INTRODUZIONE
1. Stress ossidativo
1.1. I radicali liberi
I radicali liberi possono essere definiti come molecole o frammenti molecolari contenenti uno o più elettroni spaiati nell’orbitale atomico o molecolare più esterno (Halliwell e Gutteridge, 1999). L’energia inerente a questa configurazione determina un’alta instabilità ed un’elevata reattività, per le quali i radicali liberi possono anche dare inizio a reazioni autocatalitiche, là dove le molecole con cui questi reagiscono vengano a loro volta convertite in radicali liberi che propagano la catena del danno.
I radicali liberi dell’ossigeno o, più generalmente, le specie reattive dell’ossigeno (ROS), così come le specie reattive dell’azoto (RNS), sono sottoprodotti del normale metabolismo cellulare. Nella cellula, la fonte principale della produzione di radicali liberi dell’ossigeno (ROS), è rappresentata dai mitocondri, a livello dei quali l’ossigeno molecolare è ridotto ad acqua mediante l’aggiunta in sequenza di quattro elettroni. Tuttavia, durante tale processo, vengono prodotte piccole quantità di forme reattive, parzialmente ridotte, come inevitabili prodotti collaterali della respirazione mitocondriale. Nelle cellule i radicali più comuni sono:
l’anione superossido (O2−), il radicale idrossile (OH⋅), il radicale
idroperossile (HO2.) e l’ossigeno singoletto (O⋅). In condizioni fisiologiche, i
enzimatici come la vitamina E, la vitamina C ed il glutatione, l’ubichinone ed enzimatici come la superossido dismutasi, la catalasi e la glutatione perossidasi.
L’anione superossido (O2−), un ossidante fortemente reattivo, si forma per
addizione di un singolo elettrone dall’ossigeno molecolare; è prodotto a livello dei Complessi I e III della catena di trasporto degli elettroni (Valko et al., 2006); viene considerato il ROS “primario” in grado di interagire con altre molecole e dare origine a ROS “secondari”, sia direttamente, sia indirettamente tramite processi enzimatici (Valko, Morris e Cromin, 2005); viene detossificato dalla superossido dismutasi (SOD) a perossido di idrogeno ed ossigeno (Fridovich , 1978):
O2 + 1e− → O2·− (radicale superossido) superossido dismutasi 2H+ + O2·− + O2·− ⎯⎯⎯⎯⎯⎯⎯⎯⎯→ O2 + H2O2 catalasi 2H2O2 ⎯⎯⎯⎯→ 2H2O + O2 glutatione perossidasi H2O2 + 2GSH ⎯⎯⎯⎯⎯⎯⎯⎯⎯→ H2O + GSSG
Il radicale superossido può anche reagire con l’acqua ossigenata:
O2·− + H2O2 ⎯⎯→ OH· + O2 + OH−
Il radicale idrossile (OH⋅), forma neutra dello ione idrossido, viene prodotto
il ferro, il rame, il cromo, il cobalto, etc.), che donano o accettano elettroni liberi durante le reazioni intracellulari, catalizzando la formazione di radicali liberi, come nella reazione di Fenton:
H2O2 + Fe2+ Æ Fe3++ OH. +OH-.
Il radicale idroperossile (HO2.) si forma da una protonazione dell’anione
superossido (O2−); si pensa sia in grado di attraversare le membrane
biologiche, ed ha una reattività molto maggiore rispetto al radicale da cui ha origine (Benzie, 1996).
La formazione di specie reattive radicaliche, oltre che a livello mitocondriale, può avvenire anche:
- nei perossisomi;
- a livello della membrana plasmatica (ad esempio durante il metabolismo dell’acido arachidonico ad opera della lipoossigenasi);
- a livello del reticolo endoplasmatico liscio [dove troviamo sia i citocromi b5
e P450 sia le rispettive riduttasi contenenti gruppi flavinici che, una volta
ridotti generano anione superossido (O2⎯) ed acqua ossigenata];
- nei processi infiammatori acuti e cronici (Robbins, 2000) per effetto della rapida attivazione della NADPH ossidasi [enzima presente in macrofagi e leucociti, che ossida il NADPH riducendo l’ossigeno ad anione superossido
(O2⎯)];
- per effetto dell’esposizione e dell’assorbimento di radiazioni ionizzanti od eccitanti dell’ambiente o prodotte da sorgenti artificiali.
1.2 Meccanismo d’azione dei radicali liberi
L’accumulo dei danni da radicali liberi, generato da uno scompenso tra i sistemi di produzione e di neutralizzazione, determina alterazioni a livello cellulare. In caso di sovraproduzione di radicali è più facile che si possa manifestare un danno a carico di macromolecole, quali il DNA sia nucleare che mitocondriale [soprattutto il radicale idrossile (OH⋅)], alterando le purine, le pirimidine e lo scheletro di desossiribosio (Halliwell e Gutteridge, 1999). Possono essere inoltre promotori, a livello proteico, dell’ossidazione dei residui aminoacidici, in particolar modo di cisteina e metionina (Stadmann, 2004), e quindi della formazione di legami crociati, mediati, ad esempio, da ponti disolfuro. Possono inoltre determinare una reazione a catena a carico dei lipidi, sia della membrana plasmatica che delle membrane degli organelli, chiamata perossidazione lipidica.
E’ noto che gli acidi grassi poliinsaturi sono particolarmente suscettibili all’attacco di qualsiasi specie chimica che abbia una reattività sufficiente a
“strappare” un atomo di idrogeno da un gruppo metilene (>CH2), alterando
così la natura del doppio legame. Una volta iniziato, il processo procede spontaneamente come una normale reazione a catena di radicali liberi. La perossidazione lipidica può essere schematizzata in varie fasi:
una reazione di inizio in cui un acido grasso poliinsaturo reagisce con un
radicale libero che cattura l’atomo di H per formare il radicale dell’acido grasso (L•);
una reazione di propagazione in cui i radicali L• reagiscono con O2 per formare un radicale lipoperossido che dà inizio alla propagazione reagendo con un altro acido grasso poliinsaturo e formando un lipide idroperossido (LOO•), ed un nuovo radicale libero del secondo acido grasso. Si possono, cosi, formare miscele di idroperossidi lipidici che automantengono il processo perossidativo fino a che non intervengono reazioni di
terminazione.
