POLITECNICO DI MILANO
Scuola di Ingegneria Industriale e dell’Informazione
Dipartimento di Chimica, Materiali ed Ingegneria Chimica “Giulio Natta”
Corso di Laurea Magistrale in Ingegneria Chimica
Chemo-Enzymatic Synthesis of
Enantiopure Aliphatic Amino Acids
Relatore: Prof. Davide Tessaro
Tesi di Laurea Magistrale di:
Nicolò Rossi
Matricola: 863867
A.A 2017-2018
ABSTRACT
The synthesis of aliphatic amino acids with high optical purity is of major importance in synthetic chemistry, since such compounds find use in a variety of tailor-mad oligopeptides with biological or pharmacological activity. Biocatalysis, being based on the exploitation of enzymes with intrinsical high selectivity, may represent in this view a preferential technology. In this thesis work, we carry out an enzyme-mediated selective hydrolysis of amino acid thioesters based on a thienylic core in presence of an organic base (trioctylamine), which promotes their racemization. In such conditions, a dynamic kinetic resolution takes place, and we were able to synthesize a series of variously substituted thienylglycines presenting high enantiomeric excess in high yield. The successive reduction of the thienylic core promoted by Ni Raney in presence of hydrazine permits to obtain the corresponding long-chain alkylic amino acid with retention of enantiomeric excess. In conclusion, we were able to devise and put in practice a new synthetic strategy for the obtainment of long-chain alpha amino acids, based on the consecutive action of a biocatalyst and an inorganic catalyst which furnishes products in high yield and excellent enantiomeric excess.
ABSTRACT IN LINGUA ITALIANA
La sintesi di ammonoacidi alifatici ad elevata purezza ottica presenta una grossa importanza, in quanto tali composti trovano uso, oltre che per se stessi, nella sintesi di oligopeptidi a struttura nota aventi diverse applicazioni. La biocatalisi, fondata sull'uso di enzimi ad alta selettività, può rapprentare una via preferenziale in quest'ottica. In questo lavoro di tesi si sfrutta un'idrolisi enzimatica selettiva di tioesteri di amminoacidi basati su di un nucleo tienilico in presenza di una base organica che ne favorisce la racemizzazione, in modo da essere in condizioni di risoluzione cinetica dinamica. Tramite tale metodo, una serie di tienilglicine variamente sostituite è stata ottenuta con elevato eccesso enantiomerico ed alta conversione. Una successiva riduzione del nucleo tienilico mediata di Ni Raney ha permesso di ottenere i corrispondenti amminoacidi alchilichi con ritenzione della purezza ottica. In conclusione, è stata progettata e realizzata una nuova via di sintesi per amminoacidi a lunga catena basata sull'azione successiva di un catalizzatore biologico e di un catalizzatore inorganico.
SUMMARY
ESTRATTO IN LINGUA ITALIANA _________________________________________ 10
1.1 Amino Acids ___________________________________________________________ 13
1.2 Synthesis of α-Amino Acids ______________________________________________ 15
1.2.1 Strecker Synthesis __________________________________________________ 16
1.2.2 Amination of α-Halogen Acids ________________________________________ 17
1.2.3 Amination via Molecular Rearrangement _______________________________ 19
1.3 Synthesis of Entiomerically Pure Amino Acids _______________________________ 21
1.4 Asymmetric Synthesis ___________________________________________________ 24
1.4.1 Non-enzymatic Catalytic Asymmetric Synthesis _________________________ 25
1.4.2 Enzymatic Asymmetric Synthesis _____________________________________ 26
1.5 Kinetic Resolution ______________________________________________________ 27
1.5 Deracemization Process _________________________________________________ 28
1.5.1 Dynamic Kinetic Resolution __________________________________________ 29
1.5.2 Stereoinversion of Crixivan __________________________________________ 30
1.6 Fermentation __________________________________________________________ 31
1.7 Other Resolution Methods ________________________________________________ 33
1.7.1 Crystallization Procedures ___________________________________________ 33
1.7.2 Diastereoisomeric Salts ______________________________________________ 35
1.8 Paper Chromatographic Approaches _______________________________________ 37
2.1 Introduction ___________________________________________________________ 38
2.2 Dynamic Enzymatic Resolution of Thioesters ________________________________ 38
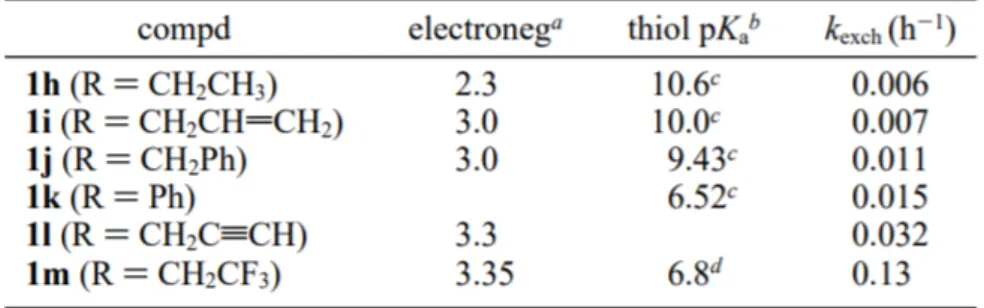
2.2.1 Kinetic α-Proton Acidity of Thioesters _________________________________ 39
2.2.2 Thioester as Substrates for Hydrolytic Enzyme __________________________ 42
2.2.3 Demonstration of Dynamic Enzymatic Resolution of Thioesters ____________ 43
2.2.4 Conclusion ________________________________________________________ 44
2.3 Previous work _________________________________________________________ 44
2.3.1 DKR of α-aryl Amino Acid Thioesters _________________________________ 47
2.3.1 DKR of aryl and aliphatic Amino Acid Thioesters ________________________ 48
3.1 introduction ___________________________________________________________ 50
3.2 Preparation of Substrates ________________________________________________ 51
3.2.1 Production of D,L-2-ThienylGlicyne ___________________________________ 52
3.2.3 Production of D,L NBoc Thinyl-Glycine thioester ________________________ 54
3.3 Dynamic Kinetic Resolution of D,L NBoc Thienyl-Glycine-Thioester Mediated by Solution of Subtilisin Carlsberg ______________________________________________ 55
3.4 Deprotection of Enantiomerically Pure Amino Acids __________________________ 57
3.5 Production of aliphatic amino acids by reduction with RaNi ____________________ 58
3.6 Conclusion & Prospects _________________________________________________ 59
4.1 Procedures for the production of enantiopure nor-Leucine _____________________ 60
4.1.1 Production of D,L-2-Thienylglycine ____________________________________ 60
4.1.2 Production of D,L-NBoc-2-Thienylglycine ______________________________ 62
4.1.3 Production of D,L-NBoc-2-Thienylglycine Thioester ______________________ 63
4.1.4 Enzymatic hydrolysis of D,L-NBoc-2-Thienylglycine Thioester _____________ 64
4.1.5 Deprotection of enantiopure NBoc-2-Thienylglycine ______________________ 65
4.1.6 Reduction of enantiopure 2-Thienylglycine______________________________ 65
4.2 Procedures for the production of Isoleucina/Alloleucina _______________________ 66
4.2.1 Production of D,L-3-Thienylglycine ____________________________________ 66
4.2.2 Production of D,L-NBoc-3-Thienylglycine ______________________________ 68
4.2.3 Production of D,L-NBoc-3-Thienylglycine Thioester ______________________ 69
4.2.4 Enzymatic hydrolysis of D,L-NBoc-3-Thienylglycine Thioester _____________ 70
4.2.5 Deprotection of enantiopure NBoc-3-Thienylglycine ______________________ 71
4.2.6 Reduction of enantiopure 3-Thienylglycine______________________________ 71
4.3 Procedures for the production of Heptanoic Amino Acid _______________________ 73
4.3.1 Production of D,L-Me-2-Thienylglycine ________________________________ 73
4.3.2 Production of D,L-NBoc-Me-2-Thienylglycine ___________________________ 75
4.3.3 Production of D,L-NBoc-Me-2-Thienylglycine Thioester __________________ 76
4.3.4 Enzymatic hydrolysis of D,L-NBoc-2-Thienylglycine Thioester _____________ 77
4.3.5 Deprotection of enantiopure NBoc-Me-2-Thienylglycine __________________ 78
4.3.6 Reduction of enantiopure Me-2-Thienylglycine __________________________ 78
4.4 Procedures for the production of Octanoic Amino Acid ________________________ 79
4.4.1 Production of D,L-Et-2-Thienylglycine _________________________________ 79
4.4.2 Production of D,L-NBoc-Et-2-Thienylglycine ____________________________ 82
4.4.3 Production of D,L-NBoc-Et-2-Thienylglycine Thioester ___________________ 83
3.4.4 Enzymatic hydrolysis of D,L-NBoc-Et-2-Thienylglycine Thioester __________ 84
4.4.5 Deprotection of enantiopure NBoc-Et-2-Thienylglycine ___________________ 84
4.5 Eluents Used for TLC ___________________________________________________ 86
4.6 Preparation OPATBC ___________________________________________________ 86
4.7 HPLC Analysis of the Biotrasformation Product _____________________________ 87
4.8 HPLC Analysis of the Deprotection ________________________________________ 91
4.9 HPLC Analysis of the Reduction __________________________________________ 98
4.10 MS Spectra __________________________________________________________ 102
INDEX OF FIGURES
