta maggiore, i contatti sinaptici sono più numerosi ed il numero degli alberi dendritici è maggiore negli animali allevati in condizioni di arricchimento.L’obiettivo principale del nostro studio è quello di sperimentare gli effetti di un intervento di arricchimento ambientale costituito da un training psico-fisico nei soggetti adulti con sindrome di Down.
6
2 La sindrome di Down
2.1 Definizione ed Epidemiologia
La Sindrome di Down (SD) è la causa più comune di aberrazione cromosomica e la più frequente forma di ritardo mentale geneticamente determinato, riconosciuta come entità clinica da più di 150 anni [1]. Interessa tutte le etnie ed entrambi i sessi, con un’incidenza complessiva di circa un caso su 733 nati vivi negli Stati Uniti [2].
A causa di una minore efficienza dei meccanismi che contrastano la non-disgiunzione meiotica e per la riduzione delle difese biologiche deputate a distruggere le uova e gli zigoti anomali, la frequenza di questa malattia aumenta con l’aumentare dell’età materna, con una rapida accelerazione dopo i 35 anni [3] passando da un’incidenza di un caso su 2500 nati da madre in età inferiore a 20 anni, a un caso su 50-100 nati da madre oltre i 40 anni. In realtà, i concepimenti di zigoti con trisomia 21 sono molto più frequenti, ma circa 3 casi su 4 evolvono in un aborto o una nascita di un bambino morto; se questo non succedesse l’incidenza della sindrome all’interno della popolazione salirebbe a circa un caso su 200.
7
Nei paesi a maggior sviluppo la SD costituisce un terzo circa dei casi di deficit intellettivo. La sua prevalenza è molto alta, ciò è dovuto sia all’elevata incidenza della sindrome, sia al notevole incremento dell’aspettativa di vita dei soggetti con SD a cui si è assistito negli ultimi due decenni. Questo dato è emerso da un recente studio condotto in Australia che ha stimato un’aspettativa di vita di 61,1 anni per i maschi e 57,8 anni per le femmine SD [4, 5].A causa di diversi fattori sociali, la diversa facilità di accesso alle cure mediche, l’aspettativa di vita è significativamente minore nei soggetti di razza diversa da quella caucasica [6]. Oggi, in Italia, gli adulti Down rappresentano il 60% di tutta la popolazione con SD [7].
2.2 Eziopatogenesi
Nel 1959 Lejeune, Gautier e Turpin scoprirono l’associazione tra la SD e un terzo cromosoma 21 [8]. La duplicazione del materiale genetico localizzato su questo cromosoma determina uno squilibrio di 200-250 geni, la maggior parte dei quali, ad oggi, è già stata identificata e studiata, la restante parte solamente ipotizzata.
La regione del cromosoma 21 responsabile delle principali manifestazioni fenotipiche della SD è rappresentata dalla banda q22, e ancor più specificamente dalla banda q22.2-22.3 che si trova nella parte terminale del braccio lungo del cromosoma 21. In questa “regione critica” si trovano i geni per il collagene tipo VI che possono avere un ruolo nella patogenesi delle cardiopatie congenite associate frequentemente alla SD; il gene codificante la proteina alfa-1 del cristallino è legato alla patogenesi della cataratta e delle opacità corneali; alcuni oncogeni che possono essere causa della maggior incidenza di leucemia in soggetti Down [9].
Altri geni soprannumerari potrebbero essere implicati nei fenomeni di precoce invecchiamento, tipici della SD: il gene APP, che codifica per la proteina precursore dell’amiloide (sostanza che si accumula in distretti cerebrali dei soggetti con malattia di Alzheimer e in adulti Down dopo i 34-40 anni) e il gene SOD-1, che codifica per l’enzima superossidodismutasi, e il cui eccesso comprometterebbe una efficace difesa cellulare contro i radicali liberi dell’ossigeno.
8
Le manifestazioni cliniche, i tratti fisici e cognitivi dei soggetti con SD, possono essere in parte assenti o presenti con stadi diversi di gravità, a dimostrazione del fatto che le suddette caratteristiche non dipendono dall’effetto isolato di singoli geni ma dalla interazione tra geni diversi [7].Le cause genetiche che determinano la SD sono diverse. La causa più frequente è una condizione definita “trisomia 21 libera” caratterizzata da un fenomeno di non disgiunzione meiotica, per cui uno dei due genitori porta nella sua cellula riproduttiva due cromosomi 21 invece di uno come di norma, e per questo lascia in eredità tre cromosomi 21 (Figura 1). Questa è la situazione di gran lunga più frequente, che interessa il 95% dei casi [10] e che nell’88% dei casi è determinata da una non disgiunzione materna [11]. Questa condizione si verifica sporadicamente e in modo imprevedibile anche in coppie senza anamnesi familiare positiva per SD o altre anomalie cromosomiche più o meno importanti.
Figura 1: Cariotipo di un uomo con trisomia 21(www.asalsido.org/down).
Un altro possibile meccanismo, che riguarda il 4% circa dei casi, è dato dal fenomeno della traslocazione: cioè il trasferimento del cromosoma 21 su un altro cromosoma acrocentrico (13, 14, 15, 21, o 22, ma più frequentemente sul 13 o sul 14). Ne consegue una conta totale di 46 cromosomi come di norma, ma con massa cromatinica del 21 rappresentata tre volte invece che due. Molto spesso (3/4 dei casi), la traslocazione rappresenta un nuovo evento, tanto è vero che, eseguendo l’analisi cariotipica dei genitori, solo nel 20-40% dei casi un genitore ha una traslocazione bilanciata [10]. In questo caso le madri portatrici della
9
traslocazione possono dare alla luce tre tipi di prole: fenotipo e cariotipo normale, fenotipo normale e cariotipo con traslocazione, trisomia 21 da traslocazione. Le possibilità dovrebbero verificarsi con la stessa incidenza, ma in realtà i figli trisomici sono solo il 10% probabilmente per l’elevata mortalità dello zigote o del feto trisomico. I padri portatori influenzano la prole di rado, sebbene possano generare sia figli normali che traslocati [8].La SD può conseguire ad un processo di mosaicismo (1%) per cui solo una percentuale di cellule dell’individuo possiede un cromosoma “libero” in più. I mosaici possono originare da una non-disgiunzione in una cellula derivata dalla prima o dalle successive divisioni dello zigote oppure dalla perdita di un cromosoma 21 nella prima o successive divisioni di uno zigote concepito trisomico. Ne risulta la formazione di due diverse linee cellulari , una euploide e una trisomica. La percentuale di cellule affette determina in modo proporzionale una più spiccata o più lieve espressione del fenotipo Down [10].
Una minima parte dei casi di SD (circa lo 0,2%) è determinata da “trisomie parziali”. Queste interessano solo una parte del cromosoma 21 e possono essere già presenti in famiglia o presentarsi de-novo.
2.3 Diagnosi e Clinica
La diagnosi della SD può essere prenatale, effettuata mediante l’amniocentesi, consigliata a tutte le donne di età superiore a 35 anni o a quelle che hanno già avuto un figlio affetto. Lo screening viene fatto nel primo trimestre di gravidanza con il tri-test (dosaggio su siero materno di alfa-feto proteina, della beta-gonadotropina corionica e dell’estriolo non coniugato) associato ad esame ecografico per la valutazione della translucenza nucale (Figura 2) (limite massimo dello spessore: 3mm). La combinazione dello screening e della traslucenza nucale permettono di rilevare l’80-85% dei casi con un 5% di falsi positivi. La positività dello screening rende necessaria una valutazione più approfondita tramite amniocentesi anche nelle gravide non considerate a rischio [9].
10
Figura 2: Translucenza nucale (www.labsanmichele.it).Il fenotipo del bambino con SD (Figura 3) è caratteristico fin dalla nascita: peso e lunghezza inferiore alla media, il volto è piccolo, rotondeggiante con profilo piatto e naso piccolo, con radice piatta e rivolta verso l’alto, epicanto (terza palpebra), rima palpebrale obliqua dall’alto in basso e dall’esterno all’interno, iride punteggiato (Brushfield spots), brachicefalia, microcranio, occipite piatto, ipoplasia mascellare e mandibolare, macroglossia per ipertrofia del muscolo con lingua prominente e con solchi trasversali (lingua scrotale), padiglioni auricolari piccoli, talora displastici, spesso a basso impianto, collo corto e tozzo con cute retronucale sovrabbondante, solco palmare unico, mono o bilateralmente, con quinto dito piccolo con o senza clinodattilia, ampio spazio tra primo e secondo dito del piede, alluce valgo, piede piatto, assenza mono o bilaterale della dodicesima costa, displasia pelvica e ginocchio valgo [8]. L’80% dei neonati trisomici presenta ipotonia marcata, notevole lassità legamentosa e riflesso di Moro debole. La loro spesa energetica nella prima settimana di vita è inferiore del 14% rispetto a quella di un neonato sano [9]. La mimica facciale del bambino con SD è caratteristica: movimenti non coordinati dei muscoli del volto associati a protrusione della lingua che si muove velocemente e senza alcuno scopo [8]. La fontanella anteriore è slargata e si chiude tardivamente, l’eruzione dentaria è rallentata e caratterizzata da pattern anomali e denti piccoli.