1.3 Perossidazione lipidica e suoi prodotti
I lipoperossidi sono composti piuttosto labili a causa della bassa energia di dissociazione dei legami O-O. Sono praticamente stabili a temperatura ambiente e corporea mentre si decompongono facilmente alle alte temperature, a concentrazioni elevate ed in presenza di ferro o rame o di semplici chelati di questi ioni metallici (Gutteridge e Halliwell, 1990). Ad esempio, il Fe(II) reagisce con idroperossido (LOOH), ma anche con
perossido di idrogeno (H2O2), determinando la rottura del legame O-O per
formare il radicale alcossile (LO•).
Si decompongono inoltre in presenza di alcune ferro-proteine come l’emoglobina e la mioglobina. I lipoperossidi possono, inoltre, subire una decomposizione ad opera di enzimi, quali la ciclossigenasi e la lipossigenasi che catalizzano non solo la perossidazione di acidi grassi insaturi, ma anche la decomposizione dei loro idroperossidi.
In corso di lipoperossidazione si producono vari metaboliti, quali aldeidi, alcoli e chetoni.
Tra le aldeidi, i principali prodotti sono la malondialdeide (MDA) e il 4-idrossi-2-nonenale (HNE). La MDA è mutagenica in cellule batteriche e di mammifero e carcinogenica nei ratti. L’HNE ha un’azione più debole come
mutagenico ma sembra essere uno dei prodotti più tossici della perossidazione lipidica (Valko et al., 2006).
La MDA deriva dalla rottura della catena carboniosa dei PUFA: essa è in grado di reagire con qualsiasi molecola che contenga un gruppo amminico. I prodotti della reazione concorrono a formare i cosiddetti “pigmenti da usura” o lipofuscine. Il loro accumulo è una delle caratteristiche dell’invecchiamento (Zs-Nagy, 1988).
L’ulteriore metabolismo della MDA porta alla formazione dell’acetaldeide (Dhnakoti e Draper, 1987).
CH3CH
O
(ACT)
Direttamente dallo scheletro del glicerolo o dai trigliceridi si origina la
formaldeide (Winters et al., 1988).
HCH
O
(FA)
Dall’estrazione di un atomo di carbonio dall’acido acetoacetico si forma infine la propionaldeide (Bagchi et al., 1995).
CH2 CH
O CH3
I prodotti aldeidici possono essere liberati dai tessuti ed infine escreti con le urine (Davie, 1987).
Il dosaggio di questi prodotti di degradazione fornisce potenziali biomarcatori, non invasivi, del danno radicalico e dello stress ossidativo (Draper et al., 2000). Un’estesa perossidazione dei lipidi di membrana è associata ad una grande varietà di effetti tossici: riduzione della fluidità e della funzione della membrana, caduta del potenziale, alterazioni delle funzioni dei mitocondri e dell’apparato del Golgi; inibizione degli enzimi associati ai vari organuli; alterazioni dell’omeostasi del calcio (Stadtman e Oliver, 1991), aumento della permeabilità a ioni idrogeno e altri ioni e può in ultimo causare rotture che possono portare al rilascio del contenuto della cellula e dei suoi organelli (Poli et al., 1987; Comporti, 1989; Van der Vliet e Bast, 1992).
1.4 Modello di danno radicalico prescelto
Durante i comuni processi metabolici cellulari, il NADPH, forma ridotta del NADP+ (nicotinammide adenin dinucleoside fosfato), è necessario nei normali processi di biosintesi riduttive. E’ considerato il trasportatore di potere riducente nelle cellule, donatore di uno ione idruro H+ (Strayer, 2001). Da tempo è noto che frazioni cellulari incubate aerobicamente con
NADPH, rilasciano sostanze come superossidi (Aust et al., 1972) e H2O2
(Ernster et al, 1982). E’ stato inoltre dimostrato che la perossidazione lipidica NADPH-dipendente è catalizzata dalla NADPH-citocromo P450-reduttasi (Svingen et al., 1979). Questo enzima, così come riduce il citocromo P450, può trasferire elettroni al ferro libero (FeIII) e quindi generare Fe(II) che stimola la lipoperossidazione (Gosh et al., 1997). Nella maggior parte dei
sistemi in vitro, il ferro è aggiunto in un complesso con l’ADP e necessita
della presenza del radicale O2., affinchè sia conservato in soluzione e
convertito nella sua forma ridotta e attiva. E’ stato proposto che la perossidazione lipidica NADPH- dipendente inizi in seguito all’astrazione di un idrogeno metilene da un acido grasso poliinsaturo (Gosh et al., 1997) e che necessiti della presenza di uno ione ADP-perferrile (Pederson et al., 1973):
O2.- ADP-Fe3+ O2 - ADP-Fe2+
E’ possibile schematizzare tale processo di lipoperossidazione, dividendolo in due fasi distinte:
NADPH-Cyt-P450-reduttasi ADP-Fe3+ I N I Z I A Z I O N E NADPH-Cyt-P450-reduttasi P R O P A G A Z I O N E NADP+ ADP-Fe2+ O2 ADP-Fe3+-O2·− ADP-Fe2+-O2 LOOH LH NADPH + H+ Fe 2+ Fe 2+ NADPH + H+ NADP+ Cyt-P450 Fe3+ Cyt-P450 degradato LOO., L. ,O2-.
Prodotti finali della perossidazione lipidica LH O2 LOOH LH ADP-Fe3+
La prima fase, di iniziazione, è caratterizzata dalla riduzione diretta,
catalizzata dalla NADPH-Cyt-P450-reduttasi, dell’ADP-Fe3+ in ADP-Fe2+,
e dalle successive reazioni di quest’ultimo con ossigeno molecolare, fino alla formazione di un intermedio, lo ione ADP-perferrile. L’ossigeno legato a tale ione catalizza rapidamente la formazione di lipidi idroperossidi. La fase di propagazione necessita dei lipidi idroperossidi formatisi durante la prima fase la cui rottura è catalizzata dal citocromo-P450-ferrico, per formare nuovi intermedi reattivi della perossidazione lipidica (Svingen et al., 1979).