Figura 1 - Substrati utilizzati _________________________________________________ 12 Figura 2 - Sintesi norLecina a partire da NBoc-2-ThGlySEt ________________________ 12 Figure 3 - Amino Acids Classification __________________________________________ 13 Figure 4 - Heminhendral Crystals _____________________________________________ 34 Figure 5 - Phase Diagrams __________________________________________________ 36 Figure 6 - Time course for α-proton exchange for thioesters of α-phenylpropionic acid as monitored by 1H NMR. Ht = proton signal intensity at each time point. H0 = initial α-proton signal intensity. ______________________________________________________ 41 Figure 7 - Proton Exchange Rate of Thioester of α-Aryl-Amino Acids _________________ 45 Figure 8 - Proton Exchange Rates of Thioesters of non-α-aryl-substituted-amino amino acids ________________________________________________________________________ 47 Figure 9 - CLEA ___________________________________________________________ 49 Figure 10 - Selected Substrates _______________________________________________ 52
INDEX OF TABLES
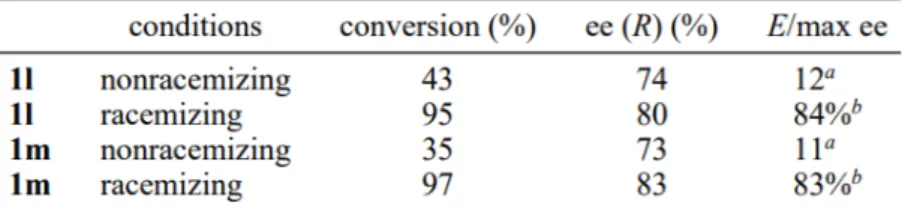
Table 1 - α-Proton Exchange Rate of Different Thioesters (Drueckhammer) ____________ 40 Table 2- Rates of R-Proton Exchange for R-Phenylpropionate Thioesters (1, X = Ph) ____ 41 Table 3 - Rates of Enzymatic Hydrolysis of Ethyl Butyrate and Ethyl Thiobutyrate
(µmol/(min*g of enzyme))____________________________________________________ 43 Table 4 - Resolution of 1l,m with Subtilisin Carlsberg under Nonracemizing and Racemizing Conditions ________________________________________________________________ 43 Table 5 summarized the dynamic kinetic resolution of compounds 1, 4-8 _______________ 48 Table 6 - Results of DKR with CLEA ___________________________________________ 50 Table 7 - Results of DKR of ThienylGlycine Thioesters _____________________________ 56 Table 8 - Measured Optical Power of ThienylGlycines _____________________________ 56
INDEX OF SCHEMES
Scheme 1 - Strecker Synthesis ________________________________________________ 16 Scheme 2 - Modified Strecker Syntheses ________________________________________ 17 Scheme 3 - Cahours synthesis, X=Cl or Br ______________________________________ 17 Scheme 4 - Hell-Volhard-Zelinsky _____________________________________________ 18 Scheme 5 - Gabriel Synthesis _________________________________________________ 18 Scheme 6 - Curtius and Sieber Synthesis ________________________________________ 19 Scheme 7 - Darapsky Synthesis _______________________________________________ 20 Scheme 8 - Huang, Lin and Li Synthesis ________________________________________ 21 Scheme 9 - Asymmetric Synthesis ______________________________________________ 22 Scheme 10 - Kinetic Resolution _______________________________________________ 23 Scheme 11 - Deracemization Processes _________________________________________ 23 Scheme 12 - Asymmetric Synthesis of L-DOPA ___________________________________ 26 Scheme 13 - Enzymatic Asymmetric Synthesis of L-t-Leucine ________________________ 27 Scheme 14 - Asymmetric Oxidation (b) _________________________________________ 27 Scheme 15 - Kinetic Hydrolysis (d) ____________________________________________ 28 Scheme 16 - Acid Hydrolysis _________________________________________________ 28 Scheme 17 - DKR by Hydantoinase/Racemase ___________________________________ 29 Scheme 18 - Stereoinversion of Hafner and Wellner ______________________________ 30 Scheme 19 - Stereoinversion of Crixivan ________________________________________ 31 Scheme 20 - Diastereoisomeric Salts ___________________________________________ 36 Scheme 21 - Resonance of Thioester ___________________________________________ 39 Scheme 22 - Racemization of Thioester _________________________________________ 39 Scheme 23 - General System Used to Measure α-Proton Exchange Rate (Drueckhammer) _ 40 Scheme 24 - Hydrolysis of Ethyl butyrate and Ethyl Thiobutyrate with various enzyme ____ 42 Scheme 25 - General System to Measure α-Proton Exchange rate by 1H NMR (Previous Work) ___________________________________________________________________ 45 Scheme 26 - System Used to Measure α-Proton Exchange Rate of Aryl and Aliphatic Amino Acid Thioesters ____________________________________________________________ 46 Scheme 27 - DKR of α-Aryl Amino Acid Thioesters ________________________________ 47 Scheme 28 - DKR of Aryl and Aliphatic Amino Acid Thioester with CLEA and DBU _____ 50 Scheme 29 - General Procedure to produce Aliphatic α-Amino Acids _________________ 51 Scheme 30 - Production of D,L-2-ThGly ________________________________________ 52 Scheme 31 - Production of Ethyl-Thienyl Aldehyde ________________________________ 53 Scheme 32 - Protection Reaction ______________________________________________ 54 Scheme 33 - Production of D,L-2-NBoc-ThGly Thioester ___________________________ 54 Scheme 34 - DKR of NBoc-2-ThGlySEt _________________________________________ 55 Scheme 35 - Racemization mechanism __________________________________________ 55 Scheme 36 - Deprotection Reaction ____________________________________________ 57 Scheme 37 - Reduction of Thiophen Ring ________________________________________ 58 Scheme 38 - Synthesis of D,L-2-ThGly (1) _______________________________________ 60 Scheme 39 - Synthesis of D,L-2-ThGly (2) _______________________________________ 61 Scheme 40 - Synthesis of D,L-2-ThGly (3) _______________________________________ 61 Scheme 41 - Synthesis of D,L-NBoc-2-ThGly _____________________________________ 62 Scheme 42 - Synthesi of D,L NBoc-2-ThGly _____________________________________ 63 Scheme 43 - Enzymatic Hydrolysis of D,L NBoc-2-ThGly Thioester ___________________ 64
Scheme 44 - Deprotection Reaction of NBoc-2-ThGly ______________________________ 65 Schema 45 - Reduction of 2-ThGly _____________________________________________ 65 Scheme 46 - Synthesis of D,L-3-ThGly (1) _______________________________________ 66 Scheme 47 - Synthesis of D,L-3-ThGly (2) _______________________________________ 67 Scheme 48 - Synthesis of D,L-3-ThGly (3) _______________________________________ 68 Scheme 49 - Synthesis of D,L-NBoc-3-ThGly _____________________________________ 68 Scheme 50 - Synthesi of D,L NBoc-3-ThGly _____________________________________ 69 Scheme 51 - Enzymatic Hydrolysis of D,L NBoc-3-ThGly Thioester ___________________ 70 Scheme 52 - Deprotection Reaction of NBoc-3-ThGly ______________________________ 71 Scheme 53 - Reduction of 3-ThGly _____________________________________________ 72 Scheme 54 - Synthesis of D,L-Me-2-ThGly (1) ____________________________________ 73 Scheme 55 - Synthesis of D,L-Me-2-ThGly (3) ____________________________________ 74 Scheme 56 - Synthesis of D,L-Me-2-ThGly (3) ____________________________________ 74 Scheme 57 - Synthesis of D,L-NBoc-Me-2-ThGly _________________________________ 75 Scheme 58 - Synthesi of D,L NBoc-Me-2-ThGly __________________________________ 76 Scheme 59 - Enzymatic Hydrolysis of D,L NBoc-Me-2-ThGly Thioester _______________ 77 Scheme 60 - Deprotection Reaction of NBoc-Me-2-ThGly __________________________ 78 Scheme 61 - Reduction of Me-ThGly ___________________________________________ 78 Scheme 62 - Synthesis of D,L Et-2-ThGly (1) _____________________________________ 79 Scheme 63 - Synthesis of D,L Et-2-ThGly (2) _____________________________________ 80 Scheme 64 - Synthesis of D,L Et-2-ThGly (3) _____________________________________ 81 Scheme 65 - Synthesis of D,L-NBoc-Et-2-ThGly __________________________________ 82 Scheme 66 - Synthesi of D,L NBoc-Et-2-ThGly ___________________________________ 83 Scheme 67 - Enzymatic Hydrolysis of D,L NBoc-Et-2-ThGly Thioester ________________ 84 Scheme 68 - Deprotection Reaction of NBoc-Et-2-ThGly ___________________________ 84 Scheme 69 - Reduction of Et-2-ThGly __________________________________________ 85
ESTRATTO IN LINGUA ITALIANA
Gli amminoacidi sono composti organici caratterizzati dalla presenza simultanea di un gruppo carbossilico (-COOH) e di un gruppo amminico (-NH2). Ne esistono diverse classi, ma la più importante è quella degli α-amminoacidi, i quali possiedono i due gruppi funzionali connessi allo stesso atomo di carbonio. La principale caratteristica che rende tali amminoacidi così interessanti è la presenza, in posizione α, di un centro chirale. Essi trovano apllicazione in diversi campi, ma vengono usati soprattutto come “building blocks” nella sintesi di svariati farmaci. Nei primi anni del XX secolo, visto il continuo aumento della richiesta di amminoacidi varie vie di sintesi furono sviluppate e migliorate. Si passò dall’estrazione alla sintesi chimica o ai primi processi fermentativi.