11
Figura 3: Rappresentazione delle caratteristiche cliniche della sindrome di Down (www.vividown.org).Il 40-60% dei bambini con SD presenta difetti cardiaci congeniti [12] tra i quali i più frequenti sono: difetti del setto atrio-ventricolare (45%), difetti del setto interventricolare (35%), dotto arterioso pervio (7%), Tetralogia di Fallot (4%). Nell’adolescente e nel giovane adulto si rilevano disfunzioni valvolari quali prolasso mitralico e rigurgito aortico [13].
I neonati trisomici presentano, seppur raramente, difetti congeniti del tubo digerente, quali atresia o stenosi duodenale, pancreas anulare, fistola tracheo-esofagea, stenosi del piloro, ano imperforato, malattia di Hirschprung [14]. Inoltre possono presentare reflusso gastro-esofageo [15] e celiachia (4-8% dei casi), che si manifesta con diarrea, calo delle performance e rallentamento di crescita [16]. Nella seconda-terza infanzia in circa il 50% dei bambini affetti è presente sovrappeso fino all’obesità per la riduzione del metabolismo basale e per la scarsa attività motoria [18].
Tra i primi mesi di vita e i tre anni e poi tra i dieci e diciassette anni si assiste nei soggetti Down ad un ritardo nella velocità di crescita [19], ne consegue una statura adulta ridotta, fondamentalmente a causa del mancato “growth-sprut” che generalmente accompagna la maturazione sessuale nei soggetti non affetti.
12
L’età d’esordio della pubertà, le caratteristiche sessuali primarie e secondarie e le concentrazioni delle gonadotropine risultano essere simili nei soggetti sani e in quelli SD, sia nelle ragazze [20] che nei ragazzi [21]. Le donne possono essere fertili, ma hanno un alto rischio di trasmettere la sindrome, i maschi sono solitamente sterili, si trovano due soli casi in letteratura di maschi Down che hanno concepito un figlio [22]. La menopausa è più precoce nelle donne SD rispetto a quelle con ritardo mentale non Down e ancor di più rispetto a quelle sane [23].I problemi oftalmologici dei bambini Down aumentano di frequenza in proporzione all’età: il 38% dei bambini al di sotto di un anno e il 80% di quelli tra i 5 e i 12 anni [24] necessitano di monitoraggio o intervento per errori di rifrazione (35-76%), strabismo (25-57%), nistagmo (20%), o anche per cataratta, blefariti o cheratocono.
Nella popolazione SD si riscontrano patologie autoimmunitarie più frequentemente che in quella generale [25] e le più comuni sono: l’ipotiroidismo, nel 15% dei Down pediatrici secondo la “Health Supervision for children with Down Syndrome [26]”; il diabete di tipo 1, dall’1 al 10% degli adolescenti Down [28]; la celiachia con una frequenza tra il 4,6 e il 7,1% [27]; la vitiligine e l’alopecia (5-9%) [29]; tra le malattie autoimmunitarie sistemiche la più comune è la sindrome da anticorpi anti-fosfolipidi [30] con frequente interessamento del SNC (strokes, TIA, trombosi venosa centrale, disfunzione cognitiva, demenza, depressione, psicosi) [31].
Rispetto alla popolazione generale i soggetti SD sono più suscettibili al verificarsi di disordini mieloproliferativi transitori (presenza di blasti nel sangue periferico); trombocitopenia o trombocitosi a risoluzione spontanea in 2 o 3 mesi o progressione a leucemia megacarioblastica acuta; leucemia linfoblastica acuta o miogena acuta, rispettivamente più frequenti in bambini >1 anno e in lattanti [32]. I bambini con SD hanno più frequentemente infezioni sistemiche e respiratorie dovute a difetti immunologici (riguardanti sia l’immunità specifica che aspecifica) e ad alterazioni del timo (che appare di piccole dimensioni e con anomalie morfostrutturali). La regolazione della risposta immune potrebbe essere influenzata anche dai geni presenti sul cromosoma 21 [33].
L’8-14% dei soggetti Down sviluppa epilessia [34] con un’incidenza che sembra essere età-correlata, con due picchi, uno entro i primi dodici mesi di vita e l’altro
13
nella quarta-quinta decade di vita [35] e rispettivamente correlati a complicanze mediche precoci (encefalopatia ipossica-ischemica o cardiopatie congenite) [36] e allo sviluppo di cambiamenti neuropatologici Alzheimer-like [37] di cui si tratterà più in dettaglio in seguito. I tipi di crisi che si riscontrano più frequentemente in soggetti SD sono: crisi generalizzate tonico-cloniche [38], crisi miocloniche e spasmi infantili con ipsaritmia. Le crisi parziali si verificano generalmente nei pazienti con eziologia identificabile. La Sindrome di Lennox-Gastaut, anche se rara, è ben documentata nella SD con caratteristico esordio de-novo intorno agli undici anni e senza essere preceduta da altre forme di epilessia [39].2.4 Profilo Neuropsicologico
2.4.1 Profilo Cognitivo
Il fenotipo clinico della SD è, quindi, facilmente riconoscibile, ma il più interessante tratto clinico di questa sindrome è un profilo funzionale caratterizzato dall’ampia variabilità dei vari gradi di ritardo mentale che possiamo riscontrare.
Il quoziente intellettivo (QI) nei soggetti con SD varia solitamente nell’ambito del ritardo mentale di grado medio-grave (QI tra 25 e 55). E’ tuttavia presente una ridotta percentuale di casi in cui il ritardo risulta di grado lieve e sono stati riportati dei punteggi di QI nel range della normalità in alcuni adulti con SD [40].
In un recente articolo, Vicari et al. [41] hanno riportato i dati del QI di un campione di 56 individui italiani con DS. Il valore medio di QI era 44,7 con un range variabile da 28 a 71.
In termini di età mentale vengono riportate età equivalenti medie di 4-5 anni [42], con punteggi che difficilmente superano i 7-8 anni d’età [43].
Il quoziente intellettivo nei soggetti con SD è influenzato da molteplici fattori genetici e ambientali l’andamento del QI risente di un progressivo declino che inizia già dal primo anno di vita e rende non costante il rapporto tra l’età mentale e quella cronologica [43]. Tale declino cognitivo non deve essere associato ad un arresto di sviluppo bensì ad un rallentamento di quest'ultimo. Il QI decresce nella prima decade di vita per raggiungere un plateau che si mantiene nell’adolescenza e nella prima parte dell’età adulta [44].
14
La traiettoria del QI tende a passare da circa 60-66 nei primi tre anni di vita a 33-38 tra i 13-18 anni. Coerentemente, Vicari et al. [41] hanno riportato valori di QI compresi tra 45 e 71 nei bambini con SD di 6,5-8 anni di età cronologica. Il QI era, invece, più basso negli adolescenti e giovani adulti (in ordine cronologico età=12,2-25,9) variando tra 28 e 47.Il ritardo è globale, ma il fenotipo comportamentale che caratterizza i soggetti con SD rispetto agli altri quadri di RM, evidenzia una compromissione selettiva a carico di alcuni aspetti verbali (fonetici-fonologici-morfosintattici e di memoria verbale) rispetto ad abilità visuospaziali che appaiono più evolute.
Abilità Visuo-Spaziali
Le abilità visuo-spaziali appaiono essere un punto di forza nei soggetti con SD in quanto solitamente in linea con la loro età mentale.