1.5 Altri modelli di danno radicalico
Per la migliore comprensione di questa ricerca è utile definire i tratti principali di altri due modelli di danno radicalico.
Le radiazioni ultraviolette (UV), sono vibrazioni del campo elettromagnetico che hanno un notevole effetto biologico mediante meccanismi diretti e indiretti(Dianzani, 1996).
Gli effetti diretti sono immediatamente dipendenti dall’assorbimento dell’energia della radiazione da parte delle strutture sensibili. Tra le strutture cellulari che mostrano massimi di assorbimento nell’UV, le principali sono gli acidi nucleici, i doppi legami degli acidi grassi insaturi e le proteine. L’azione indiretta è mediata attraverso sostanze fotodinamiche o fotosensibilizzatrici. La differenza di energia fra i fotoni della luce incidente e quelli della luce fluorescente, tipica dei raggi UV, rappresenta la quota di energia trattenuta dalla molecola della sostanza fotodinamica, la quale passa allo stato eccitato metastabile di singoletto; nella sua tendenza a
tornare allo stato normale, la sostanza ha molte probabilità di reagire con altre presenti nei tessuti. La reazione avviene con produzione di ossigeno singoletto (O2.), che concorre alla successiva formazione di radicali liberi. In questo modello di danno radicalico la produzione di sostanze ossidanti avviene in maniera uniformemente distribuita nello spazio.
L’esposizione ad agenti esterni chimici, come ad esempio il tetracloruro di
carbonio (CCl4), un idrocarburo alogenato, può favorire e incrementare la
produzione di forme radicaliche dell’ossigeno. Il CCl4 è metabolizzato, nel
fegato, dal sistema di biotrasformazione citocromo P450-dipendente localizzato a circa 20Å dalla superficie idrofobica nella membrana del
reticolo endoplasmatico liscio. Una volta legatosi al sito attivo, il CCl4 attiva
la NADPH-citocromo P-450 reduttasi che determina il trasferimento elettronico del legame dell’agente citotossico, portando alla formazione di due radicali liberi fortemente reattivi, cioè l’anione cloruro (Cl•) e il radicale
triclorometile (CCl3•). Il Cl• tende a captare un elettrone trasformandosi in
ione, il CCl3•, è dà inizio alla cascata perossidativa dei PUFA (Comporti et
al., 1965).
In questo sistema di aggressione radicalica la produzione di radicali liberi si concentra in prossimità del sito attivo del citocromo P450, prossimo alla regione centrale del bilayer lipidico, dove si colloca il dolicolo.
2. Sistemi antiossidanti
Per proteggersi dal danno ossidativo, gli organismi viventi, tra cui anche gli esseri umani, utilizzano una serie di sistemi di difesa, dislocati in modo strategico, nei vari distretti cellulari. I sistemi di difesa includono difese fisiche e meccanismi antiossidanti e operano con azioni difensive e riparatrici.
Gli enzimi specifici che interagiscono con le specie reattive dell’ossigeno sono: la superossido dismutasi (SOD) e la glutatione perossidasi (GPx), presenti nel citosol e nei mitocondri (Utsunomiya et al.,1991) e la catalasi (CAT) localizzata nei perossisomi. Nell’uomo esistono tre forme di SOD (una citosolica, una mitocondriale ed una extracellulare), codificate e regolate indipendentemente (Stryer, 2001).
Solitamente questi enzimi sono localizzati in prossimità dei siti di produzione dell’anione superossido e del perossido d’idrogeno sui quali agiscono (Robbins, 2000). La maggior parte della protezione delle membrane deriva dall’azione degli antiossidanti che possono influire anche sulla stabilità delle membrane stesse (Giammarioli, Filesi, Sanzini, 1998).
Gli antiossidanti possono essere distinti in primari e secondari.
Gli antiossidanti primari interferiscono con l’inizio della catena lipoperossidativa ed includono chelanti dei metalli di transizione, ortoidrossibenzofenoni (che proteggono le sostanze organiche dalla degradazione fotochimica), composti solforati che reagiscono con gli idroperossidi.
Gli antiossidanti secondari reagiscono con i radicali propagatori, attraverso reazioni di trasferimento di idrogeno, di trasferimento elettronico o di addizione, terminando la catena lipoperossidativa o producendo un nuovo radicale meno reattivo e più stabile. Tra gli antiossidanti secondari sono compresi i fenoli, le ammine aromatiche, i dialchilnitrossidi, i diarilnitrossidi e idrossilamine.
Alcuni antiossidanti vengono sintetizzati direttamente nel nostro organismo altri vengono assunti con la dieta, come i tocoferoli, i carotenoidi e l’acido ascorbico.
L’acido ascorbico (Vitamina C) è un composto idrosolubile e nelle diete occidentali la maggior parte proviene da alimenti di origine vegetale, principalmente agrumi, vegetali a foglia verde, pomodori, peperoni e patate. (Giammarioli et al. 1998). L’acido ascorbico è considerato il più importante antiossidante nei fluidi cellulari ed è in grado di funzionare da “scavenger” per numerosi radicali liberi, rappresentando una delle più importanti difese antiossidanti nel plasma umano nei confronti dello stress ossidativo. Studi in vitro hanno evidenziato che l’acido ascorbico, bloccando i radicali liberi in fase acquosa, prima che abbia inizio la perossidazione lipidica, può proteggere le membrane cellulari dalle modificazioni ossidative, probabilmente anche rigenerando la forma ridotta dell’α-tocoferolo (anche se tale attività sinergica non è ancora stata completamente dimostrata nei sistemi biologici) (Giammarioli et al., 1998).