Negli anni cinquanta il caso del talidomide, un farmaco usato dalle donne in gravidanza come sedativo e anti-nausea, determinò un punto di svolta per la sintesi dei farmaci. Il talidomide veniva fino a quel momento venduto in forma racema, solo che ci si accorse, dopo la nascita di 10'000 bambini malformati, che effettivamente un enantiomero agiva da farmaco mentre l’altro incideva negativamente sulla crescita del feto. Quindi furono imposti maggiori controlli sui farmaci ed iniziò di conseguenza la ricerca a nuovi metodi per sintetizzare composti enantiomericamemte puri.
Tra le tante tecniche scoperte una è particolarmente interessante: la risoluzione cinetica dinamica. Essa si basa su un sistema composto da un substrato chirale capace di racemizzare spontaneamente nelle condizioni di reazione e da un enzima capace di convertire selettivamente uno solo dei due enantiomeri. In sostanza, da una miscela racema è possibile ottenere un solo enantiomero convertendo tutto il composto iniziale.
Questo lavoro di tesi si inserisce nell’ampliamento della risoluzione cinetica dinamica sviluppata dal gruppo di ricerca del prof. S. Servi. Tale tecnica è basata sull’uso di tioesteri di amminoacidi N-protetti, i quali hanno l’idrogeno in posizione α sufficientemente acido da permettere la racemizzazione tra i due enantiomeri del composto in presenza di una base. Inizialmente tale tecnica venne provata su tioesteri di α-amminoacidi aromatici con ottimi risultati: grande resa ed elevato eccesso enantiomerico. Essa fu condotta tramite l’utilizzo di un sistema bifasico (acqua/MTBE), di una base organica idrofobica (TOA, triottilammina) capace di estrarre il protone in posizione α e dell’Alcalasi© come enzima. Purtroppo, per quanto riguarda i tioesteri di amminoacidi alifatici, tale sistema non risulta funzionare. Infatti,
a causa della minore acidità dell’idrogeno in posizione α (l’anione corrispondente è meno delocalizzato), la TOA non si rivela abbastanza forte per estrarre il protone. Si decise allora di usare la DBU (1,5-diazabiciclo(5.4.0)undec-7-ene) la quale, essendo una base decisamente più forte, era in grado di promuovere la racemizzazione del composto, come venne dimostrato da dati NMR. Tale base, però, a contatto con l’acqua si protona per la maggior parte, rendendo cosi impossibile un’efficiente estrazione del protone dal composto e quindi la sua racemizzazione. Si decise allora di eliminare la maggior parte dell’acqua dal sistema di reazione passando ad un sistema di solvente monofasico (tert-butanolo) che potesse dissolvere allo stesso tempo il substrato, la base, l’acqua necessaria all’idrolisied il prodotto. A tali condizioni venne ritenuto più adatta una forma immobilizzata dello stesso enzima, detta CLEA© (Cross-Linked Enzyme Aggregates). La risoluzione cinetica dinamica di tioesteri di ammino acidi alifatici attraverso un sitema composto da DBU, CLEA e tert-butanolo come solvente, portò effettivamente alla sintesi dei corrispondenti L-amminoacidi N-protetti con alta resa ed elevato eccesso enantiomerico. La stessa tecnica è stata poi estesa alla sintesi delle corrispondenti ammidi enantiomericamente pure partendo sempre dai tioesteri racemi, impiegando diverse ammine come nucleofilo.
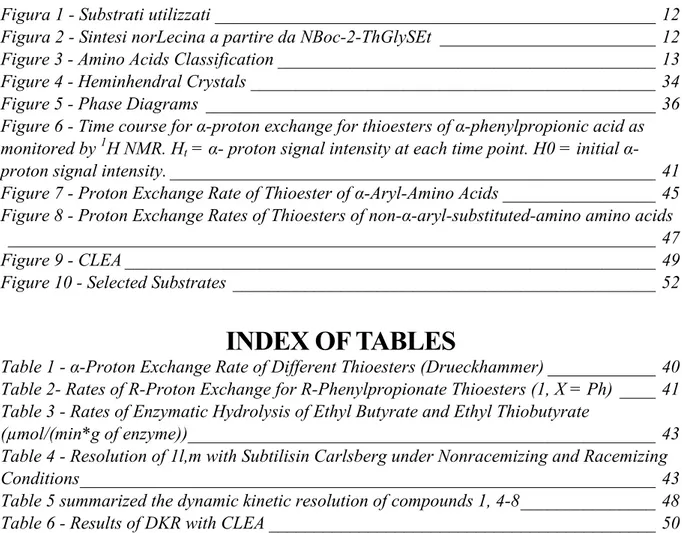
In questo lavoro di tesi è stata ideata e messa in pratica una via alternativa alla produzione di amminoacidi alifatici, sempre basata però sulla risoluzione cinetica dinamica di tioesteri. Nello studio precedente si è usato un approccio del tipo “reaction engineering” andando a modificare le condizioni di reazione per ottenere gli amminoacidi alifatici, mentre nel nostro caso è stato utilizzato un approccio di “substrate engineering”. L’idea è di “mascherare” la catena alifatica attraverso un anello tienilico, anche sostituito, il quale, essendo aromatico, possiede un’elevata acidità dell’idrogeno in posizione α cosi da permettere l’utilizzo del sistema bifasico; in un secondo momento l’anello tienilico viene ridotto e desolforato, trasformandosi nella corrispondente catena alchilica. Sono stati usati i seguenti substrati:
Figura 1 - Substrati utilizzati
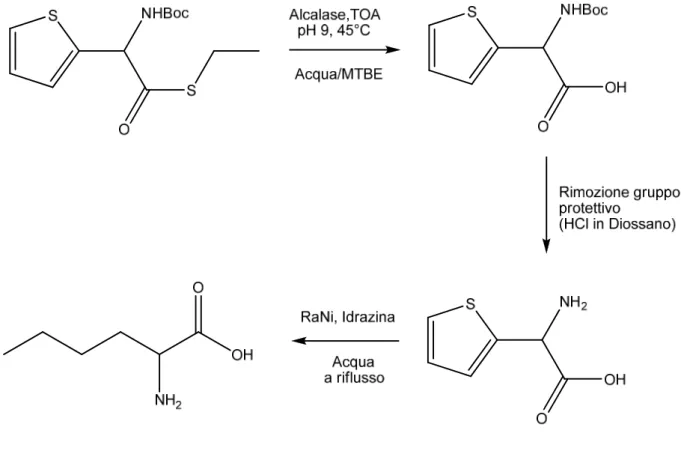
Quindi, la risoluzione cinetica dinamica è stata eseguita a pH 9 a 45°C in un sistema bifasico (acqua/MTBE) in presenza di TOA e Alcalasi. Dopo deprotezione dell’azoto, la riduzione dell’anello è stata condotta in acqua a riflusso, catalizzata da Nichel Raney e impiegando idrazina come fonte di idrogeno.
Figura 2 - Sintesi norLecina a partire da NBoc-2-ThGlySEt
La risoluzione cinetica dinamica così sviluppata ha ottimi risultati, resa superiore al 99% e eccesso enantiomerico maggiore del 97%. Anche la riduzione finale conduce a ottimi risultati, con la conservazione dell’eccesso enantiomerico e elevata resa.