Da recenti dati clinici e sperimentali relativi alle abilità visuo-spaziali in individui con SD, emerge che questi soggetti non differiscono dai controlli nei compiti che implicano il processamento di materiale spaziale [45], mentre conseguono esiti insufficienti quando ad essere elaborato è il materiale visivo come forme e colori. In uno studio molto recente [46] viene descritta una discrepanza nel livello di prestazioni raggiunte dagli individui con DS nell’ambito visuo-spaziale. Il gruppo dei soggetti con DS erano significativamente più deficitari rispetto ai soggetti con sviluppo tipico di pari età mentale nel processare il materiale percettivo e le immagini. Tuttavia, quando il compito richiesto è l’elaborazione delle informazioni spaziali, le prestazioni dei soggetti con SD sono simili a quelle dei soggetti con sviluppo tipico.
Memoria
Diversi studi hanno dimostrato nei soggetti con SD una riduzione della memoria verbale a breve termine e della memoria di lavoro rispetto ai controlli di pari età mentale [41]; inoltre il ritmo di sviluppo della memoria verbale è più lento rispetto al ritmo dello sviluppo mentale [49].
L’interazione reciproca tra deficit di memoria verbale e difficoltà linguistiche al momento è ancora controversa. Secondo alcuni autori il deficit di memoria verbale a breve termine non si correla con il livello di sviluppo del vocabolario recettivo che appare più evoluto [50]. Per Chapman [51] sarebbe il deficit a
15
carico dei processi linguistici che comprometterebbe l’efficacia della memoria verbale. Infatti lo span di memoria verbale dei soggetti con SD risulta analogo a quello dei controlli pareggiati per età verbale piuttosto che per età mentale. Nell’ambito della memoria a lungo termine emergono maggiori difficoltà nella memoria esplicita rispetto alla memoria implicita. Processi automatici che richiedono un basso livello di attenzione supportano la memoria implicita, mentre la memoria esplicita necessita di un apprendimento consapevole, della codifica delle informazioni, di strategie di recupero e di un alto grado di attenzione. I soggetti con SD hanno normale capacità di apprendimento per le attività che richiedono una memoria implicita ma presentano una compromissione selettiva della memoria esplicita, con informazioni di codifica povere e un deficit nelle capacità di recupero e di attenzione [52, 53]. Le ridotte capacità della memoria a lungo termine possono essere attribuite a disfunzioni a livello temporale e ippocampale. Nell’esecuzione di prove specifiche per la valutazione della funzionalità dell’ippocampo e del sistema prefrontale, si osserva una importante compromissione sia negli adulti che nei bambini con SD [54].2.4.2 Profilo metacognitivo
Nei soggetti affetti da SD, come anche in molti di coloro che presentano un ritardo cognitivo di differente origine, esiste una particolare compromissione degli aspetti metacognitivi che, sovrapponendosi al disturbo intellettivo di base, determinano un ulteriore peggioramento delle prestazioni. La compromissione metacognitiva è in parte dovuta alle caratteristiche neurobiologiche del ritardo mentale, ma non solo.
Schematicamente possiamo riconoscerne tre dimensioni che contribuiscono al funzionamento meta cognitivo nei soggetti con ritardo mentale:
1) ridotta consapevolezza che il soggetto ha del proprio funzionamento mentale generale e specifico (come la memoria) e dei processi che possono favorire ed ostacolare tale funzionamento.
2) ridotta capacità di controllo che si riferisce alla capacità di attivare strategie adeguate di autoregolazione durante la risoluzione di un problema, come l'analisi, la pianificazione, il monitoraggio, la verifica del
16
compito, i meccanismi che consentono la generalizzazione di un apprendimento.3) la componente emotivo-motivazionale che fa riferimento alla scelta degli obiettivi che un soggetto si pone, l'atteggiamento di fronte ai compiti, di fronte ai successi o fallimenti. Nelle persone con SD la maggior parte delle esperienze portano alla costituzione del loro senso di incompetenza. Questo induce ad un atteggiamento di rinuncia e di evitamento del compito, o di accorciamento del tempo di confronto con una prova.
2.4.3 Profilo Linguistico
Il linguaggio è forse il dominio comportamentale meglio analizzato nella SD. Le componenti indicate come maggiormente compromesse sono l’articolazione [55, 56], la fonologia [57] e l’organizzazione morfo-sintattica [55] che raggiungono prestazioni inferiori rispetto a quelli attese per l’età mentale. Il corso evolutivo dello sviluppo linguistico dei bambini con SD appare caratterizzato da un ritardo nel primo sviluppo lessicale che si accompagna ad un ritardo più marcato della produzione frasale [58].
Generalmente nei bambini con sviluppo linguistico normale intorno ai 16-20 mesi si assiste ad una accelerazione rapida della crescita del lessico, la così detta “esplosione del vocabolario”, cui si associa una parallela emergenza della capacità combinatoria [58]. Uno studio di Caselli et al. [58] ha evidenziato che nel gruppo di bambini con SD da loro esaminati il fenomeno dell’esplosione del vocabolario avviene tardivamente (5,6 anni) dopo un periodo di incremento lessicale lento. Inoltre gli autori hanno rilevato una ulteriore asincronia di sviluppo tra i bambini normali e i bambini con SD nell’ampiezza del vocabolario raggiunto dal momento in cui iniziano a produrre le prime frasi: i bambini normali iniziano a presentare la combinatoria sintattica quando raggiungono un vocabolari medio di 126 parole mentre i bambini con SD quando hanno raggiunto un vocabolario di circa il doppio (227 parole). La disomogeneità del profilo linguistico dei soggetti con SD nella prima e seconda infanzia è documentata da numerosi studi della letteratura che evidenziano come aree di “forza” le acquisizioni lessicali e le competenze pragmatiche e come aree di “debolezza” quelle relative al dominio fonologico e morfo-sintattico [59, 60, 61, 62]. Inoltre diversi studi hanno
17
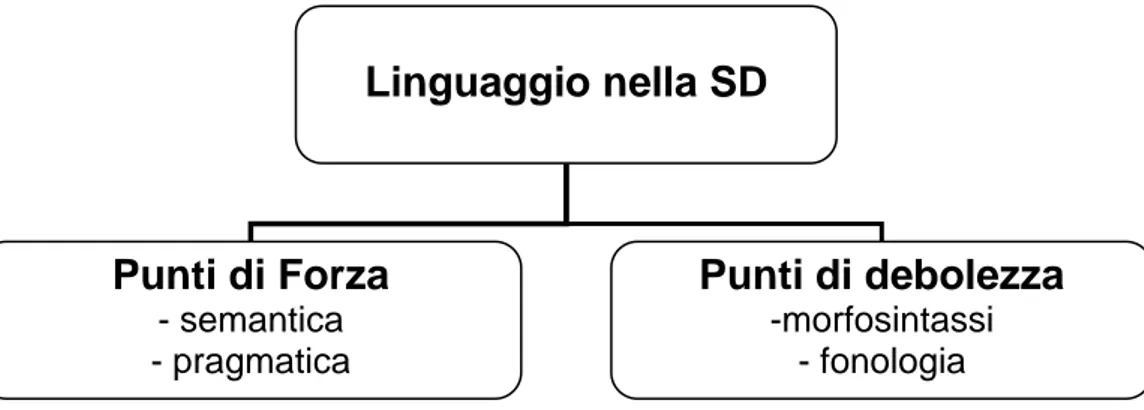
evidenziato una discrepanza tra le abilità di comprensione lessicale che risultano preservate rispetto al livello di età mentale raggiunto, rispetto alla capacità di comprensione morfosintattica in cui i soggetti con SD hanno una prestazione inferiore a quella attesa in base all’età menale [63].Figura 4: Profilo linguistico nella SD: punti di forza e punti di debolezza.
Due sono le principali ipotesi avanzate per spiegare le compromissioni linguistiche selettive riscontrate in questa sindrome: da un lato le difficoltà potrebbero originare da un disturbo intrinseco al sistema linguistico o per deficit di competenza (ridotta conoscenza delle regole) o per un problema di organizzazione dell’output (“output, implementazione, Fowler), dall’altro il disturbo linguistico viene ricondotto ad un disturbo di memoria a breve termine che renderebbe il recupero e la selezione delle informazioni più difficile e dunque ostacolerebbe il processo di acquisizione del linguaggio [64].
2.4.4 Profilo adattivo
L’analisi del Quoziente Intellettivo non è un indicatore sufficiente del funzionamento generale di un individuo. Le abilità di un soggetto dipendono infatti dalle caratteristiche proprie dell’individuo e dagli scambi che esso ha con l’ambiente in cui vive. Questa dimensione relazionale è indagabile attraverso l’analisi delle capacità d’adattamento intese come l’insieme delle abilità concettuali, sociali e pratiche che le persone imparano per vivere la propria quotidianità.