I carotenoidi sono un gruppo di pigmenti, chimicamente correlati, distribuiti abbondantemente in natura; possono essere suddivisi in caroteni, costituiti solo da un atomo di carbonio ed idrogeno, ed ossicarotenoidi, che contengono anche ossigeno. Il β-carotene, in particolare, esercita funzioni antiossidanti nelle fasi lipidiche, funzionando da scavenger di radicali liberi. La reazione del β-carotene con i radicali perossidici avviene in modo non convenzionale, mediante una reazione di addizione e ed è stato suggerito
(fisiologiche) di ossigeno (Giammarioli et al., 1998). La sua attività antiossidante può contribuire alla protezione delle membrane dalla perossidazione lipidica, tuttavia, allo stato attuale, sembra che il β-carotene abbia un effetto protettivo inferiore all’α-tocoferolo e funzionerebbe da antiossidante di riserva (Giammarioli et al., 1998).
Il resveratrolo (3,4’,5-trans-tri-idrossi-stilbene) è uno stilbene di origine vegetale che si trova in alcune spermatofite, principalmente nell’uva rossa, nelle noccioline americane e nei pinoli, essendosi evoluto nelle piante come molecola-segnale capace di interagire con strutture biologiche, attivando una serie di processi di riparo e meccanismi difensivi. E’una molecola naturale facente parte della classe delle fitoalexine dotate di azione difensiva contro infezioni da microrganismi patogeni, da radiazioni UV, esposizione ad ozono o ad ioni di metalli pesanti (Caruso et al., 2004).
Il Resveratrolo è un efficace antiossidante a doppio meccanismo d’azione perché agisce sia come chelante che come “radical scavenger”. Studi in diversi sistemi modello hanno dimostrato che le proprietà del Resveratrolo sono superiori a quelle delle Vitamine C, E e del β-carotene e che esso è in grado di sviluppare un’azione sinergica con queste molecole. Alcuni lavori hanno dimostrato che il trasferimento dell’atomo di idrogeno è il meccanismo principale per gli antiossidanti fenolici come il Resveratrolo e mettono in luce l’importanza strutturale del gruppo 4’-OH della molecola per le sue attività biologiche. (Caruso et al., 2004). Il Resveratrolo può, per la sua proprietà antiossidante, ridurre l’incidenza di malattie coronariche cardiache inibendo la per ossidazione dei lipidi delle lipoproteine a bassa densità (LDL) e prevenire la citotossicità delle LDL ossidate.
3. Antiossidanti lipofili
La membrana cellulare, per sua stessa natura, è la componenente cellulare più sensibile al danneggiamento. Il doppio strato lipidico che la costituisce, è protetto da antiossidanti lipofili che giacciono tra i lipidi di membrana: il dolicolo (alcol lipofilo), la vitamina E, gli stessi acidi grassi poliinsaturi e l’ubichinone. Generalmente si ritiene che i PUFA diano un contributo piuttosto debole e siano inclini alla perossidazione, la vitamina E, invece, e’ considerata uno “scavenger” che protegge i PUFA dalla perossidazione lipidica stessa. E’ stato ipotizzato che il dolicolo possa funzionare da nuovo scavenger endogeno lipofilo in grado di proteggere i PUFA dalla perossidazione lipidica in cooperazione con la vitamina E. Infine l’ubichinone (CoQ), che svolge un ruolo chiave nei processi di trasferimento elettronico, pur essendo un potenziale prossidante, nella sua forma ridotta, è in grado di interrompere la catena lipoperossidativa, proteggendo i fosfolipidi di membrana ( James et al., 2005).
3.1 Vitamina E
Il termine vitamina E indica propriamente un gruppo di potenti antiossidanti naturali, comprendente 4 tocoferoli (α,β,γ,δ) e 4 tocotrienoli (α,β,γ,δ) (IUPAC-IUB, 1982). Rappresentano costituenti ubiquitari tra i lipidi di membrana e le lipoproteine.
La vitamina E è stata scoperta nel 1922 in ratti che mantenuti con una dieta contenente tutte le vitamine fino ad allora conosciute, non riuscivano a riprodursi (Evans et al.,1922): la sostanza attiva è stata isolata nel 1936 da Evans e chiamata tocoferolo dal significato greco “portare frutto”.
3.1.1. Struttura e metabolismo
Il nome “tocoferolo” attribuito da Evans a questa sostanza, è stato completato con i prefissi α, β, γ, δ per distinguere nell’ambito della stessa famiglia di composti le diverse sostanze.
La forma più abbondante in natura e con la più alta attività biologica è l’α-tocoferolo (Sheppard et al., 1993);
HO
O
Fig.3.1.1.1 - α-tocoferolo.
I suoi derivati differiscono solo nel numero e nella posizione dei gruppi metilici legati al gruppo cromanolo. Da un punto di vista strutturale, la molecola di tocoferolo consiste di due domini funzionali: una catena idrocarburica principale C16 responsabile della lipofilicità della molecola e che spiega la sua collocazione all’interno della membrana plasmatica, ed un gruppo cromanolo responsabile dell’attività antiossidante: per questa seconda capacità, la parte del cromanolo funziona come un “rompicatena” capace di terminare le catene radicaliche cedendo un atomo di idrogeno.
Fig 3.1.1.2.- Schematizzazione della localizzazione della Vitamina E nel bilayer lipidico (tratto da Afri et al., 2004).
I tocotrienoli differiscono dai tocoferoli per la presenza di doppi legami presenti all’interno della catena laterale.
La vitamina E è assunta esclusivamente con la dieta è assorbita a livello intestinale, entra in circolo attraverso il sistema linfatico e viene trasportata fino al fegato insieme ai chilomicroni e poi nell’organismo come parte delle lipoproteine del plasma.
Tra le proteine trasportatrici, l’ LDL può contenere da 6 a 12 molecole di α-tocoferolo (Stocker et al., 1996). Per la sua natura lipofilica, la vitamina E si localizza nei lipidi tissutali oppure nei domini idrofobici delle lipoproteine. La distribuzione della vitamina E nel “core” idrofobico delle membrane cellulari varia a seconda della specie molecolare: l’ α- tocoferolo si distribuisce in gran parte nell’apparato del Golgi e nei lisosomi. In questi siti il rapporto α-tocoferolo/fosfolipidi è dell’ordine 1:65, molto più alto di quello trovato in altre membrane subcellulari (Wang e Quinn, 1999).
