1 SOMMARIO
1. INTRODUZIONE ... 2
2. SCOMPENSO CARDIACO... 9
2.1 - Definizione e inquadramento clinico ... 9
2.2 - Cause, forme e diagnosi d’insufficienza cardiaca... 11
3. Fisiopatologia dello scompenso cardiaco congestizio ... 13
3.1 - Adattamento cardiaco... 15
3.2 – Adattamenti extra-cardiaci ... 16
4. Terapia... 22
4.1 – Terapia Standard dello Scompenso Cardiaco ... 22
5. Effetti collaterali della terapia diuretica... 26
5.3 – Squilibri elettrolitici ... 31
5.4 – Alterazione dell’equilibrio acido-base... 35
5.5 – Ototossicità ... 35
5.6 – Resistenza ai diuretici ... 35
6. SCOPO DELLA TESI... 37
7. MATERIALI E METODI... 38
8. RISULTATI... 43
8.1 – Test T a campioni appaiati... 43
9. CONCLUSIONI ... 48
BIBLIOGRAFIA... 51
1. INTRODUZIONE
Negli Stati Uniti l’insufficienza cardiaca rappresenta uno dei principali problemi di salute pubblica.
Il numero di persone affette da questa patologia si aggira infatti intorno a 5 milioni, mentre ogni anno vengono diagnosticati circa 550.000 nuovi casi. Lo scompenso cardiaco è una condizione morbosa che colpisce principalmente gli anziani (≥ 65 anni) e il ben conosciuto fenomeno dell’invecchiamento della popolazione ne fa aumentare ulteriormente l’incidenza 1.
In Italia, dall’analisi dei ricoveri eseguita nel triennio 2001-2003, emerge che sono circa 3 milioni le persone affette da questa patologia.
Solo nel 2003, il numero dei ricoveri per insufficienza cardiaca ammontava a 211.183, con una degenza media sensibilmente più alta rispetto a quella della casistica trattata. Questa differenza risultava evidente sia nei ricoveri effettuati in regime ordinario, che nei ricoveri per acuti dove, ai 6.7 giorni di degenza media per tutti i ricoveri, si contrapponevano i 9.3 per i pazienti con insufficienza cardiaca.
In particolare, per gli ultrasessantacinquenni, la degenza media si attestava intorno ai 9.4 giorni per i dimessi con insufficienza cardiaca, contro gli 8.6 per tutti gli anziani dimessi da reparti per acuti. La più lunga degenza mediamente rilevata per questo gruppo di pazienti è da attribuire alla peculiare ‘fragilità’ della popolazione con scompenso cardiaco: la popolazione anziana infatti, è spesso affetta da patologie croniche concomitanti, che possono determinare l’allungamento della degenza 2. La mortalità annua nei pazienti con scompenso cardiaco varia a seconda del tipo di popolazione presa in esame, della gravità clinica e della terapia farmacologica attuata, oltre che delle eventuali comorbidità: i dati oscillano
3
dal 2.3% dei pazienti arruolati negli studi clinici controllati, al 19% dei registri ospedalieri 3.
Uno dei principali motivi di ricovero per i pazienti scompensati è la ritenzione di liquidi, che si manifesta con dispnea, edemi agli arti inferiori e ascite nelle forme più gravi. L’approccio terapeutico messo in atto nella pratica clinica è la somministrazione, spesso con dosaggi saturanti, di diuretici dell’ansa, in particolare della Furosemide.
A un iniziale miglioramento della sintomatologia, coincidente con la somministrazione di questo farmaco, segue un peggioramento delle condizioni cliniche del paziente.
La furosemide contribuisce all’attivazione del sistema renina-angiotensina-aldosterone e del sistema simpatico 4, già sollecitati dallo scompenso, portando a una progressiva riduzione della funzionalità renale, all’ulteriore ritenzione di liquidi, alla necessità di un nuovo ricovero ed al peggioramento della prognosi.
Uno studio retrospettivo, eseguito da Cooper et al. su 6797 pazienti dello studio SOLVD (Studies of Left Ventricular Dysfunction), ha evidenziato un aumentato rischio di morte per aritmia associato all’uso dei diuretici non risparmiatori di potassio [rischio relativo (RR)1,85, p=0,0001], rischio che è rimasto significativo anche dopo la correzione per importanti fattori di base (RR 1,37, p=0,009) 5.
Basandosi sullo stesso gruppo di pazienti SOLVD, Domarski et al., hanno dimostrato che il trattamento a base di diuretici dell’ansa o di tiazidici si associa a un incremento sia della mortalità [RR corretto 1,28, con il 95% d’intervallo di confidenza (IC) 1,19-1,49], che dell’ospedalizzazione (RR corretto=1,38, 95%IC 1,11-1,71] 6.