Linguaggio nella SD
Punti di Forza
- semantica
- pragmatica
Punti di debolezza
-morfosintassi
- fonologia
18
Vari studi hanno evidenziato come le capacità d’adattamento nei soggetti con SD risultino maggiori rispetto sia a quelle riscontrabili in soggetti sani con pari età mentale, che a quelle raggiunte da soggetti con pari grado di ritardo mentale determinato da una differente origine (Figura 5) [65]0 10 20 30 40
abilità motorie autonomia quotidiana controllo emotivo comunicazione
Client Development Items Differentiating Subjects
do
ma
ins
capacità adattive a confronto
RM SD
puteggio massimo
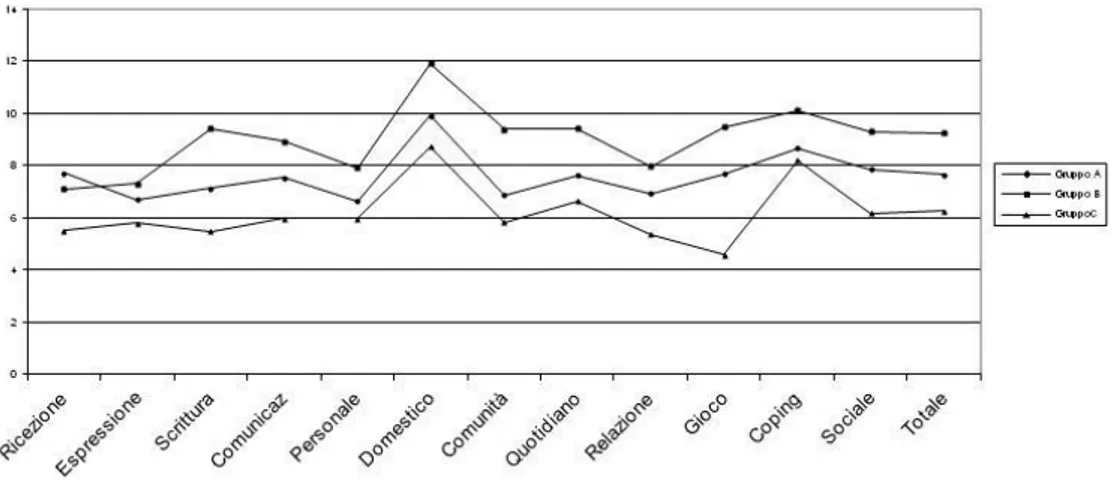
Figura 5: Risultati ottenuti dalla somministrazione di una scala sulle funzioni adattive in un gruppo di 413 soggetti con SD, istituzionalizzati da circa 20 anni, con età media di 30 anni e ritardo mentale di grado grave, appaiati con soggetti presentanti analoghe caratteristiche, ma con ritardo dipendente da diversa eziologia [65].
Lo studio di Dykens et. al [66] ha utilizzato le categorie della scala Vineland per definire le competenze adattive meglio sviluppate nella SD. Le abilità quotidiane rappresentano l’area con punteggi più alti seguite dalla socializzazione e poi dalle capacità comunicative. La cosa non dovrebbe sorprenderci, data l’influenza che le capacità linguistiche (punto di debolezza nella SD) hanno nella comunicazione.
Inoltre il buon funzionamento adattivo permette un miglior inserimento sia nell’ambiente scolastico, dove influenza significativamente i livelli di educazione raggiungibili, che in quello lavorativo. Queste ricadute sono particolarmente significative dato l’aumento dell’aspettativa di vita registrato nell’ultimo decennio per i soggetti con SD.
19
La modificabilità del comportamento adattivo si basa sul livello delle abilità cognitive proprie del soggetto, ma è anche fortemente influenzata da fattori esterni quali: la compliance familiare, la scolarità, il trattamento.Vianello et al. [67], in una sintesi della letteratura sul profilo cognitivo e comportamentale della SD evidenzia come, rispetto alle prestazioni intellettive e linguistiche, vi sia un “surplus” nelle abilità sociali e nelle prestazioni scolastiche. La discrepanza tra lo sviluppo cognitivo e quello adattivo nell’età adulta è stata evidenziata anche dallo studio di Bargagna et al. [68] effettuato su 34 adulti con SD. I risultati di tale studio hanno mostrato che l’età equivalente totale alla scala Vineland è risultata pari ad 8,0 mentre l’età equivalente alle matrici di Raven è risultata tra i 5 e i 6 anni e quella della produzione grammaticale tra 3 e 4 anni. Interessante è valutare l’evoluzione del funzionamento adattivo in correlazione con l’età mentale e quella cronologica dei soggetti con SD. Sebbene alcuni autori sostengano che il comportamento adattivo presenta nel corso del suo sviluppo, periodi di avanzamento durante la prima infanzia, seguiti da periodi di plateau o di rallentamento durante la seconda infanzia e l’adolescenza (Figura 6).Altri studi, dimostrano la possibilità di potenziamento delle funzioni adattive anche in soggetti adolescenti e giovani adulti [69].
Figura 6: Studio condotto in un gruppo di 80 bambini con sindrome di Down di età compresa tra 1 e 11,5 anni. Si osserva un miglioramento del funzionamento adattivo fino a 6 anni, con una fase successiva di plateau. Tuttavia nei soggetti di età superiore vi è una maggiore variabilità [69].
20
Bargagna et. al. [68] hanno suddiviso 34 soggetti in tre gruppi: 19-24 anni (gruppo A), 25-31 anni (gruppo B) e 37-51 anni (gruppo C) ed hanno osservato come le capacità comunicative si mantengono stabili nel tempo mentre le capacità quotidiane e la socializzazione aumentano con l’età fino ai 30 anni potendo raggiungere per alcuni individui un età equivalente pari a 8 anni o più. Segue una fase di plateau o di lieve progresso o regresso tra i 30 e i 50 anni.Figura 7: Profilo per Età Equivalente alla Vineland in gruppi di età cronologica diversa.
2.5 Disturbi psichiatrici
La prevalenza dei disturbi psichiatrici nei soggetti con DS è del 20-30% e tale percentuale risulta essere meno comune della prevalenza di disturbi psichiatrici nei soggetti con ID di diversa eziologia [70, 71, 72]. Tale aspetto può essere biologicamente determinato, ma vi è anche un’influenza dell'ambiente e delle competenze adattive nella SD.
Nei soggetti con SD di età inferiore ai 20 anni la prevalenza dei disturbi psichiatrici è del 17,6%. I disturbi più frequenti in questa età sono: disturbo da deficit di attenzione con o senza iperattività e disturbo del comportamento (6,1%), disturbo della condotta od oppositivo-provocatorio (5,4%), comportamenti aggressivi (6,5%). In età adulta la prevalenza dei disturbi psichiatrici è del 25,6%;
21
sono maggiormente frequenti i disturbi depressivi (12%) e i comportamenti aggressivi (6,1%) [73]. La doppia diagnosi di Autismo e SD è fatta con difficoltà; recenti studi riportano una frequenza variabile tra il 1% e il 7% [74]. Rara è anche la comorbidità della SD con schizofrenia [71, 72]La depressione è, quindi, la diagnosi psichiatrica più frequente nei soggetti con DS. Le caratteristiche comportamentali maggiormente presenti sono il rallentamento psicomotorio, perdita dell'appetito e calo ponderale, mentre le caratteristiche cognitive quali ad esempio la perdita della concentrazione o l'ideazione suicida possono non essere sempre ben evidenti.
Ulteriori criteri diagnostici oggettivamente evidenziabili sono: deterioramento della cura della persona, ritiro sociale, variazione nei tempi dei pasti con aumento o perdita di peso, scarso eloquio, disturbi del sonno e comportamento aggressivo. È possibile osservare anche sintomi depressivi “atipici” quali irritabilità e aggressività sia etero che auto-diretta.