1.Introduction
1.1 Amino Acids
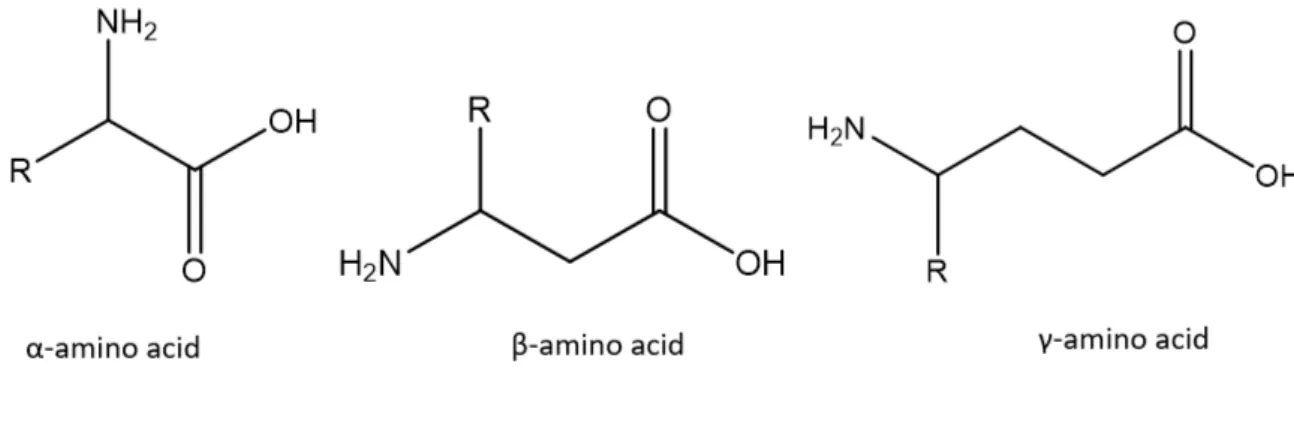
Amino acids are organic compounds characterized by the simultaneous presence of amino (-NH2) and carboxyl (-COOH) functional groups together with an organic side chain, specific to each amino acid. In Nature, these chemicals are the monomers of proteins, very complex molecules that play crucial functions in living organisms. They can be classified according to the core structural functional groups locations in alpha- (α-), beta- (β-), gamma- (γ-) amino acids.
Figure 3 - Amino Acids Classification
The α-amino acids, usually bearing four different substituents in position 2 (namely: the carboxy-group, the amino group, a hydrogen and the side chain), are intrinsically chiral, with the sole exception of glycine. It is notable that, even if there is the possibility of the existence of two enantiomers, the D-isomers are uncommon in living organisms. In fact, the α-amino acids include 22 proteinogenic amino acids, which are the building blocks of proteins and are all in the form. The proteogenic amino acids are: Alanine, Asparagine, Arginine, L-Aspartic acid, L-cysteine,L-Glutamine, L-Glutamic acid, Glycine , L-Proline , L-Serine , L-Tyrosine , L-Histidine ,L-Isoleucine ,L-Leucine ,L-Lysine ,L-Valine ,Methionine , L-Phenylalanine ,L-Threonine ,L-Tryptophan, L-Selenocysteine, L-Pyrrolysine (see Figure 1).
Twenty of the proteinogenic amino acids are encoded directly by triplet codons in the genetic code and are known as "standard" amino acids.
Amino acids were discovered in 19th century, but only in the 1935 it was proven that nine of them are “essential” to man because they can’t be synthesized by the human organism. This shows that amino acids are necessary first of all as nutrients for the production of proteins. Furthermore, many proteinogenic and non-proteinogenic amino acids have important biological functions. For example, glutamate (standard glutamic acid) and γ-amino-butyric acid ("GABA", non-standard γ-amino acid) are, respectively, the main excitatory and inhibitory neurotransmitters1. Nowadays about 500 amino acids are known.
Historically, amino acids are mainly used as additives to animal feed due the fact that such preparations have low levels or even lack some essential amino acids. For example, soybean is poor of methionine. In the same field they are also used to chelate metal cations in order to improve the absorption of minerals from supplements, which may be required to improve the health or production of these animals2.
This last technology is used also in fertilizers for agriculture to facilitate the delivery of minerals to plants in order to correct mineral deficiencies.
Amino acids find their use also in the food industry, not only to overcome the shortage of minerals, as previously described, but also employing some of them, in particular glutamic acid, aspartame (a dipeptide formed by L-Asp and L-Phe) and tryptophan, for particular applications. The first one is used as flavour enhancer and the second as artificial sweetener. The third, in association with histidine acts as an antioxidant to preserve milk powder. Another simple amino acid, cysteine, is used for the preservation of fruit juices3,4.
In recent years amino acids are used more and more in pharmaceutical industry due to their structural diversity and functional versatility. In particular, nonnatural amino acids find application in the synthesis and improvement of drugs or to favour drug-delivery. Many pharmaceuticals contain α-amino acids as critical components, such as L-Dopa (used for Parkinson disease) and Taxol (an anticancer drug). For example, Taxol is loaded into protein nanoparticles, appearing to be less toxic and more effective when used for metastatic breast cancer treatments5,6. Amino acids can be also used as chemical building blocks for chiral catalyst their stereogenic properties7.
The use of proteins and peptides as human therapeutics has recently expanded due to: discovery of novel peptides and proteins
a better understanding of the mechanism of action in vivo
improvements in expression or synthesis of proteins and peptides that closely resemble fully human proteins and peptides
improvements in formulation or molecule-altering technologies that have the ability to deliver polypeptides in vivo with improved pharmacokinetic and pharmacodynamic properties8.
Amino acids find application also in the field of biodegradable plastics as component of polymer. They can be part of the main chain or bonded as side chain and these alter the physical properties and reactivities of the polymers. Polyaspartate, for example, is a poly(amino acids) with free carboxylic group and when is produced higly linear is fully biodegradable. TPAs (thermal polyaspartate) are being targeted for three global markets: performance chemicals, diapers, and agriculture. In agriculture, PAs have been found to stimulate crop growth by enhancing root development9.
In conclusion, amino acids, in particular α-amino acids, are used for a variety of applications in industry. In order to correspond at the market demand were developed vary different methods were developed to efficiently produce theythem, from the chemical synthesis to the extraction from natural sources
1.2 Synthesis of α-Amino Acids
Although isolations from acidic, alkaline, or enzymic digest of proteins, or from other natural sources, have sufficed to supply a goodly portion of the amino acids available in the past, and are in fact still exploited for the large-scale procurement of arginine, asparagine, cystine, glutamic acid, histidine, hydroxyproline, proline, and tyrosine, such methods none the less do not currently serve as major source of supply for most of the protein-derived amino acids. However, a number of practicable chemical synthesis are presently known which permit ready access to alanine, aspartic acid, glycine, isoleucine, leucine, lysine, methionine, phenylalanine, serine, threonine, tryptophan, and valine, as well as to a host of other α-amino acids, on both an industrial and laboratory scale. The more important procedures are as follows:
Strecker Synthesis
Amination via Molecular Rearrangement
1.2.1 Strecker Synthesis
In the 1850, Strecker, when he tried to prepare lactic acid via the acid hydrolysis of the amino-nitrile intermediate CH3CH(NH2)CN, formed from the interaction of acetaldehyde first with ammonia, then with hydrocyanic acid, produced alanine instead. So Strecker accidentally reported the first route for α-amino acids synthesis that could be generally applied.
Scheme 1 - Strecker Synthesis
Already in 1900 many modifications of the Strecker procedure were introduced for the dual purpose of generally increasing the relatively poor yields secured by the original procedure and obviating the technical difficulties imposed by handling the higly toxic hydrogen cyanide. In fact, the main modifications were the use of alkaline cyanides and ammonium salts instead of hydrogen cyanide and ammonia.
An interesting modification was described in 1889 by Pinner and Spilker10, who obtained the 5-alkylhydantoin (VII) by digestion of the corresponding cyanohydrin (VI) with urea and treating the reaction mixture with hydrochloric acid. Alkaline hydrolysis of (VII) then yielded the desired amino acid (IV). This procedure was indeed ignored until 1934 when Bucherer and his collaborator11,12 demonstrated that 5-substituted hydantoins (VII) could be readily prepared in high yield upon heating an α-amino-nitrile (III) or a cyanohydrin (VI) with ammonium carbonate. 5-substituted hydantoins (VII) with an alkaline hydrolysis were converted into the corresponding amino acids.
During the 1940’s this procedure was extended to a wide variety of α-amino acids due to commercial aviability of a number of hitherto inaccessible precursor aldehydes. Since the Bucherer modification of the Strecker procedure has proved economical from the standpoints of time, labor, and material, it constitutes one of the principal synthetic means currently employed for large-scale production of α-amino acids13.
Scheme 2 - Modified Strecker Syntheses
The reaction mechanism of the Strecker synthesis start with the carbonyl oxygen of an aldehyde which is protonated, followed by a nucleophilic attack of ammonia to the carbonyl group. After subsequent proton exchange, water is cleaved from the iminium ion intermediate. A cyanide ion then attacks the iminium carbon yielding an aminonitrile. In the second step instead the nitrile nitrogen of the aminonitrile is protonated, and the nitrile carbon is attacked by a water molecule. A 1,2-diamino-diol is then formed after proton exchange and a nucleophilic attack of water to the former nitrile carbon. Ammonia is subsequently eliminated after the protonation of the amino group, and finally the deprotonation of a hydroxyl group produces an amino acid.