Ancora una volta partendo dallo studio SOLVD, Knight et al. hanno scoperto come l’uso dei diuretici sia un fattore indipendente nel determinare la riduzione della funzionalità renale, mostrando così un terzo meccanismo, oltre ai disturbi elettrolitici ed all’attivazione neurormonale, attraverso cui i diuretici dell’ansa possono peggiorare la prognosi dei pazienti 7.
Una riduzione clinicamente significativa del Filtrato Glomerulare (GFR≤60 mL/min) è un predittore indipendente di tutte le cause di morte. Si pensa che i diuretici dell’ansa possano ridurre il GFR, principalmente attraverso la deplezione di volume e l’attivazione dei sistemi vasopressori e che una loro sospensione migliori la funzionalità renale in pazienti con insufficienza sistolica stabile 8.
I risultati ottenuti da un’analisi retrospettiva eseguita su 7788 pazienti arruolati nello studio DIG (Digitalis Investigation Group), confermano quanto affermato precedentemente.
La terapia diuretica si associa a un aumento di tutte le cause di morte (RR 1,31, 95% IC 1,11-1,55; p=0,002) e di ospedalizzazione per scompenso cardiaco. I dati sono indipendenti da variabili concomitanti, incluse quelle relative alla severità dell’insufficienza cardiaca ed al trattamento 9. Questo studio rappresenta un ulteriore passo avanti se confrontato con le indagini fatte sulla popolazione SOLVD. Primo perché fa uso di una migliore analisi statistica e corregge i risultati sulla base di un maggior numero di variabili, secondo perché >90% dei pazienti sono trattati contemporaneamente anche con ACE inibitori e circa l’80% dei soggetti appartiene alla classe NYHA I e II. Ciò a dimostrazione di come la terapia con ACE inibitori possa non essere sufficiente a proteggere dagli effetti negativi dei diuretici e come gli stessi possano avere effetti avversi anche negli stadi iniziali della malattia 10.
5
Risultati simili sono stati ottenuti da altre analisi multivariate eseguite sulla medesima popolazione DIG:
- Domanski et al., hanno individuato come i soggetti in terapia con diuretici non risparmiatori di potassio (diuretici dell’ansa), vadano incontro a un aumentato rischio di morte per tutte le cause (RR 1,36, 95% IC 1,17-1,59, p<0,0001), di morte per cause cardiovascolari (RR 1,38, 95% IC 1,17-1,63, p=0,0001), per progressione dell’insufficienza cardiaca (RR 1,41, 95% IC 1,06-1,89, p=0,02), di morte cardiaca improvvisa (RR 1,67, 95% IC 1,23-2,27, p=0,001) e di ospedalizzazione (RR 1,68, 95% IC 1,41-1,99, p< 0,0001) rispetto ai pazienti che non fanno terapia diuretica 11;
- Ahmed et al., con la loro analisi multivariata, attribuiscono all’uso di diuretici un significativo aumento del rischio di morte per cause cardiovascolari [hazard ratio(HR)=1,50; 95% IC 1,15-1,96, p=0,003], e di ospedalizzazione per aggravamento dell’insufficienza cardiaca (HR=1,48, 95% IC 1,13-1,94, p=0,005). In questo caso sono stati presi in considerazione solo pazienti con età ≥ 65 anni 12.
Due studi eseguiti nel 2004 evidenziano l’azione che i diuretici dell’ansa hanno direttamente sul cuore:
- Lopez et al., hanno preso come campione pazienti cronicamente scompensati appartenenti alle classi II, III e IV della NYHA che, oltre a fare la terapia standard per lo scompenso cardiaco, hanno ricevuto per otto mesi una terapia diuretica giornaliera a base di furosemide (20-40 mg/die) o di torasemide (20-40 mg/die). Mentre nel gruppo trattato con torasemide la quantità di collageno rilevata dalla biopsia settale ed il peptide sierico del pro collagene tipo I diminuiva (p<0,01), nel gruppo
che riceveva furosemide il quadro rimaneva immutato, senza miglioramento della fibrosi cardiaca 13.
- McCurley et al., somministrando furosemide e placebo in maniera randomizzata a 32 maialini Yorkshire, li sottoponevano successivamente ad uno stimolo tachicardico al fine di indurre insufficienza sistolica. Il gruppo a cui era stata somministrata la furosemide andava più rapidamente incontro a disfunzione ventricolare sinistra (21,4 ± 3,2 giorni contro i 35,1 ± 5,1 giorni degli animali facenti placebo; p=0,038). Inoltre, nel gruppo trattato, al quattordicesimo giorno, i livelli di aldosterone erano significativamente più alti rispetto al placebo (43.0 ± 11,8 ng/dl contro 17,6 ± 4,5 ng/dl; p<0,05). Quindi nei miaialini tachicardici riceventi furosemide, si verificava una progressione significativa dell’insufficienza cardiaca ed un più rapido instaurarsi della disfunzione ventricolare sinistra 14.