Myers and Pueschel [75] riportano 22 casi di soggetti con SD affetti da depressione. Il sintomo più frequentemente riportato è la riduzione degli interessi nella vita del soggetto (95,4%) seguito dai disturbi del sonno e la riduzione dell'appetito o il calo ponderale (entrambi 81,8%), agitazione (72%), ansia (40,9%), astenia (36,3%) e deficit di memoria (22,7%). L'analisi della sintomatologia sopra riportata pone l'accento sull'importanza rivestita dalle modificazioni neurobiologiche oggettivamente osservabili nei soggetti con ritardo mentale. Dal punto di vista neuroanatomico soggetti depressi non affetti da disabilità intellettiva presentano un aumento delle dimensioni dei ventricoli celebrali e atrofia corticale [76]. Nei soggetti con SD l'intero volume celebrale risulta essere ridotto e sproporzionato. Queste alterazioni sono riconducibili ad un'aumentata espressione genica dovuta alla presenza di un cromosoma soprannumerario. Il rischio di sviluppare la patologia depressiva è maggiore nei soggetti che mostrano atrofia alla RM [77] ed in particolare un ridotto volume ippocampale. Le radici della depressione in questi soggetti risiedono in un' alterazione del substrato anatomico (ridotto volume ippocampale), in alterazioni della concentrazione di alcuni neurotrasmettitori (in particolare della serotonina) e in un pattern cromosomico soprannumerario. A questi si aggiunge un fattore di rischio comune a tutte le psicopatologie presenti in soggetti con SD: il ritardo mentale. Quest'ultimo condiziona l'interazione del soggetto con l'ambiente
22
rendendola un'esperienza difficile e ricca di frustrazioni. L'esplorazione cognitiva dell'ambiente inizia ad essere difficoltosa già nei primi anni di vita durante i quali l'ambiente risulta di difficile comprensione e poco prevedibile nei suoi sviluppi. Ne deriva una costruzione disarmonica del proprio Io e una scarsa fiducia nei propri mezzi cognitivi. Ne possono conseguire una chiusura relazionale e un isolamento dagli altri che possono esitare in un disturbo depressivo. Dall'altro lato la percezione di un ambiente ingannevole e imprevedibile può costituire il substrato per l'insorgenza di psicosi e tratti persecutori. Quanto detto pone le basi per una riflessione circa l'importanza, per questi soggetti, di un corretto intervento cognitivo che si possa rivelare efficace sia dal punto di vista psicopatologico che prestazionale.Nelle forme più gravi di ritardo mentale sembrano prevalere manifestazioni di tipo simil-autistico. Diversamente da quanto alcuni stereotipi possono far credere i soggetti con SD non sono sempre socievoli e con un temperamento affabile,spesso presentano tratti ossessivi, resistenza al cambiamento e isolamento sociale. La letteratura riporta un'incidenza di comorbidità tra SD e autismo compresa tra 1 e 7%. La predisposizione a sviluppare autismo sembra essere correlata con una maggiore incidenza di patologie cliniche di varia natura: patologie cardiache congenite, anomalie del tratto gastroenterico, patologie oftalmiche e apnee del sonno. La diagnosi di autismo in questi soggetti è resa difficile dal fatto che i segni con cui si manifesta sono in parte sovrapponibili a quelli presenti nel ritardo mentale: stereotipie, tratti ansiosi e ritiro sociale. Uno studio recente di Dressler et al. [78] eseguito su 24 pazienti ha dimostrato come il profilo adattivo di pazienti con SD affetti da autismo fosse più affine a quello di pazienti con SD senza psicopatologia associata piuttosto che a quello di soggetti autistici. Dal punto di vista neurologico nei soggetti con SD e autismo si osserva un aumento della sostanza bianca a livello cerebellare e un'ipoplasia del verme cerebellare e una riduzione nella concentrazione delle cellule di Purkinjie che sembra essere correlata con una scarsa efficienza dei circuiti cerebello-talamo-corticali.
23
2.6 Deterioramento Cognitivo
I soggetti affetti da SD sviluppano frequentemente un deterioramento cognitivo precoce, legato ad un pattern neuroistopatologico del tutto simile a quello che si ritrova in adulti affetti da malattia di Alzheimer (MA).
Le cause di questo dato clinico dimostrano un forte legame su base genetica tra le due patologie e rendono lecito parlare di demenza Alzheimer-like nel soggetto con SD.
Tra il corteo sintomatologico del deterioramento si ritrovano: riduzione delle funzioni cognitive, cambiamenti di personalità (nel 100% dei casi), perdita dell’autonomia e delle abilità della vita quotidiana. Questi sintomi sono di gran lunga i più frequenti, ma possono comparire anche crisi parossistiche (53%), deterioramento del cammino (73%), incontinenza sfinterica (40%) e riflessi patologici (67%) [79].
Studi recenti [80, 81] hanno suggerito che lo stadio pre-clinico della demenza Alzheimer-like in soggetti SD è caratterizzato da cambiamenti della personalità e comportamentali e compromissione delle funzioni esecutive, a differenza di quanto accade nella MA, in cui si riscontra in una fase precoce un declino della memoria episodica.
La demenza nei soggetti Down insorge circa 20-25 anni prima rispetto alla MA, ad un’età media di circa 50 anni [82] e da diversi studi se ne evince la altissima incidenza: il 50-70% dei soggetti Down sviluppa MA entro i 60-70 anni [83, 84]; la prevalenza della demenza è del 55% tra i 40 e i 52 anni, del 33% tra i 30 e i 39 anni e dello 0% tra 20 e 29 anni [85]. Questi dati coincidono con un più recente studio di Roizen del 2003 [24] che stima nella popolazione Down di più di 60 anni che il 75% dei soggetti sia affetto da MA. Tra i fattori che influenzano l’età di esordio della demenza in questi soggetti, quello più significativo sembra essere la presenza dell’allele epsilon4 dell’apolipoproteina E (ApoE), di cui è accettato l’effetto sulla suscettibilità per l’AD.
Come nella popolazione generale la diagnosi di demenza è una diagnosi di esclusione. Nei soggetti con SD questa diagnosi è particolarmente difficile per il fatto che l'esplorazione cognitiva del soggetto è spesso limitata a criteri osservazionali riferiti dai care-givers. La diagnosi di demenza entra in diagnosi differenziale con quella di depressione anche se spesso coesistono. Una
24
diagnosi di pseudodemenza può impedire il corretto trattamento di una depressione trattabile. Un'altra patologie con cui entra in diagnosi differenziale è l'ipotiroidismo, patologia particolarmente frequente in questi soggetti.Le cause della stretta associazione tra SD e MA sono attribuibili a geni soprannumerari, contenuti nel cromosoma 21, e implicati nel meccanismo neurodegenerativo:
APP: gene precursore della proteina dell’amiloide.
SOD1: gene della Cu/Zn superossido dismutasi.
DSCR1: Regione Critica della sindrome di Down 1.
Ets2: gene per il fattore di trascrizione omonimo.
Il gene APP sembra avere un ruolo critico per lo sviluppo di MA in SD, è situato nella regione cromosomica che deve essere trisomica per la piena espressione del fenotipo Down [86] ed è stato descritto il caso di un soggetto con trisomia 21 parziale, che mancava del terzo gene APP e che non mostrava né demenza né caratteristica neuropatia Alzheimer- like [87].
La APP (Amyloid precursor protein, Proteina Progenitrice dell'Amiloide) viene prodotta è degradata durante il processo di trasporto sulla superficie cellulare (processo di degradazione della APP) e vede coinvolti tre enzimi che operano tagli proteolitici: la α-secretasi e la β-secretasi in un primo momento e successivamente la γ-secretasi. Attraverso due tagli successivi operati prima dall'α-secretasi e poi dall'γ-secretasi, viene prodotto un peptide innocuo chiamato p3. La β-secretasi opera un taglio differente che, in seguito al successivo taglio da parte della γ-secretasi, porta alla produzione di due peptidi di 40 e 42 aminoacidi, chiamati beta-amiloide (Aβ 1-40 e Aβ 1-42): il secondo (Aβ 1-42) è considerato il più tossico a livello neuronale. Nei soggetti sani il processo di degradazione della APP sembra essere operato principalmente dalla α-secretasi. Per motivi non totalmente chiariti, nei soggetti malati l'enzima che interviene sull'APP non è l'α-secretasi ma la β-secretasi, con una larga produzione di proteina beta-amiloide.
25
Figura 8: La proteina precursore dell'amiloide (APP) è una proteina transmembrana scissa dagli enzimi secretasi. Nel che non porta alla formazione dell’amiloide, l’ APP viene scissa preferenzialmente da una α-secretasi. Nel percorso che porta alla formazione dell’amiloide, peptidi neurotossici Aβ aggregati, vengono rilasciati a seguito della scissione sequenziale di APP da β-secretasi, e si accumulano in aggregati oligomerici.Tale beta-amiloide non presenta le caratteristiche biologiche della forma naturale, ma tende a depositarsi in aggregati extracellulari sulla membrana dei neuroni. Tali placche neuronali innescano un processo infiammatorio che attiva una risposta immunitaria richiamando macrofagi e neutrofili, i quali produrranno citochine, interleuchine e TNF-alfa che danneggiano irreversibilmente i neuroni.