Gli studi del catabolismo della vitamina E si sono focalizzati sui metaboliti prodotti dall’ossidazione della componente aromatica, promossi dal forte interesse per la sua attività antiossidante. Il primo prodotto di degradazione a livello epatico è l’α−tocoferolo chinone derivato dalla reazione dell’ α-tocoferolo radicale; quest’ultimo è ridotto a α−tocoferil idrochinone da enzimi mitocondriali e microsomiali NADPH dipendenti. I prodotti metabolici liberati nelle urine (chiamati metaboliti di Simon) sono l’acido α−tocoferonico e il suo lattone.
3.1.2. Ruolo biologico
La localizzazione della vitamina E nei depositi di grassi e nelle membrane cellulari è certamente dovuta al carattere marcatamente idrofobico della molecola. Per queste caratteristiche, si ritiene che essa abbia un ruolo antiossidante protettivo rispetto alle membrane, preservandone la stabilità e l’integrità dei lipidi. L’α-tocoferolo e il γ-tocoferolo costituiscono componenti essenziali dei meccanismi cellulari di difesa contro ossidanti
dell’α-tocoferolo sono di natura non enzimatica e, soprattutto, molto veloci. Il ruolo principale dell’α-tocoferolo sembra essere quello di scavenger dei radicali perossilipidici, le specie reattive responsabili della propagazione della reazione perossidativa (Burton et al., 1986; Liebler, 1993).
L’ossidazione dei lipidi procede a partire da una reazione a catena mediata da radicali liberi dove un lipide perossido è l’iniziatore e la propagazione avviene per eliminazione di un atomo di idrogeno dal target lipidico da parte del radicale:
LO2. + LH LOOH + L.
Dove, LO2. è il lipide perossiradicale, LH è il lipide bersaglio, LOOH è un
lipide idroperossido e L. il lipide radicale. Quest’ultimo, è la specie
reattiva che porta avanti la reazione, che è a questo punto in grado di rialimentarsi.
Il ruolo della vitamina E in questo contesto, riguarda l’intervento sul perossiradicale prima che questo sia in grado di attaccare il suo bersaglio lipidico.
LO2. + TOH LOOH + TO.
Dove, TOH e TO. rappresentano rispettivamente α-tocoferolo e il suo
radicale. Studi in vitro, hanno dimostrato che la rigenerazione dell’α-tocoferolo a partire dal suo radicale, è mediata dalle vitamine A (Bohm et al, 1998) e C (Chan et al, 1991) e dalla forma ridotta del coenzima Q
(CoQH2) (Beyer, 1994; Kagan et al., 1998). L’interazione del tocoferolo con altri costituenti delle membrane influenza la stabilità delle stesse, ma come questo avvenga di preciso è ancora da chiarire. Alcuni dati sperimentali indicano che la formazione di complessi della vitamina E con certi componenti tende a destabilizzare i bilayer, mentre altre suggeriscono che il tocoferolo destabilizzi le membrane e promuova la fusione. Altri studi sulla stabilità dei fosfolipidi di modelli di membrana in presenza di differenti livelli di α-tocoferolo, inducono invece a ritenere che la vitamina E stabilizzi i doppi strati lipidici e prevenga la fusione. La vitamina E potrebbe stabilizzare le membrane formando complessi con i lipidi.
In vivo la vitamina E può diminuire i livelli della perossidazione lipidica delle lipoproteine a bassa densità (LDL). La vitamina E ha un ruolo inibente nella trasmissione dei segnali molecolari, soprattutto in relazione all’attività della proteina chinasi C, forse attribuibile, in parte, all’ attenuazione della formazione del diacilglicerolo. Probabilmente l’azione non è dovuta alle capacità antiossidanti dell’α-tocoferolo ma dipende dalla sua integrazione direttamente nella struttura della membrana.
L’aumento dell’attività di due enzimi come la fosfolipasi A2 e la
cicloossigenasi, implicati nella cascata dell’acido arachidonico, rafforza l’idea che la vitamina E in rapporto alla sua dose, favorisca il rilascio di prostaciclina, un potente vasodilatatore e inibitore dell’aggregazione piastrinica (Ahkong et al. 1972; Nevril, 1994). Studi epidemiologici, inoltre, hanno rivelato che un elevato apporto di vitamina E in virtù della sua attività antiossidante potrebbe essere correlato con la riduzione del rischio di malattie cardiovascolari.
3.2 Ubichinone
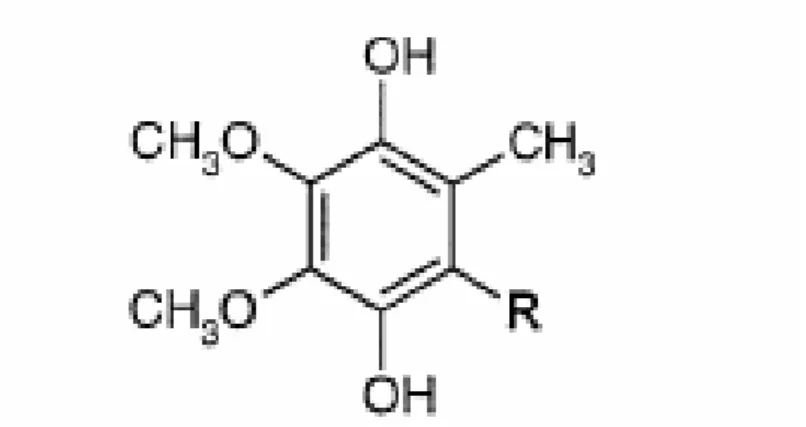
L’ubichinone (2,3,-dimetossi-5-metil-6-multiprenil-1,4-benzochinone) o coenzima Q (CoQ) è un lipide endogeno con funzioni redox, presente nello spazio idrofobico del doppio strato lipidico di tutte le membrane cellulari (Kwong et al., 2002).