1.2.2 Amination of α-Halogen Acids
In 1858, Cahours14 treated α-chloroacetic acid with ethanolic ammonia thereby accomplishing the first synthesis of glycine. Further investigation on this reaction and similar ones conducted by others demonstrated that the formation of an α-amino acid, via the action of aqueous, alcoholic, or liquid ammonia on the corresponding α-chloro or α-bromocarboxylic acid, is a general pathway.
More comprehensive study revealed that α-bromo acids are more active than the corresponding α-chloro analogs15. The ammoniolysis, due to its conveniency and respectable over-all yields, remains a highly preferred method for the synthesis of α-amino acids. Furthermore, if the requisite α-halo acids are unavailable, they may be synthesized through the use of standard organic procedures, such as bromination of the appropriate malonic ester or the Hell-Volhard-Zelinsky procedure.
Scheme 4 - Hell-Volhard-Zelinsky
Gabriel in the 1889 proposed a method in which ammonia is not used. In this regard, the method involves the condensation of potassium phthalylamino acid ester with an α-halo ester, by thermal fusion in the absence of solvent16. The so derived pththalylamino acid ester usually yields the desired amino acid in good amount upon removal of the blocking substituents by drastic acid hydrolysis, or by a two-step alkaline-acid hydrolysis. This method is applicable not only to the introduction of α-amino moieties but to that of ω-amino substituents as well. It has been extensively employed in the preparation of α,ω-diamino acids17.
1.2.3 Amination via Molecular Rearrangement
The well-known rearrangement of acid azides to isocyanates, with the concomitant elimination of nitrogen, was discovered by Curtius in 1890. Utilization of such reaction for the preparation of α-amino acids was described by Curtius and Sieber18 in 1921 and extended by the former19 nine years later.
Scheme 6 - Curtius and Sieber Synthesis
This sequence involves the rearrangement of an alkyl- or aryl-malonylazidic acid to the corrensponding isocyanate upon heating in ether or chloroform solution. The latter, which undergoes ready cyclization to the amino acid N-carbonic acid anhydride, is then acid-hydrolyzed to the pertinent amino acid. Preparation of the crucial azidic acid intermediate is affected by the action of nitrous acid on the half -hydrazide of the corresponding malonic acid which, in turn, is derived from the treatment of the malonic half-ester with hydrazine. As the preparation of the substrate from the suitably substituted malonic ester represents a formidable chore in itself, it becomes apparent that such prolonged and tedious procedure, in its present form, is of little value for the practicable preparation of α-amino acids20.
Ten years later, Darapsky proposed the use of substituted cyanoacetic esters instead of the corresponding malonic esters. This permitted to him and coworkers21 to devise a comparable, but more conveniently accomplished, synthesis of amino acids than that described by Curtius. Thus, the requisite alkylcyanoacetic ester, which is easily accessible from the sodium
ethoxide-catalyzed condensation of cyanoacetic ester and an alkyl halide, readily affords the monohydrazide analogous to on interaction with hydrazine in ethanol solution. Subsequent treatment of with nitrous acid converts it to the azide, which upon heating in ethanol undergoes a Curtius rearrangement to the corresponding urethane, presumably via a transitory isocyanate intermediate. Acid hydrolysis of the latter then yields the desired amino acid.
Scheme 7 - Darapsky Synthesis
A procedure related to that just given, as described by Huang, Lin and Li22, is founded on the use of a Hofmann degradation rather than a Curtius rearrangement as the key reaction. Thus, saponification of thesuitably alkylated cyanoacetic ester results in the production of the monoalkylcyanoacetic acid which, in turn, is transformed into the corresponding malonamic acid by treatment with hot, concentrated sulfuric acid. The action of bromine on an alkaline solution of then leads to a Hofmann rearrangement with the concominant formation of the desired amino acid.
N OEt O N OEt O R N COOH H2NOC COOH R R H2N COOH R RX NaOEt OH -H2SO4 Br2,OH
-Scheme 8 - Huang, Lin and Li Synthesis
1.3 Synthesis of Entiomerically Pure Amino Acids
In the XX century, due to the rapid advances in medicine, a strong struggle to develop convenient synthetic routes to yield the desidered new complex drugs took place. An aspect, in particular, was studied as a crucial aspect for the synthesis of pharmaceutical compounds: the difference of biological activity between enantiomers.
Two enantiomers have the same physical properties, except for their effect on theon the plane polarized light, but not have the same biological activity. In 1962 Thalidomide, a racemic drug used by pregnant women against nausea and to alleviate morning sickness, had to be removed by the market because while an enantiomer had a sedative effect the other had a strong teratogenic side effect. In fact, between 1957 and 1962, 10’000 infants were born with malformation of the limbs. Events like this have stimulated legislation on drug regulation including highly restrictive guidelines for the marketing of synthetic chiral drugs. The marketing of racemates was indeed not completely interrupted, but rather more controlled with the introduction, by the FDA (The United States Food and Drug Administration), of mandatory investigations on the bioavailability and pharmacological effects of the drug with the collection of all background information on each enantiomer and, possibly, on the racemic mixture.
In order to avoid these costly investigations, the possibility to synthesize only a single enantiomer was investigated. In particular, time has been spent on the synthesis of optically pure amino acids due to the fact that they are important chemical building block of pharmaceutical compounds, as described in chapter 1.1. Different procedures have been proposed and developed; these, can be summarised as follows:
Asymmetric synthesis or desymmetrization Kinetic resolution
Deracemization Fermentation
Other resolution methods
Desymmeritrization is a chemical reaction (or reaction sequence) in which one or more new elements of chirality are formed in a substrate molecule and which produces the stereoisomeric (enantiomeric or diastereoisomeric) products in unequal amounts. This type of reaction starts from a pro-chiral or meso- compound and can obtain a theoretical yield of 100%. M P Q kp kQ M = meso-substrate
kP,kQ= stereodivergent reaction rate
Scheme 9 - Asymmetric Synthesis
Kinetic resolution, instead, is based on the use of an enantioselective reaction so one entiomer is not affected while the other is converted into the desired product. In this case, the theoretical maximumyield is 50% due to the fact that only half of the starting material reacts.
Scheme 10 - Kinetic Resolution
Deracemization can be divided in three protocols and rely on the transformation of both substrate enantiomers A and B into a single product of different structure (i.e., P or Q) via independent enantioconvergent pathways through retention and inversion of configuration (Scheme 11, enantioconvergent process). Alternatively, the non-reacting substrate enantiomer may be racemized (in situ) to furnish a dynamic kinetic resolution (DKR) (Scheme 11, dynamic kinetic resolution). In contrast, deracemization by enantioselective stereo inversion23 of enantiomer a yields substrate enantiomer B as the sole product. This process is sometimes also denoted as enantiomerization. Stereoinversion is any reaction that inverts the chiral centre of a compound.
Enantioconvergent process
AR BS PR QS kR kS Retention InversionA,B = Substrate enantiomers P,Q = Product enantiomers
Dynamic kinetic resolution
A B P Q Racemization
Stereoinversion
A B InversionScheme 11 - Deracemization Processes
In a fermentation the desired product is produced from microorganisms. Nowadays it is an important route to produce complex optically pure molecule because micorganisms can be genetically modified with recombinant methods. The idea is to modify their genetic code to obatain the well-defined coumpound in great quantity and high yield.
To the techniques just described other less important resolution methods can be added and be summarised as follows:
Spontaneous crystallization procedures Diastereoisomeric salts
Paper chromatographic approaches
Before moving on, it is useful to state that many of these procedures can involve biotrasformations. Such reactions can be defined as the specific modification of a definite compound to a distinct product with structural similarity, by use of biological catalysts (including microorganisms). The biological catalyst can be described as an enzyme, or a whole, inactivated microorganism containing a single active enzyme or several enzymes working together24. The essential difference between fermentation and biotransformation is that in fermentation there are generally several catalytic steps between the substrate and the product while in a biotrasformation there is only one or two steps. The distinction is also in the fact that the chemical structures of the substrate and the product resemble one another in a biotransformation, but not necessarily in fermentation. The use of biotrasformation have many advantages as high performances beacause enzyme are very selective for reactions and are able to work in mild conditions. This opportunity has been deeply investigated in the last fifty years in particular due to development of recombinant method in which recombinant DNA, molecules of DNA engineered in laboratory, is introduced in a microorganism in order to improve and modify its activity. So, the application of recombinant DNA technology can allow to rapidly screen and design biocatalysts for new application.
1.4 Asymmetric Synthesis
Asymmetric synthesis is the process by which a substrate containing no stereogenic elements is transformed by means of an asymmetric step into the desired chiral product. The advantage of this technique is the theoretical yield of 100%; obviously, it is necessary to consider also the cost of the starting material and of the operating conditions.
There are many approaches to achieve the desired final chiral product, such as enantioselective catalysis, the use of chiral auxiliaries, biotacatalysis, enantioselective organocatalysis or chiral pool synthesis.