Figura 9: Rappresentazione delle placche amiloidee e dei grovigli neurofibrillari nella Malattia di Alzheimer.
26
Lo stress ossidativo, ha inoltre, un ruolo determinante nello sviluppo della MD nella SD. Il SOD1 è localizzato sul cromosoma 21 subito al di sopra della Regione Critica minima [86], è un enzima citosolico che catalizza la dismutazione di radicali superossido in ossigeno e perossido di idrogeno, il quale a sua volta viene rimosso da una catalasi o dalla glutatione perossidasi. Qualsiasi perturbazione dell’equilibrio tra il primo e il secondo passaggio può portare ad un eccesso di radicali liberi. Nel cervello di feti con SD è stato evidenziato un’eccesso di attività del SOD1 senza un incremento correlato dell’attività della glutatione perossidasi [89] e questa disfunzione potrebbe spiegare la precocità d’insorgenza della MA in SD. La sovraespressione di SOD1 è stata rilevata anche in neuroni in fase di degenerazione in soggetti affetti da MA [90].Il DSCR1 è un fattore stress inducibile, sovraespresso nella SD, coinvolto nella regolazione della trasmissione del segnale [91], e che a sua volta stimola la produzione di SOD1. L’attività della SOD1 è quindi coinvolta nell’effetto acuto protettivo e cronico dannoso di DSCR1 [92]. Quindi nella SD la alterata espressione di DSCR1 determina un aumento dello stress ossidativo. Lo stesso danno ossidativo può essere spiegato anche dai numerosi deficit mitocondriali associati alla SD ed evidenziati da diversi studi sia nell’uomo che nel modello animale [93, 94, 95]. Busciglio e collaboratori [96] inibendo la fosforilazione ossidativa mitocondriale in astrociti e colture neuronali di feti non Down hanno ottenuto lo stesso danno individuato su astrociti in coltura derivati da feti Down, dimostrando così che un ridotto funzionamento mitocondriale prenatale aumenta il rischio di insorgenza di lesioni degenerative a carico del SNC simili a quelle della MA.
Un altro fattore rilevante per la patogenesi di MA in SD è l’Ets2, fattore di trascrizione coinvolto nella differenziazione e maturazione cellulare [97], sovraespresso in caso di stress ossidativo. Ets2 risulta, quindi, essere aumentato nelle cellule cerebrali corticali di soggetti con SD e MA e questo promuove l’apoptosi neuronale mediata dai mitocondri per aumento di alcune sostanze pro-apoptiche [116].
Altri meccanismi ipotizzati essere alla base dell’apoptosi neuronale nella SD sono: aumento di espressione della proteina S100beta (aumentata anche nella MA); alterata produzione della proteina Bcl-2, soprattutto nei lobi temporali, dove si registra la maggior quota di perdita neuronale [117]; disregolazione del fattore
27
di trascrizione REST [118]; aumento di proteine correlate all’apoptosi quali Bim/BOD e p21.Diversi studi di neuroimaging suggeriscono che, così come avviene nella MA, anche nella SD il processo di invecchiamento si accompagna ad atrofia cerebrale regionale, comprendente ippocampo, paraippocampo e aree associative corticali, e tale atrofia si evidenzia prima dell’insorgenza della demenza clinica. La neuropatologia Alzheimer-like, che determina la perdita di neuroni e sinapsi è alla base di questi cambiamenti macroscopici. [119].
2.7 Aspetti neuro-anatomici
A partire dalla fine degli anni ’80, soprattutto in seguito all’avvento della Risonanza Magnetica, sono state condotte molte ricerche finalizzate allo studio delle correlazioni neurobiologiche sottese al peculiare pattern neuropsicologico presentato dagli individui con SD. Molte sono le alterazioni della morfologia cerebrale riscontrabile nella SD. Tra queste quella più frequentemente messa in rilievo nei diversi studi sono relative ad una riduzione volumetrica che può interessare tutto il volume cerebrale o essere limitato ad alcune strutture
2.7.1 Anatomia Macroscopica
Il profilo cognitivo descritto nelle persone con SD presumibilmente deriva da anomalie specifiche riscontrabili nello sviluppo cerebrale. Tuttavia ogni tentativo di identificare quali strutture neuroanatomiche siano specificatamente coinvolte nel profilo tipico di tale sindrome è basato necessariamente su confronti qualitativi con le diverse tipologie di deficit presentate da pazienti a seguito di lesioni cerebrali acquisite.
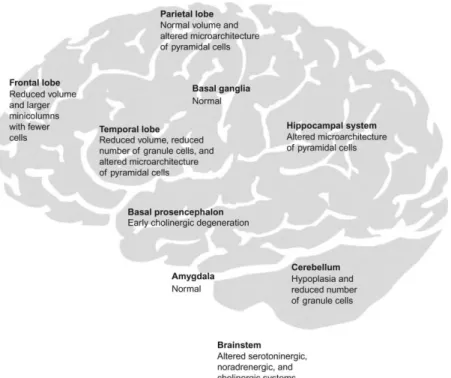
L’alterazione morfologica più frequentemente evidenziata, sia al riscontro autoptico che durante studi volumetrici di risonanza magnetica, è che i soggetti affetti da SD presentano un ridotto volume cerebrale, con una diminuzione sproporzionata a livello del cervelletto e dei lobi frontali e temporali (con inclusione dell’uncus, dell’amigdala e dei giri ippocampale e paraippocampale). Per contro il cervello dei soggetti con SD mostra un volume relativamente preservato delle aree sottocorticali, quali i nuclei lenticolari, così come dei lobi
28
parietali ed occipitale [120, 121]. L’atrofia del corpo calloso è stata riscontrata in adolescenti con SD [122] e negli adulti può essere associata anche ad atrofia della commissura anteriore [124] (Figura 10).Figura 10: Strutture coinvolte dalle alterazioni morfologiche e microscopiche nella sindrome di Down. Tratto da Lott et al. [123].
La riduzione del volume cerebrale può essere un segno molto precoce nello sviluppo cerebrale della SD; essa compare già a 4-5 mesi dell’età gestazionale e progredisce durante gli ultimi 3 mesi [125]. Studi di risonanza magnetica mostrano una riduzione di circa il 17% del volume cerebrale in soggetti con SD tra 10 e 20 anni [120] confermata nei soggetti adulti con SD in assenza di segni di demenza [126].
Difetti nella composizione biochimica della mielina ed un ritardo nella formazione della mielina sono inoltre molto diffusi nella SD [121, 127].
Se prima della nascita i livelli di mielinizzazione sono normali nel 100% dei casi e tali rimangono nel 75% dei casi per tutta l’infanzia, intorno ai 6 mesi dalla nascita nel 25% dei bambini con SD si manifesta un ritardo nella mielinizzazione che interessa dapprima tutto il cervello e in un secondo momento prevalentemente gli assoni che collegano i lobi frontali con quelli temporali [127, 128].
29
Tuttavia, alcuni autori hanno effettuato una correzione per il volume generale, evidenziando come alcune strutture cerebrali siano di dimensioni simili a quelle dei controlli ed, in alcuni casi, addirittura maggiori. Ad esempio, i lobi frontali dei soggetti con SD, che prima della correzione per il volume totale del cervello apparivano più piccoli rispetto a quelli dei controlli, dopo la correzione, avevano un volume paragonabile a quello dei controlli. Resta comunque la possibilità che un loro incompleto sviluppo possa spiegare molte delle carenze cognitive riportate in letteratura (memoria di lavoro fonologica e/o funzioni esecutive deficitarie). Inoltre, dopo la correzione per il volume totale della sostanza grigia del loro cervello, il volume della sostanza grigia delle strutture sottocorticali dei soggetti con SD appariva superiore a quello dei soggetti di controllo [128, 129]. L’anomalo volume delle strutture sottocorticali potrebbe riflettere i tempi diversi della maturazione di strutture corticali e sottocorticali nel cervello. I cervelli di individui con SD non presentano differenze volumetriche né anomalie neuropatologiche fino al sesto mese, periodo in cui i gangli della base hanno terminato la loro maturazione mentre la corteccia cerebrale invece è ancora in piena maturazione con i processi di arborizzazione dendritica, sinaptogenesi e mielinizzazione. Una spiegazione alternativa dell’anomala grandezza dei gangli della base nei pazienti con SD potrebbe essere dovuta a una insufficiente capacità di eliminare i neuroni e le sinapsi in eccesso. La mancata eliminazione delle cellule in eccesso potrebbe condurre allo sviluppo di gangli della base grandi ma non funzionali. Dopo la correzione per il volume totale, anche il volume dei lobi parietali e temporali è risultato essere maggiore nei pazienti con SD che nei controlli. In particolare, nei lobi temporali dei pazienti con SD il volume della sostanza bianca era maggiore che nei controlli con l’unica significativa eccezione del Giro Temporale Superiore (GTS) in cui il volume della sostanza bianca era inferiore rispetto a quella presente nei controlli [131].Correlati neuropsicologici
Seppure non si possano estrapolare relazioni di tipo causa- effetto tra anomalia morfologica cerebrale e deficit funzionale, le frequenti alterazioni cerebrali riscontrate negli individui con SD fanno comunque ipotizzare che esista una correlazione tra pattern neuropsicologico e linguistico e alterazioni neuro anatomiche.