Altri studi si sono concentrati su un possibile ruolo del dosaggio della furosemide come fattore prognostico indipendente. La somministrazione di alte dosi di diuretico si associa a un incremento della mortalità nei pazienti con insufficienza cardiaca avanzata 15,16, nei pazienti anziani e nei pazienti ospedalizzati per scompenso cardiaco acuto 17. Queste relazioni restano statisticamente significative anche dopo essere state associate ad altre variabili all’interno di analisi multivariate. Alte dosi di furosemide possono quindi avere un ruolo nei modesti risultati ottenuti con i pazienti scompensati 10. Lo studio condotto da Mielniczuk et al., suggerisce però che la dose di furosemide debba essere considerata non come causa, ma come marker d’instabilità clinica 18. Una recente review 10 analizza tutti questi risultati proprio con lo scopo di individuare un trattamento più
7
efficace e meno lesivo dell’insufficienza cardiaca. La Tabella 1 mostra alcuni degli studi osservazionali sopra citati.
Tab.119
Solo pochi studi si sono concentrati sul cambiamento dei parametri clinici successivi alla sospensione della terapia con diuretici dell’ansa. Van Kraaij et al., hanno osservato un gruppo di pazienti anziani con insufficienza cardiaca senza disfunzione ventricolare sinistra, individuando, a distanza di tre mesi dalla sospensione del farmaco, una persistente riduzione dell’attivazione del sistema renina angiotensina (-1,61 ±0,71 nmol/l/h, p<0,05) 20. Un altro studio invece, ha preso in considerazione 26 pazienti affetti da scompenso cardiaco stabilizzato con disfunzione sistolica. In questo caso, la sospensione della furosemide era associata a un miglioramento significativo dei parametri di funzionalità renale (urea p=0,014; creatinina p=0,013), un miglioramento del metabolismo del glucosio ed una riduzione dell’attività reninica plasmatica (p=0,026), mentre il peptide natriuretico atriale aumentava in maniera significativa (p=0,004)21.
La parte sperimentale di questa tesi è dedicata allo studio della variazione dei parametri clinici conseguente la sospensione della terapia diuretica in una popolazione di pazienti anziani affetti da scompenso cardiaco. L’invecchiamento coincide con un deterioramento della funzione renale, la
Observational Studies ofDiuretics and Outcomes in Heart Failure
Study Population N Comparison Endpoint Risi.: 9S~.CI
SOLVD19 LV dysfunction with or without HF 6791 Oral diuretics vs. none Morta1ity 1.31 1.08-1.13
DIG21 chronic HF 2782 Oral diuretics vs. none Mortality 1.31 1.11-1.55
Butle,22 ADHF 382 Dose ofIV loop diuretics worsening renal function(f.O.3mgldl) 1.04 per 20 mg increment of 1.004--1.076 furosemide
ESCAPE23 Advanced HF inpts 395 Dose ofIV loop diuretics Morta1ity 1.15 per doubling of dose 1.025--1.28
Eshaghian24 Advanced HF outpts 1354 Dose of oral diuretics Morta1ity 3.4 per quartile of dose 2.4-4.7
Neurerg25 chronic HF 1153 Diuretic oral dose(O80 mg Morta1ity 1.31 for dose above median nOIprovided, p=O.OO4
furesemide)
Philbin26 ADHF 1150 # ofIV diuretic doses In-hospital mortality 1.11 per #of doses 1.06-1.17
compromissione dell’attitudine a concentrare le urine e quindi alla conservazione di volume.
Il mantenimento della funzione renale necessariamente richiede incremento delle resistenze renali, pre-requisito per la generazione del volume idrostatico associato alla generazione del GFR.
L’impiego di diuretici dell’ansa, ancorché necessario, insiste sia sui meccanismi di diluizione che di concentrazione urinaria e può generare un bilancio negativo di acqua che, riducendo eccessivamente il volume intravascolare, innesca un circuito perverso che insiste sulla possibilità renale di generare un Filtrato Glomerulare adeguato in presenza di resistenze vascolari aumentate.
Quindi il nostro studio rappresenta un ulteriore approfondimento sugli effetti negativi della terapia diuretica quando, applicata in quantità saturanti in una popolazione di pazienti già prona alla disidratazione, rende di fatto impraticabile la terapia dello scompenso in tutti i suoi effetti favorevoli, ivi compreso l’effetto diuretico.