Fig. 3.2.1. -Ubichinone
3.2.1 Struttura e metabolismo
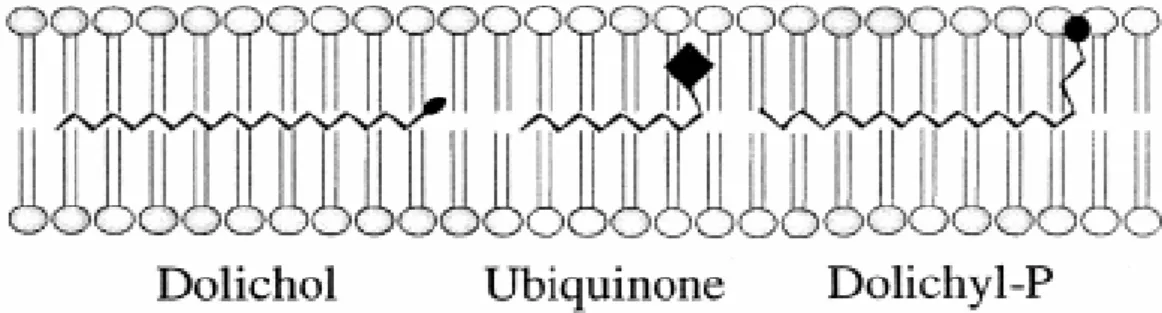
La via del mevalonato è una sequenza di reazioni cellulari, che portano alla formazione del farnesil-pirofosfato, substrato comune alla sintesi di dolicolo, dolicolo fosfato, colesterolo e ubiquinone, oltre che alla prenilazione delle proteine.
Fig. 3.2.1.1. -Via di biosintesi dell’ubichinone (tratto da Dallner et al., 2000)
A differenza di altri lipidi isoprenoidi, l’ubichinone contiene un anello benzenico, ereditato da una molecola di tirosina durante il processo di biosintesi. E’ dotato di una lunga catena laterale isoprenoide formata da 6 (nei lieviti) a 9-10 (nei mammiferi) unità isopreniche. Grazie a questa catena laterale alifatica, estremamente idrofobica e non ancorata ad alcuna proteina, l’ubichinone gode di ampia mobilità nell’ambito fosfolipidico della membrana interna.
Fig. 3.2.1.2.- localizzazione dei lipidi polisoprenoidi all’interno del doppio strato lipidico. (tratto da Dallner et al., 2000)
In virtù di questa mobilità può venire a contatto da un lato con la
NADH(H+) deidrogenasi e con le varie flavoproteine da cui riceve
elettroni, dall’altro con il citocromo b cui li cede (Chance et al., 1961). Va tuttavia sottolineato che il CoQ può anche formare un semichinone relativamente stabile e partecipare quindi, oltre che al trasferimento simultaneo di due elettroni, anche al trasferimento di un solo elettrone. Nell’uomo il CoQ10 (la numerazione indica le unità isopreniche presenti nella catena laterale) è la forma dominante in tutti i tessuti, il CoQ9, invece, rappresenta solo il 2-7% del totale; nel ratto, al contrario, il CoQ9 risulta essere prevalente, ma il CoQ10 costituisce il 10-30% del totale (Aberg et al., 1992). Utilizzando un precursore dell’ubichinone marcato radioattivamente, due diversi studi sono giunti alla conclusione che il metabolismo del CoQ integrato nella dieta, mima quello del CoQ endogeno (Imada et al., 1970; Nakamura et al., 1999). Nei ratti, il CoQ esogeno, viene trasportato attraverso il sangue in concentrazione
maggiore al fegato e alla milza, e si distribuisce in quantità minori a tutti gli altri organi (Dallner et al., 2000). I due principali metaboliti identificati
nelle urine corrispondono alle forme coniugate dell’idrochinone (QH2), e
presentano l’anello benzenico intatto ma la catena laterale drasticamente ridotta. É interessante notare che la somministrazione di CoQ esogeno non influenza l’escrezione dei due metaboliti derivanti da quello endogeno, suggerendo l’ipotesi che le due fonti di CoQ possano rappresentare due pool separati (Nakamura et al., 1999).
3.2.2 Ruolo biologico
L’ubichinone è parte integrante del sistema mitocondriale di trasporto
degli elettroni, che ha come effetto il movimento di H+ dalla matrice al
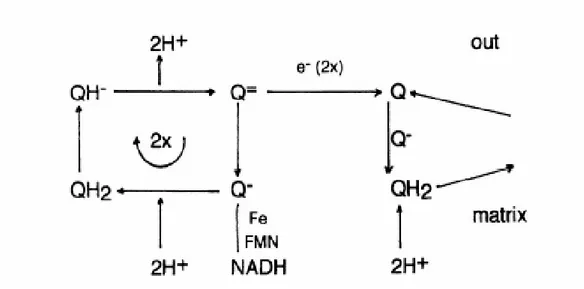
compartimento intermembrana e l’instaurarsi di un gradiente protonico che fornisce l’energia necessaria alla sintesi di ATP. Tale sistema è costituito da quattro complessi proteici orientati (I, II, III, IV) che lavorano in sequenza per trasferire all’ossigeno gli elettroni trasportati dal NAD(P)H e da due trasportatori mobili, che fanno la spola per spostare gli elettroni da un complesso all’altro; uno di questi è l’ubichinone, l’altro è il citocromo c. I gruppi prostetici di ciascun complesso, i citocromi e il CoQ passano ciclicamente dallo stato ridotto allo stato ossidato mano a mano che gli elettroni vengono prelevati e ceduti. L’ubichinone, l’unico trasportatore non strettamente legato a proteine, trasporta sia elettroni che idrogenioni, e può presentare quindi due livelli redox nei quali accetta o rilascia un elettrone ed un idrogenione, o una coppia di elettroni e di idrogenioni (Brandt, 1999).