In the enantioselective catalysis, usually is based on a catalyst characterized by chiral ligands, and this technique is often suitable for industrial scale synthesis. An alternative is constituted
by the use of chiral auxiliaries, which are organic compounds which couple to the starting material to forman intermediate which can in turn then undergo enantioselective reactions via intramolecular asymmetric induction. Chiral auxiliariesd must be used in stoichiometric amounts to be effective and require additional synthetic steps to apply and remove the auxiliary.
Biocatalysis makes use of biological compounds, ranging from isolated enzymes to living cells, to perform chemical transformations. Enzymes are often very selective reagent and work at mild condition with low environmental impact.
Chiral pool synthesis is one of the simplest and oldest approaches for enantioselective synthesis. A readily available chiral starting material is manipulated through successive reactions, often using achiral reagents, to obtain the desired target molecule. This route is attractive in particular if the starting material is inexpensive and synthethic path is not tortuous.
In general, the asymmetric synthesis can be split in two families: non-enzymatic methods and enzymatic methods.
1.4.1 Non-enzymatic Catalytic Asymmetric Synthesis
Non-enzymatic catalytic asymmetric synthesis is based on the generation of chirality by the interaction between a chiral ligand and the starting substrate. The first industrial non-enzymatic catalytic asymmetric synthesis was developed by Dr William S. Knowles, at Monsanto Company, who in the 2001 won the Noble Nobel Prize in Chemistry for his work on asymmetric hydrogenation reactions. Knowles in the 1974 made up synthesis of L-DOPA, an important amino acid used in the treatment of Parkinson’s desease, using catalytic asymmetric hydrogenation. He discovered that a cationic rhodium complex containing DiPAMP, a chelating diphosphine with two chiral phophorus atoms, catalyzes highly enantioselective hydrogenations of enamides such as A, the enamide precursor of L-DOPA. In the key step of the syntheses of L-DOPA enamide A is hydrogenated in the presence of a catalytic amount of [Rh(R,R)-DiPAMP)COD]+BF4 affording the protected amino acid B in quantitative yield and in 95% ee. A simple acid-catalyzed hydrolysis step completes the syntheses of L-DOPA25,26.
Scheme 12 - Asymmetric Synthesis of L-DOPA
1.4.2 Enzymatic Asymmetric Synthesis
In recent years, the enzymatic asymmetric synthesis of chiral amino acids has attracted much attention due to the high atom economy and brief reaction steps in fact using a chiral biocatalyst, the prochiral starting material is directly transformed into a single enantiomer in one step. Generally, four major routes for the enzymatic asymmetric synthesis of chiral amino acids can be followed: (1) asymmetric reductive amination of keto acids; (2) asymmetric transfer of an amino group to keto acids; (3) enantioselective addition of ammonia to a,b-unsaturated acids; and (4) aldol condensation of an amino acid to aldehydes.
An important enzymatic asymmetric synthesis is the reductive amination that produce L-tert-leucine27. This amino acid has been successfully produced on an industrial scale by Degussa in an enzyme membrane reactor using LeuDH, isolated from Bacillus cereus, with formate dehydrogenase (FDH), from Candida boidinii, for the regeneration of cofactor at a high conversion rate (93%) and, enantiomeric excess (ee) (99%).
Scheme 13 - Enzymatic Asymmetric Synthesis of L-t-Leucine
1.5 Kinetic Resolution
In the kinetic resolution the starting material is a racemic mixtures, only one enantiomer is transformed into the desired product, so a 50% conversion constitutes the maximum yield. The selectivity of the reaction towards one enantiomer depends on the performances of the used biocatalyst.
There are many biological procedures that can be applied: (a) the use of an animal to whom a racemic mixture is fed, followed by the isolation from the urine of one or the other antipode; (b) an asymmetric oxidation or decarboxylation through the action of microorganism, whereby one of the enantiomorphs is unaffected and the other is metabolized; (c) the asymmetric synthesis through the action of protease on N-acylated racemic amino acids, whereby only the L-antipode in the most favourable cases partakes in a synthetic reaction with a base to form an insoluble and hence separable derivative, leaving most of if not all the D-antipode in solution; (d) asymmetric hydrolysis through the action of amidases, esterases and acylases on the appropriately substituted racemic amino acids28.
Scheme 14 - Asymmetric Oxidation (b)
This last procedure for the resolution of amino acids is a very promising way, it is based on the presence on the amino acid of a group which may be asymmetrically removed by hydrolysis. The enzymes used for this purpose can be purified, immobilized, cell free, or in whole-cell form. Several studies have been made and typically proteins such as amidases or acylases, lipases and esterases were proven effective in the resolution of racemic mixtures for the purpose of production of optically active α-amino acids. With a few exceptions, the
method of kinetic hydrolysis could be successfully extended to the resolution of some 60 racemic amino acids by the following procedure:
Scheme 15 - Kinetic Hydrolysis (d)
Where R is H, CH3, etc. Acid hydrolysis of the respective acylated D-amino acids or D-amino acid amides yields the D-amino acids:
Scheme 16 - Acid Hydrolysis
Among the avantages of this procedure is the fact that it is applicable to the large-scale production of both optical isomers27,29.
1.5 Deracemization Process
As it previously, deracemization can be split in enantioconvergent process, dynamic kinetic resoluition and stereoinversion. In all the cases only one enantiomer is in the end obtained. In order to achieve this purpose, the enantioconvergent procedure needs biocatalysts able to convert the racemic starting mixture only in the desired enantiomer. On the other side, apparently, a dynamic kinetic resolution needs a more complex system because, togeter with an enzyme converting only one of the two enantiomers, an in situ racemization of the not consumed enantiomer must occur.
Concerning steoreoinversion, this is a very attractive procedure since involves the complete conversion of a stereisomer into the other. This is useful when in a racemic mixture only one enantiomer is needed and there is a simple method to convert the other enantiomer into the valuable one.
Even if, compared with the procedures just described, the traditional KR seems to belong to an old chemistry era, the majority of chiral molecules of industrial interest are still prepared in
this way. This is mainly due to the cost associated to the replacement of an established industrial process by a new one, even if intrinsically more convenient.
1.5.1 Dynamic Kinetic Resolution
A successful DKR must posses some fundamental characteristics. Other than a selective enzyme and a substrate configuration that permits the in situ racemization, the interconversion rate between the substrate enantiomers must be higher than the biotrasformation rate. Moreover, the final product must not be prone to racemization.
One of the most interesting and famous DKR was employed by Dagussa after 70’s and todays it’s commercially applied at a scale of >1000 tons per year for the production of D-phenylglycine and p-OH-D-phenylglycine. The reaction starts from a racemic mixture of 5-monosubstituted hydantois that can racemize spontaneously and, trough enantioselective biostrasformations, can be converted to the desired amino acid with high yield and high ee. 5-monosubstituted hydantois can be easily prepared from an aldehyde and isocyanate, or by the Bucherer–Berg synthesis and similar methods. The system is of special interest because the proton in the 5-position in the hydantoin ring (it will become the α-hydrogen in the α-amino acid) is quite more acidic then conventional protons in amino acid esters and amides and much more acidic then amino acid itself. Thus, the hydantoin can often be racemized in situ at slightly basic pHs where the enzymes are still stable and active. If these conditions are met, the amino acid can be obtained in one single enantiomeric form in yields higher than 97%.
NH N H O O R NH N H O O R NH2 N H O R COOH NH2 N H O R COOH N H2 R COOH N H2 R COOH NH N H O OH R L-AA D-AA pH > 8 or racemase D-hydantoinase L-hydantoinase D-carbamoylase L-carbamoylase Scheme 17 - DKR by Hydantoinase/Racemase
Theoretically, both the enantiomers can be produced but in reality the commercial application of the hydantoinase process is still limited to the production of D-amino acids. Processes for the production of L-amino acids are limited by low space-time-yields and high biocatalyst costs. It is useful to notice that, due to the attractiveness of the hydantoinase process, Dagussa has started a R&D program to establish a commercially feasible process for the production of natural and nonatural L-amino acids. Recently, a new generation of an L-hydantoinase process was developed based on a tailor-made recombinant whole-cell biocatalyst. Further reduction of biocatalyst cost by use of recombinant Escherichia coli cells over expressing a hydantoinase, a carbamoylase and an hydantoin racemase from Arthrobacter sp. DSM 9771 was achieved. To improve the hydantoin-converting pathway, expression levels of the different genes were balanced on the basis of differences in the specific activities of the enzymes. This has been accomplished by using different gene dose coding for the respective enzymes. This biocatalyst, has been successively modify to obtain very high performances for the L-amino acids production. So, recombinant methods can be very useful to improve existing biotrasformations30,31.
1.5.2 Stereoinversion of Crixivan
In 1971, Hafner and Wellner reported the generation of L-alanine 1a and L-leucine 1b from the corresponding D-enantiomers through the use of porcine kidney D-amino acid oxidase (DAAO) and NaBH4 (Scheme 18). Although the yield was low, the principle of stereoinversion was established; the enzyme catalysed oxidation of the D-enantiomer to the corresponding achiral imino acid 2 followed by in situ reduction by the borohydride generating a mixture of D- and L-amino acids32.