30
Da un punto di vista clinico l’ipoplasia cerebellare sembra essere la causa di alcuni dei segni clinici costantemente presenti in questa sindrome, quali l’ipotonia muscolare, le difficoltà di coordinazione motoria e le difficoltà fonoarticolatorie. È stato ipotizzato inoltre che il volume considerevolmente ridotto del cervelletto associato alla riduzione di altre strutture coinvolte nel processamento di informazioni verbali, quali il giro superiore della corteccia frontale, le strutture limbiche, potrebbero essere i correlati neurobiologici del disturbo a carico degli aspetti morfosintattici e mnesici. [98]. Pazienti con lesioni cerebellari possono presentare deficit non solo di natura motoria ma anche nelle funzioni esecutive, nella fluenza verbale e in alcune specifiche elaborazioni linguistiche (es., elaborazione di materiale morfosintattico) [99, 99, 100, 102].Il profilo neuropsicologico dei pazienti con SD in cui sono evidenti deficit prevalentemente nel dominio linguistico (inclusa la memoria a breve termine verbale) con relativo risparmio delle competenze visuo-spaziali, inclusa la memoria a breve termine visuo-spaziale [103, 104, 105, 106] è compatibile con un aspetto invariato dei lobi parietali, importanti per l’elaborazione visuo-spaziale [107].
L’atrofia dei lobi temporali interessa in particolar modo l’ippocampo, dando quindi una compromissione delle capacità di apprendimento e memoria, e il Giro Temporale Superiore (GTS) in cui il volume della sostanza bianca era inferiore rispetto a quella presente nei controlli. Visto l’importantissimo ruolo svolto dal GTS bilaterale nella percezione acustica e dal GTS dell’emisfero sinistro, in particolare, per la codifica acustica e fonologica delle parole [108], è plausibile ipotizzare che un anomalo sviluppo di queste strutture del lobo temporale possa essere collegato alle difficoltà nell’acquisizione linguistica riscontrate nei pazienti con SD.
L’ipotrofia dei lobi frontali è stata frequentemente associata nella SD ad un deficit delle funzioni esecutive, difficoltà di attenzione e tendenza alla perseverazione. [109].
Occorre tuttavia cautela nel correlare ridotti volumi cerebrali con specifici deficit cognitivi; il volume dei lobi frontali nella SD non è sempre ridotto rispetto ai controlli [110, 111, 112]. Inoltre, l’attività metabolica nei lobi frontali non è necessariamente diversa rispetto a quella presente nei lobi frontali in bambini con normale sviluppo nell’esecuzione dei medesimi compiti [113]. Ad esempio, in
31
uno studio PET Schapiro e colleghi [113] misurano l’attività metabolica dei lobi frontali in un gruppo di 10 soggetti con SD e 20 soggetti di controllo con 28 anni di età mentale durante l’esecuzione del Wisconsin Card Sorting Test (WCST). Nei soggetti normali durante l’esecuzione del WSCT si registra un aumento dell’attività metabolica nella corteccia prefrontale [114].Nel complesso, nonostante il fatto che la performance comportamentale dei soggetti con SD fosse significativamente inferiore a quella dei controlli, l’aumento di attivazione della corteccia prefrontale durante l’esecuzione del WCST era simile in entrambi i gruppi. Naturalmente, è possibile ipotizzare che l’attivazione prefrontale nei soggetti con SD non sia collegata alla performance al WCST ma potrebbe semplicemente riflettere la messa in atto di diverse strategie cognitive impiegate durante il compito oppure una caratteristica strutturale del compito come lo sforzo cognitivo da esso richiesto (es., il ricorso alla memoria di lavoro, 115).
2.7.2 Anatomia Microscopica
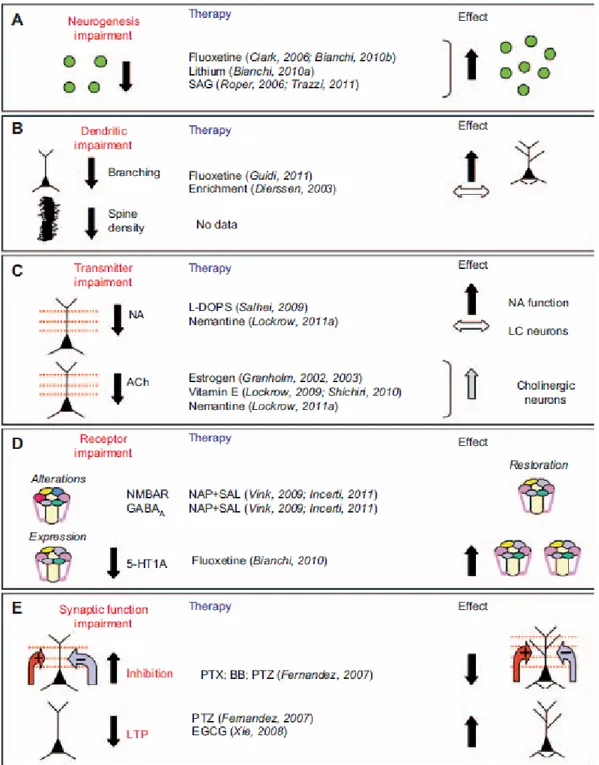
Nella descrizione degli aspetti microscopici ci soffermeremo sull’analisi delle cause di Ipotrofia cerebrale e su altri aspetti microscopici della SD come la funzione sinaptica, l’alterazione dei neurotrasmettitori e del sistema recettoriale, la riduzione di espressione dei fattori trofici.
Cause dell’Ipotrofia cerebrale
Abbiamo visto come uno degli aspetti predominanti della anatomia cerebrale nella SD sia l’ipotrofia cerebrale. Le cause dell’ipotrofia cerebrale nella SD possono essere :
1. Disordine della Neurogenesi 2. Aumento dell’Apoptosi cellulare 3. Neurodegenerazione
4. Ipotrofia dendritica
1. Disordine della neurogenesi. Poche informazioni sono disponibili sulla neurogenesi nel periodo fetale, data la difficoltà ad avere delle informazioni in
32
questo periodo. Recenti studi hanno dimostrato che nel feto con SD la proliferazione cellulare è severamente ridotta a livello del giro dentato, della matrice germinale del corno inferiore del ventricolo laterale, della zona germinale dell’ippocampo [132; 133] e dello strato granulare esterno del cervelletto [134].Figura 11: Rappresentazione della riduzione della Neurogenesi nella Sindrome di Down (DS) rispetto alla popolazione normale (NB, normal brain). Tratto da Bartesaghi R et al. [135].
2.
Aumento dell’apoptosi cellulare. L’ apoptosi cellulare è un fenomeno fisiologico che si verifica durante la neurogenesi, che consente di regolare il numero di finale di neuroni. L’apoptosi e/o neurodegenerazione in fasi più avanzate della vita porta ad una riduzione patologica del numero dei neuroni. La coltura di neuroni del feto con SD mostra un incremento dell'incidenza di apoptosi [136, 137, 138, 139]. Dagli studi autoptici dell’encefalo di soggetti con SD si evidenziano mutazioni del gene bcl-2 che determinano una ridotta attività della proteina antiapoptoica a livello del lobo temporale [140] e dell’ippocampo [141] e alterazioni del gene p55 e CD95 che determinano una aumentata attività della proteina proapoptoica [142, 143]. L’attivazione della caspasi-3 rappresenta un passo irreversibile nel percorso di morte cellulare [144] ed è stata osservata nella regione ippocampale dei feti [133] e nel cervello di adulti [145] con SD. D'altra parte, una espressione normale della caspasi-3 è presente nel cervelletto e nella corteccia dei feti con SD [134, 146], suggerendo che l’apoptosi non è un fenomeno diffuso nel cervello dei soggetti con SD in particolare nel periodo fetale.3. Neurodegenerazione Gli Adulti con SD hanno una predisposizione genetica a sviluppare una neuropatologia Alzheimer-like (AD) e un declino nelle funzioni adattive e sociali.