Fig. .3.2.2.1- Ciclo redox dell’ubiquinone. (tratto da Crane, 2001)
Il CoQ è presente, oltre che nelle membrane plasmatica e mitocondriale, anche in altre membrane interne, come l’apparato del Golgi, il reticolo endoplasmatico e i lisosomi (Ernster, 1993). Si localizza in prossimità degli acidi grassi insaturi e la catena laterale polisoprenoide potrebbe influenzare l’interazione del CoQ con il bilayer fosfolipidico (Katsikas e Quinn, 1982). L’ipotesi che la forma ridotta dell’ubichinone (CoQH2) ed in
particolare del CoQ10 che nell’uomo è la forma dominante, in quanto
donatore di ioni idrogeno possa avere una funzione antiossidante sui lipidi di membrana, fu suggerita per la prima volta nel 1961 da Kaufmann e Garloff (Kaufmann e Garloff, 1961). La caratteristica per cui il CoQ si differenzia dalla vitamina E o dal β-carotene è la capacità di non esaurirsi in seguito allo stress ossidativo (Stocker, Bowry, Frei, 1991), ma di passare
rapidamente dallo stato ossidato a quello ridotto, ad attività antiossidante, durante i normali processi cellulari. Il CoQ sarebbe in grado di ostacolare i fenomeni di perossidazione lipidica attraverso questo meccanismo:
QH2 + LOO. Q.- + H+ +LOOH
in cui LOO. rappresenta un radicale perossile e Q.- il radicale
ubisemichinonico; quest’ultimo può sia essere utilizzato per rigenerare l’ubichinolo:
2Q.- + 2H+ QH 2 + Q
sia funge da scavenger per un altro radicale perossile:
Q.- + H+ + LOO. Q+ LOOH
L’ubichinone e l’intermedio ubisemichinonico possono anche andare incontro a fenomeni di autossidazione con formazione di radicali superossidi ad azione proossidante (Frei, Kim, Ames, 1990). La percentuale di CoQred rispetto all’CoQtot varia negli animali di specie diverse e anche rispetto al tessuto considerato (Ikenoya, et al., 1981).
Il Q10H2 partecipa anche alla rigenerazione dell’ α-tocoferolo (α-TOH) a
partire dal suo radicale (α-TO.) (Kagan et al., 1998);
α-TO. + QH2 α-TOH + QH.
A livello della membrana plasmatica, il CoQ potrebbe anche essere
meccanismo di antiporto transmembrana (Crane et al., 1991). Infatti, questo sistema è inibito quando il trasporto di elettroni, mediato dal CoQ, dal NADH ad accettori esterni alla cellula come ferro o ossigeno, è inefficiente a causa della presenza di inibitori specifici dell’ ubichinone. Il meccanismo attraverso il quale ciò avvenga è sconosciuto.
3.3. Dolicolo
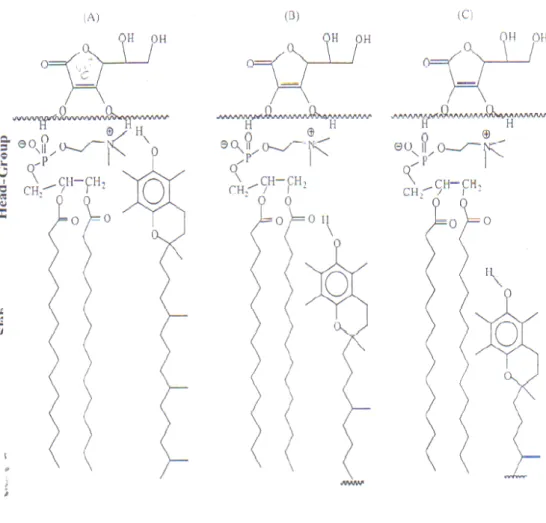
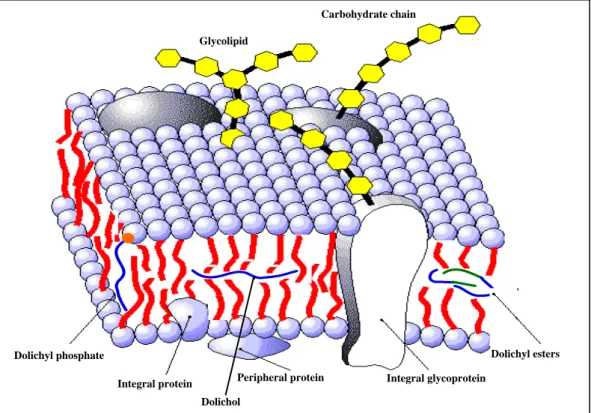
Il dolicolo, fu scoperto da Hemming e colleghi nel 1960 (Burgos et al., 1963) che identificarono questa molecola durante esperimenti di biosintesi dell’ubichinone. Il dolicolo, anche se sarebbe più corretto dire “i dolicoli” in quanto si tratta di una famiglia di derivati contenenti da 16 a 23 unità di isoprene (Chojnacki e Dallner, 1988), è un elemento costitutivo delle membrane cellulari, che si colloca nella porzione centrale del doppio strato lipidico; in particolare, si trova tra le code non polari idrofobiche degli acidi grassi dei fosfolipidi (Bergamini et al. 2004).
Glycolipid
Carbohydrate chain
Dolichol
Dolichyl esters Integral protein Peripheral protein Integral glycoprotein
Dolichyl phosphate
Fig 3.3.1.- Schematizzazione della localizzazione del Dolicolo nel bilayer lipidico.
3.3.1 Struttura e metabolismo
Il dolicolo è un alcol lipofilo formato da più unità isopreniche di cui quella terminale è satura e lega un gruppo idrossilico. L’assetto stereochimico dei
doppi legami lungo la catena è “ω - E2- poli(Z)” e l’unità α-satura possiede
un centro asimmetrico con configurazione S (Adair, 1980). La lunghezza media della catena del dolicolo varia a seconda della specie: in genere il dolicolo dei mammiferi è costituito da 16-23 unità isopreniche e il gruppo idrossilico terminale può esistere sia come gruppo libero (alcol), sia esterificato con acidi grassi e sia fosforilato.