A recent stereoinversion has been investigated by Beard and Turner33: the deracemisation of DL-piperazine-2-carboxylic acid. It is a component of the HIV-protease inhibitor Crixivan, and can be obtained in 86% yield and 99% ee with the use of NaCNBH3 rather than NaBH4
and DAAO.
Scheme 19 - Stereoinversion of Crixivan
1.6 Fermentation
Amino acids can be produce in large quantities with the use of microorganism. Historically, man has used always microorganism for the fermentation in the production of wine and beer but nowadays we can use them as an alternative to traditional chemical synthesis. In the 1950s Dr Kinoschita discovered that Corynebacteriam glutamicum can produce easily large quantities of amino acids. This microorganism was applied mainly for the production of L-glutamic acid, used as food additive and flavour enhancer in the form of its sodium salt MSG. Even if fermentation process may appear an easy task there are many aspects that must be considered. Usally it occurs in batch reactors, and in the production of amino acids are used bioreactor with sizes from 50 to 500 m3. Before reaching the production reactor a few other steps have to be carried out. Preparation of inocula is the first crucial step in most bioprocesses. The purpose of this lab procedure is to obtain a huge amount of stable inocula, which can be used to run dozens of production batches under the same defined conditions finally reaching the same result or yield. Inocula have to be validated after preparation in terms of sterility and productivity before being transferred to production, and often influence productivity and yield of the bioprocess significantly. Due to the importance of inocula quality (inoculum size and stability) and quantity (amount of inocula and cell titer), they have to be tested regularly during their use in order to avoid decrease in productivity. The inoculum is afterwards propagated in a so called seed train. In the case of bioprocesses with Coryneform bacteria this means usually 1:10 steps from seed step to seed step. Depending on the scale of the production, this is performed in shaking flasks or more preferably in bioreactors of different scale.
The fermentation is much affected by operating condition as pH, temperature, feed rate carbon source and nirogen sources. For example, addition of CO2 to the process increased the biomass yield and considerably decreased formation of organic acids in L-lysine production with leucine and homoserine auxotrophic C. glutamicum. In particular selection of raw material is essential not only for the final quality and quantity of the product, but also for the economy of amino acids production. Especially the carbon source represents a major part of variable production costs. So, an optimization of this parameter is always necessary.
Another aspect must be taken in account, namely the downstream technology. A cost-efficient downstream process is crucial to reduce investment and production costs in amino acid production. The separation of biomass is usually the first step of amino acid purification. Removal of the cell mass is accomplished either by gravitation-based techniques (centrifugation or decantation) or by filtration. A significant amount of product might be lost in this biomass removal step which has furthermore the disadvantage of (A) high investment costs and (B) biomass waste streams. Once the biomass is removed from the product stream the purification of the product begins. Typical purification steps are chromatographic technologies, combined concentration/crystallisation steps or combinations of both. What method or what sequence of methods is used depends on a couple of effectors: the physico-chemical ties of the amino acid (solubility, isoelectric point), composition of the process liquid (quality and quantity of by-products), environmental regulations and on the application of the product (feed or pharmaceutical use). Ion exchange resins are the base for chromatographic methods. Disadvantages of this common technique are lower concentrations and increased waste streams leading to higher costs in wasteliquor treatment. On the other hand chromatographic methods provide usually higher product qualities. After treatment with ion exchange resin, a further step usually consists in a single crystallisation. If only crystallisation is applied in product purification, often two or more subsequent crystallisation steps are necessary. Crystallisation techniques like cooling or vacuum crystallisation are economically favourable but cannot be applied for all amino acids34.
Like in other processes that used biocatalysts, these procedures can be nowadays applied for large vatiety of amino acids due to the possibility of genetically modified microorganism with the recombinant method.
1.7 Other Resolution Methods
The term “resolution” may be defined as a procedure whereby both optical isomerides are separated and prepared in pure form from a racemic mixture or compound. All the resolution procedures stem from the studies of Louis Pasteur that are based on the following principles:
The mechanical separation of crystals possessing requisite hemihedrism
The differential solubility of diastereoisomeric salts of the racemate with optically active compounds
The action of living organisms or of enzymes derived from living organisms which utilize or attack preferentially one of the two optical antipodes or its derivatives
To the latter principle belong for example Kinetic Resolution and stereo inversion procedure that are previous described.
1.7.1 Crystallization Procedures
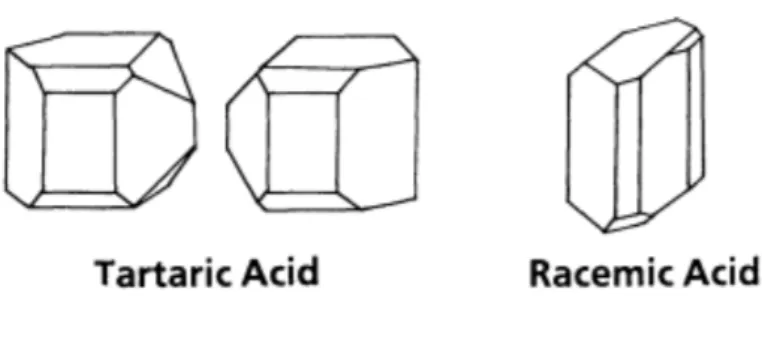
It is the oldest method to separate two enantiomers one from the other. It was discovered by Louis Pasteur in the 1848 with the separation of the two forms of tartaric acid through spontaneous crystallization. In his publication he investigated the crystallographic properties of tartaric acid beacause it can exist in three different forms and, as many other organic compounds, when it is dissolved in a solvent, has the ability to rotate the plane of linearly polarized light. Pasteur observed that while tartaric acid crystals exhibited hemihendral faces, crystals of racemic acid did not. He extended his studies to include many simple and double salts of the same acids and verified that every case optical activity was accompanied by the existence of hemihendral facets on the crystals. Pasteur then examined the crystals of optically inactive sodium ammonium paratrate under the microscope and was very surprised to note that these crystals did contain hemihendral facets. Moreover, he observed that the overall assembly appeared to contain equivalent numbers of left-handed and right-handed crystals. He manually separated the two crystal types and examined these using a polarimeter. His observation was that the crystals heminhendral to the right rotated the place of linearly polarized light in a clock-wise manner, while crystals hemihendral to the left rotated the linearly polarized light in the opposite fashion. Pasteur concluded that while racemic acid appeared to be a unique species, paratartaric acid actually consisted of a mixture of the two optically active tartaric acid forms.
Figure 4 - Heminhendral Crystals
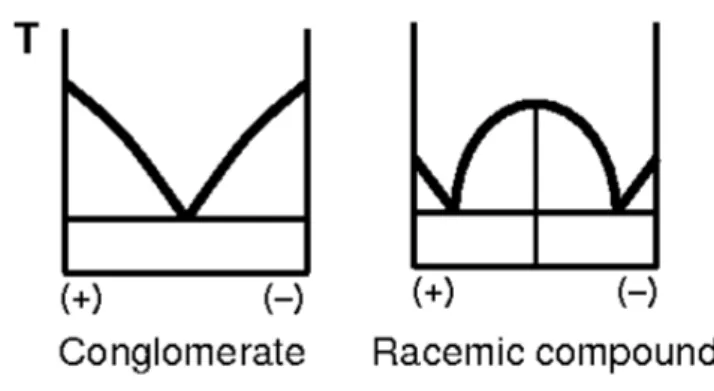
It usefull to note that today it’s known that there are four ways in which a racemate can be crystallized, depending on the substance:
Conglomerate: If the 'molecules' of the substance have a much greater affinity for the same enantiomer than for the opposite one, a mechanical mixture of enantiomerically pure crystals will result. Roughly 10% of racemic chiral compounds crystallize as conglomerates
Racemic compound: If molecules have a greater affinity for the opposite enantiomer than for the same enantiomer, the substance forms a single crystalline phase in which the two enantiomers are present in an ordered 1:1 ratio in the elementary cell.
Pseudoracemate: When there is no big difference in affinity between the same and opposite enantiomers, then in contrast to the racemic compound and the conglomerate, the two enantiomers will coexist in an unordered manner in the crystal lattice.
Quasiracemate: A quasiracemate is a mixture of two similar but distinct compounds, one of which is left-handed and the other right-handed. Although chemically different, they are sterically similar (isosteric) and are still able to form a racemic crystalline phase.
The type of crystallization may be known from the phase diagram.
In particular compounds known to crystallize as conglomerates are much easier to resolve than true racemates, since the resolution step takes place spontaneously upon crystallization. There is no need to rely on the use of resolving agents, chromatographic solid supports, or the use of biological or chemical activation process. When possible, the direct crystallization of one or both enantiomers represent the most economically attractive method for generation of enantiomerically pure substances.