33
La presenza dei segni distintivi dell’AD, le placche amiloidi diffuse e i grovigli neurofibrillari, si riscontra a circa 40 anni [147] e portano alla degenerazione neuronale in particolare dei sistemi colinergico e noradrenergico [147].Il coinvolgimento del sistema colinergico può portare ad un danno dei neuroni colinergici dei basal forebrain che innervano l’ippocampo e la corteccia, essendo così responsabili per la disfunzione della memoria esplicita e dell’attenzione osservate all'inizio della demenza DS [149, 150]. Il coinvolgimento del sistema noradrenergico può essere responsabile della perdita di neuroni a livello del Locus coeruleus che è un evento precoce in AD e DS [151]. L’alterato metabolismo dei radicali liberi e l’alterata funzione mitocondriale possono essere collegati alla degenerazione neuronale [136, 137] e possono essere associati sia al ritardo mentale sia alla patologia AD.
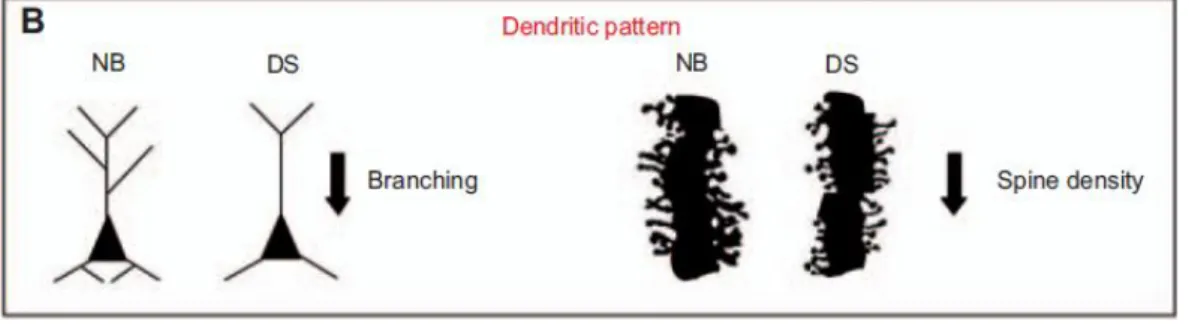
4. Ipotrofia dendritica Diversi studi hanno evidenziato delle alterazioni specifiche della arborizzazione dendritica nella SD.
Figura 12: Rappresentazione della atrofia dendritica e della riduzione della densità delle spine dendritiche nella Sindrome di Down (DS) rispetto alla popolazione normale (NB, normal brain). Tratto da Bartesaghi R et al. [135].
I neuroni della corteccia visiva del feto [152] e della corteccia prefrontale dei bambini fino a 2,5 mesi [153] non mostrano alterazioni dendritiche. Queste, però, cominciano a comparire nei bambini con SD a partire da 3-4 mesi di età in particolare a livello dei neuroni della corteccia motoria [154], della corteccia visiva [152,. 155] e della corteccia parietale [156]. L’ipotrofia dendritica continua in età adulta, con una riduzione della ramificazione dendritica [157]. Questa evidenza mostra che, a differenza dello sviluppo normale, dove la maturazione dendritica avviene nella prima infanzia, nel cervello dei soggetti con SD l’albero dendritico
34
comincia ad essere atrofico nella prima infanzia. Responsabili della alterazione dendritica possono essere varie proteine che formano il citoscheletro dei neuroni o il reticolo endoplasmatico, che sono down-regolate nel cervello dei soggetti con SD [135].Alla riduzione dendritica si associa una riduzione del numero e una alterata morfogenesi delle spine dendritiche che compare nei neonati e nei bambini con SD, mentre è assente nella corteccia visiva del feto con la SD [152]. In particolare, una riduzione della densità delle spine dendritiche si osserva a livello dei neuroni piramidali dell’ippocampo e della corteccia cingolata [157, 158, 159]. Un ulteriore riduzione della densità delle spine si verifica in pazienti con SD associata ad AD rispetto ai controlli di pari età senza AD [157, 160]. La morfologia delle spine dendritiche nella SD è molto alterata: le spine sono piccole, hanno steli corti e si mescolano con spine insolitamente lunghe a partire dalla prima infanzia [161, 162].
Alterazione delle funzioni sinaptiche.
Non ci sono numerosi dati sulla densità sinaptica e sulla relativa prevalenza delle sinapsi eccitatorie o inibitorie, tuttavia è stato dimostrato una riduzione del livello degli amminoacidi eccitatori che può suggerire una riduzione delle sinapsi eccitatorie [163].
Figura 13: Rappresentazione dell’incremento dell’attività inibitoria nella Sindrome di Down (DS) rispetto alla popolazione normale (NB, normal brain). Tratto da Bartesaghi R et al. [135].
35
Alterazioni dei neurotrasmettitori e del sistema recettoriale
Nel cervello dei soggetti con SD vi è una alterazione diffusa dei diversi sistemi di trasmissione e ridotta espressione e/o ridotta funzionalità dei recettori eccitatori e inibitori. Il risultato di questi difetti possono essere: alterazioni della neurogenesi, difettosa maturazione dei neuroni, deficit di trasmissione sinaptica e difficoltà comportamentali.
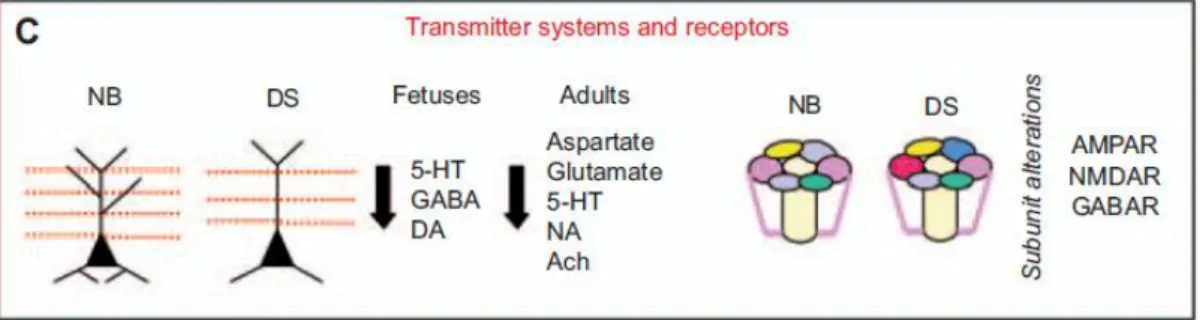
Figura 14: Rappresentazione delle alterazioni dei neurotrasmettitori e del sistema recettoriale nella Sindrome di Down (DS) rispetto alla popolazione normale (NB, normal brain). Tratto da Bartesaghi R et al. [135].
Nella corteccia frontale del feto con SD è stata riscontrata una riduzione dei livelli di serotonina (5-HT), del GABA e della dopamina (DA) , rispetto al feto normale e livelli simili di glutammato, aspartato e noradrenalina (NA) [164].
In considerazione del ruolo chiave della 5-HT nella neurogenesi, nella differenziazione neuronale, nello sviluppo dendritico, nella mielinizzazione degli assoni e nella sinaptogenesi [165], i suoi livelli ridotti nel cervello fetale con SD possono essere alla base delle numerose alterazioni di sviluppo.
Anche il GABA ha un ruolo importante nel cervello in via di sviluppo in quanto agisce come un fattore epigenetico che controlla la proliferazione cellulare, la migrazione dei neuroblasti e la maturazione dendritica [166]. I ridotti livelli di GABA nel feto con SD sono contribuiscono, quindi, all’alterazione dello sviluppo del cervello.
Nel cervello degli adulti con SD, aspartato, glutammato, livelli di NA e 5-HT e l’attività della colina acetiltransferasi (ChAT) sono ridotti rispetto ai controlli [163, 167]. I ridotti livelli di glutammato (il neurotrasmettitore eccitatorio più abbondante nel cervello) suggeriscono una compromissione della neurotrasmissione eccitatoria. I ridotti livelli di NA, 5-HT e ChAT nel cervello dell’ adulto con SD