ω - unit E - unit α− unit Z - units CH3 CH2OH n
Il dolicolo è ampiamente distribuito in tutte le cellule e nelle membrane biologiche di tessuti animali (Choinaki e Dallner, 1988; Carroll et al., 1992; Crick e Carroll, 1987). L’assorbimento nella dieta e la distribuzione nel torrente circolatorio sono molto limitati ( Keller e Nellis, 1986).
La sintesi del dolicolo, avviene nel reticolo endoplasmatico, nei perossisomi e nei mitocondri: essa segue la via comune degli isoprenoidi a partire dall’AcetilCoA per arrivare, passando attraverso mevalonato e farnesil pirofosfato, alla formazione di dolicolo, ubichinone, colesterolo e proteine isoprenilate (Beytia,1976: Grunler 1994).
Acetil CoA 3- HMG CoA 3- HMG CoA reduttasi Mevalonato
Isopentenil pirofosfato (IPP) DMAPP
Geranil difosfato (GPP)
FPP sintetasi
Farnesil difosfato (FPP) proteine isoprenilate
trans-
preniltransferasi cis preniltransferasi
squalene sintasi Solanesyl –PP Poli-prenil difosfato Ubiquinone squalene α−saturasi dolicolo colesterolo
Nella sintesi del dolicolo, l’oligomerizzazione delle unità Z-isopreniche è catalizzata da una trasferasi presente sulla membrana del reticolo endoplasmatico e dei perossisomi e il passo finale, cioè l’attacco dell’unità
satura, è mediato da una α−saturasi dotata di stereoselettività.
La sintesi del dolicolo è stata dimostrata sia in vivo che in vitro in molti tessuti (Elmberg et al., 1987) tuttavia il suo metabolismo rimane in buona parte sconosciuto (Bizzarri et al., 2003; Cavallini, 2003).
Infatti, sembra che non esista una via enzimatica precisa per quel che riguarda la degradazione del dolicolo (Choinaki e Dallner 1988); in un recente studio è stata dimostrata la suscettibilità della molecola alla degradazione ossidativa (Nanni et al., 2000). Inoltre, è stato approfondito lo studio della degradazione per via non enzimatica mediata dai radicali
liberi, stimolando epatociti isolati, con un agente xenobiotico, il CCl4 e con
i raggi ultravioletti, UVB. I risultati hanno dimostrato che entrambi i tipi di aggressioni radicaliche causano deplezioni di dolicolo tempo e dose
dipendenti, e che l’aggiunta di CCl4 al sistema di incubazione avrebbe
l’effetto più intenso e rapido (Parentini et al.,2003).
3.3.2 Ruolo biologico
Sebbene il processo di biosintesi del dolicolo sia ampiamente caratterizzato, non è possibile ancora fare considerazioni precise sul suo ruolo biologico. Tuttavia, la larga distribuzione di questa sostanza nelle membrane cellulari e la necessità della forma fosforilata del dolicolo durante lo sviluppo embrionale, suggeriscono che questo composto lipofilo sia un componente essenziale dei tessuti (Carroll et al.,1992).
È stato dimostrato che il dolicolo fosforilato, è coinvolto nella formazione delle glicoproteine, avendo, per struttura, la capacità di legare tra loro segmenti oligosaccaridici ( Leloir, 1977; Hemming, 1977; Tanner et al., 1987; Burda et al., 1999).
Tuttavia, nelle cellule di mammifero, la frazione di dolicolo-fosfato è assai bassa (1-20%) mentre la maggior parte del dolicolo è presente in forma libera o esterificata con acidi grassi, con funzioni che restano ancora non completamente chiarite (Eggens et al., 1983; Choynaki e Dallner, 1988; Marino et al., 1998).
Molti studi hanno dimostrato che il dolicolo ha un’influenza significativa sull’organizzazione delle membrane cellulari (De Ropp et al., 1984; Valtersson et al., 1985); potrebbe inoltre favorire la formazione di “micelle” (strutture globulari in cui le teste polari sono localizzate sulla superficie e le code idrocarburiche sequestrate all’interno. Stryer ,2001) e promuovere la fusione delle vescicole in modo concentrazione-dipendente (Valtersson et al., 1985; Vigo et al., 1984). Il dolicolo può incidere su un ampio numero di funzioni delle membrane stesse, quali la fluidità, la permeabilità, la capacità di fusione, ed anche sulla sensibilità allo stress ossidativo (Jakobsson-Borin et al., 1994). La stretta associazione degli esteri del dolicolo con le lipoproteine del siero, ha fatto ipotizzare un ruolo come mezzo di trasporto vescicolare (Choynaki 1988). È stata dimostrata l’esistenza, in alcune linee cellulari tumorali, di proteine modificate legate covalentemente con il dolicolo, il cui coinvolgimento nella trasformazione o nella progressione neoplastica non è però chiaro (Hjertman e al. 1997). È stato ipotizzato che il dolicolo, possa avere un ruolo determinante nel metabolismo dei radicali liberi, agendo in concerto con i PUFA (polyinsaturated fatty acids) e la vitamina E e che possa quindi essere considerato come uno scavenger lipofilo endogeno. Acidi grassi e dolicolo funzionerebbero come una sola unità che intrappola i radicali liberi
proteggendo così da eventuali danni le proteine di membrana; in particolare, il dolicolo potrebbe avere il ruolo di immagazzinare gli elettroni, dando ai trasportatori e alla vitamina E il tempo di “spegnerli ” indirizzandoli al compartimento idrofilico della cellula.
In questa prospettiva, gli acidi grassi non sarebbero più un punto debole e il dolicolo nel tentativo di proteggerli dalla perossidazione rappresenterebbe egli stesso un bersaglio, per essere risintetizzato poi, a partire dal mevalonato. È probabile che la riduzione dell’efficienza di questo meccanismo configuri un orologio biologico che potrebbe concorrere all’invecchiamento delle citomembrane (Bergamini, 2003) e alla patogenesi di numerose patologie associate all’invecchiamento, in particolare quelle di carattere neurodegenerativo, quali, ad esempio, il morbo di Alzheimer (Koumalova et al., 2003; Perry et al., 2002).