Nowadays three procedures of separation can be used. The first method involves the mechanical separation of enantiomorphic crystals, formed simultaneously while the mother
liquor remains racemic. This method is extremely time-consuming to perfom, and impossible unless the crystals form with well-defined hemihendral faces but usaually is used to obtain the seed crystals required for other direct crystallization process. In fact, a second method for enantiomer separation entails the localized crystallization of each enantiomer from a racemic, supersatured solution. Seed crystal are placed in geographically separated locations in the crystallization vessel, and these serves as nuclei for the further crystallization of the like enantiomer.
Another method used on the large industrial scale is known as resolution by entrainment. It is based on the condition that solubility of a given enantiomer be less than that of the corresponding racemate. To begin, a solution is prepared which contains a slight excess of one enantiomer. Crystallization is induced, whereupon the desired enantiomer is obtained as a solid and the mother liquor is enriched in the other isomer. In a second crystallization step, the other enantiomer is obtained35,36.
1.7.2 Diastereoisomeric Salts
The previous technique can be applied only at a small quantity of compounds, for example doesn’t work for chemical forming true racemates. So, another method was developed based on transformation of the racemic compounds in a salt in which the enantiomers are more easily separable. The idea is to perform a derivatization reaction that involves the formation of dissociable diastomer species, which is often a simple salt. The produced salts have different physical properties in particular, their solubility so that they can be resolved by differential crystallisation. After the separation, the salt is decomposed to regenerate the enantiomerically pure starting compound.
This method was applied for the first time again by Pasteur that used alkaloid bases to separate the optical isomers of D,L-tartaric acid. Racemic bases have in turn been resolved into their isomerides by the use of optically active acids.
The object is the formation of salts of optically active bases with racemic acids or of optically active acids with racemic bases leads to diastereomeric mixtures which may be resolved by the differential solubility of the two components of such mixtures.
(+B) + (D,L) A (+B)(L)A + (+B)(D)A
or
(+A) + (D,L) B (+A)(L)B + (+A)(D)B
Scheme 20 - Diastereoisomeric Salts
Although these may be separated on a small scale by preparative chromatography, the most generally used method is the fractional crystallization because diastereoisomeric salts usually present a binary melting point phase diagram with only one eutectic point not at equimolar point (figure). If the eutectic point is close or at equimolar point can be applied entrainment procedure due to the fact that diastereoisomeric salt froms a conglomerate.
Figure 5 - Phase Diagrams
After separation the salts may be decomposed by a metathetical reaction involving a stronger base than (+)B or a stronger acid than (+)A, to yield thereby the free, optically active isomerides of (D,L)A or (D,L)B.
In any case by means of these reciprocal procedures, a wider variety of optically active acids and bases has become available as tools for resolution of racemates than were hitherto on hand from natural sources.
In practical terms, this type of resolution is not so easy to perform. None of the conditions necessary for a successful resolution can be predicted a priori, and the resolution of each individual racemate constitutes a separate experimental problem. Moreover, complete purification of each salt is thus accomplished by tedious sequence of crystallization and polimetric observations, supplemented wherever possible by examination of crystal forms. Despite these formidable difficulties, the procedure has in one form or another been successfully applied to the resolution of a variety of α-amino acids. Emil Fischer converted, for example, racemic alanine, glutamic acid and aspartic acid to their N-benzoyl derivatives and mixed them with brucine or strychnine in molar proportions37. The diastereoisomeric salts
separted. The alkaloid was removed by alkaline treatment, and the enantiomorphic benzoylamino acids were converted to the free amino acids by prolonged refluxing with strong HCl. The hydrolysis of the benzoyl group occurs at drastic condition and can leads to some racemization of the product so, Fisher, subsequently employed the more easily hydrolazyble N-formyl group as substituent in the racemic amino acids.
The gerneral procedure, wether the free, racemic amino acid or some derivative therefor is employed, is as follows. The racemate is dissolved or simply suspended in the desired solvent, usually water or an alcohol-water mixture, and the solution warmed. If the racemate is monobasic or monoacid, 1 molar equivalent of the optically active base or acid, is added. The mixture is warmed until solution occurs, filtered clear and allowed to cool to some desired temperature over a period of time. When the precipitation appears to have come to an end, the crystals are filtered off and washed briefly with cold solvent, dried, and weighed. The method just is described it’s a simple way to achieve good resolution of racemic mixture and until today a large number of reactant and conditions have been proposed and investigated36,38.
1.8 Paper Chromatographic Approaches
A very particular way to achieve a resolution is based on the possible different affinity between enantiomer with the solid phase or with the solvent in a chromatography. Several studies have been made on this procedure, one of the first was that of Mason and Berg. They noted that, with Whatman paper No.1 and solvent mixture of four volumes of methanol plus two volumes each of n-butanol, benzene, and water, L-kynurenine migrated somewhat faster than did D-kynurenine, and that DL-kynurenine yielded two fluorescing spots of the same color39.
2. DKR of Substituted and Unsubstituted Amino Acid
Thioesters
2.1 Introduction
One of the most interesting ways to match the market demand of enantiomerically pure amino acids is the dynamic kinetic resolution. Enzymatic dynamic kinetic resolution is a very attractive possibility due to environmental and economic reasons and especially because nowadays it is easy to improve and modify the properties of enzyme by the use of recombinant method.
In this chapter, a new path to produce enantiopure amino acids is reported, based on the use of thioesters as substrates. They possess particular ability like D,L-5-substituted hydantoin used by Degussa to produce unnatural amino acids at 100% chemical and optical yield (chapter 1.5.1).
2.2 Dynamic Enzymatic Resolution of Thioesters
40A successful DKR is based on the following boundaries: the substrate must be able to racemize in the reaction conditions; the selective agent (e.g. the enzyme) must react with only one of the two starting enantiomers; the racemization rate must be higher than the enzymatic reaction rate.
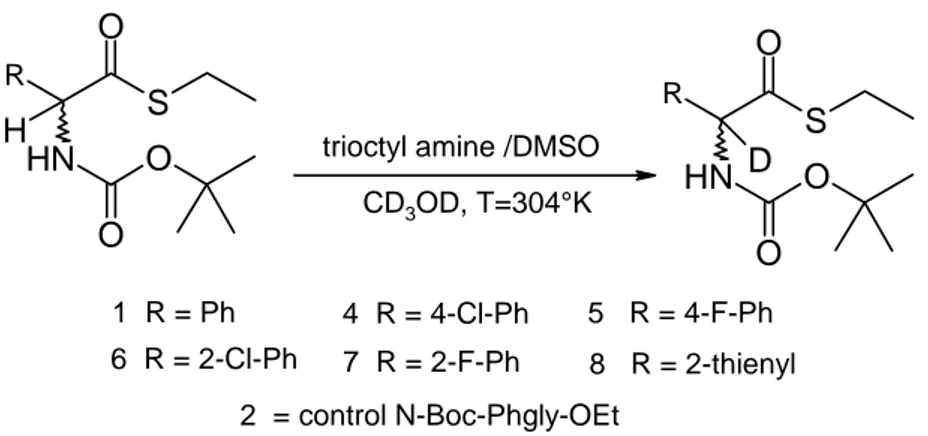
Drueckhammer first proposed the thioesters of carboxylic acids having chiral centers at the α-carbon as possible substrates for DKR. In contrast to an oxoester, the α-proton of the thioester was sufficiently acidic to permit continuous racemization of the substrate by base-catalysed deprotonation. This concept was demonstrated by the work of Tina L. Amyes and John P. Richard, in which the exchange rate for deuterium of the α-protons of ethyl thioacetate and of acetone in 3-quinuclidinone buffers in D2O at 25°C was followed by 1H-NMR spectroscopy.
From these experiments it was found that the resonance overlap of the lone-pair electrons on the sulphur atom with the carbonyl group of the type Z, which would tend to decrease the ease of formation of an adjacent carbanion, is an important factor41.
RS R' O
RS+ R' O
-Scheme 21 - Resonance of Thioester
The difference between thioester and oxoester was also initially demonstrated by Drueckhammer with an α-phenylthio propionate thioester 1a (R = Et, X = PhS), the phenylthiol group also contributing to the acidity of the α-proton (Scheme 22). So, it was expected that this procedure could be applicable to a variety of carboxylic acids having a chiral center and a proton at the α-carbon.
Scheme 22 - Racemization of Thioester
However, several factors regarding the general applicability of this procedure were unknow, then Drueckhammer started to study the real acidity of the α-proton of thioesters of other carboxylic acids and the general utility of hydrolytic enzymes as catalysts for enantioselective thioester hydrolysis.
2.2.1 Kinetic α-Proton Acidity of Thioesters
The major goal of the work of Drueckhammer has been to investigate the enforceability of this procedure to thioesters of acids having inherently lower αproton acidity. Studies of α -proton exchange with deuterated solvent were undertaken with the ethyl thioesters of several carboxylic acids to evaluate their potential for racemization during the course of enzymatic