9
2. SCOMPENSO CARDIACO
2.1 - Definizione e inquadramento clinico
Lo scompenso cardiaco è una sindrome eterogenea caratterizzata da un’anomalia della struttura o della funzione miocardica responsabile dell’incapacità, da parte del cuore, di pompare sangue o di riempirsi a una velocità adeguata alle esigenze metaboliche dei tessuti 22.
La gravità della patologia è descritta comunemente secondo i criteri sviluppati dalla New York Heart Association. Ogni paziente viene assegnato ad una delle quattro classi funzionali che sono individuate sulla base del grado di sforzo fisico necessario per evocare la sintomatologia (Tab 1) 1.
- Classe I – Assenza di limitazioni all’esercizio fisico. La normale attività fisica non causa eccessiva fatica, palpitazioni, dispnea o dolore anginoso.
- Classe II – Lievi limitazioni all’esercizio fisico. I pazienti sono asintomatici a riposo, ma una normale attività fisica provoca l’insorgenza di fatica, palpitazioni, dispnea o dolore toracico.
- Classe III – Marcata limitazione all’esercizio fisico. I pazienti sono asintomatici a riposo, ma anche una lieve attività fisica determina affaticamento, palpitazioni, dispnea o dolore anginoso.
- Classe IV – Incapacità completa all’esercizio fisico. I sintomi dello scompenso cardiaco o della sindrome anginosa sono presenti anche a riposo e qualunque tipo di attività fisica intrapresa intensifica la sintomatologia già presente 23.
Tuttavia questa classificazione funzionale riflette una valutazione medica soggettiva e variabile, che è utile per confrontare le condizioni cliniche di un
paziente, o di gruppi di pazienti, in tempi differenti.
La stadiazione dell’insufficienza cardiaca rappresenta una metodica oggettiva che permette di identificare le varie fasi di progressione della malattia, associando ad ogni stadio il trattamento più adeguato (Tab. 2) 1,23.
Tabella 2. Stadi dell’Insufficienza Cardiaca 24
Stadio Descrizione Esempi
A Pazienti ad alto rischio di sviluppare Insufficienza Cardiaca a causa della presenza di una condizione che è strettamente associata con lo sviluppo della malattia. Questi pazienti non hanno anormalità strutturali o funzionali identificate del pericardio, del miocardio, o delle valvole cardiache e non hanno mai mostrato segni o sintomi d’insufficienza cardiaca.
Ipertensione sistemica; malattia arterie coronarie; diabete mellito; storia di cardiotossicità ai farmaci o di abuso di alcohol; storia personale di febbre reumatica; storia familiare di cardiomiopatia.
B Pazienti che hanno sviluppato una malattia strutturale cardiaca che è fortemente associata con lo sviluppo di insufficienza cardiaca, ma che non ha mai mostrato segni o sintomi di insufficienza cardiaca.
Fibrosi o ipertrofia ventricolare sinistra; dilatazione o ipocontrattilità del ventricolo sinistro; malattia cardiaca valvolare asintomatica; precedente infarto del miocardio. C Pazienti che hanno avuto, o
attualmente presentano, sintomi di insufficienza cardiaca associati con sottostante malattia strutturale cardiaca.
Dispnea o fatica dovuti a disfunzione ventricolare sinistra; pazienti asintomatici che però sono sotto trattamento per precedenti sintomi d’insufficienza cardiaca.
D Pazienti con una malattia cardiaca strutturale avanzata e con sintomi marcati d’insufficienza cardiaca a riposo nonostante la somministrazione della terapia medica massimale e che richiedono un intervento specializzato.
Pazienti che sono frequentemente ricoverati per insufficienza cardiaca o che non possono essere dimessi tranquillamente dall’ospedale; pazienti ospedalizzati che aspettano il trapianto cardiaco; pazienti che a casa ricevono un continuo supporto endovenoso per ridurre i sintomi o che sono supportati da un dispositivo meccanico di circolazione assistita; pazienti localizzati in un ospizio organizzato per il trattamento dell’insufficienza cardiaca.
11
2.2 - Cause, forme e diagnosi d’insufficienza cardiaca
La sindrome clinica che caratterizza l’insufficienza cardiaca può derivare da alterazioni del pericardio, del miocardio, dell’endocardio o dei grandi vasi, ma il quadro clinico più diffuso deriva dal malfunzionamento del ventricolo sinistro. E’ ampio lo spettro di disfunzioni ventricolari associato a questa patologia: si va da un normale volume ventricolare con conservata frazione d’eiezione, a una dilatazione del ventricolo sinistro con frazione d’eiezione marcatamente ridotta 1. In particolare, in base all’anomalia anatomica si distinguono due tipi di scompenso:
- il sistolico è caratterizzato da ipertrofia eccentrica e progressiva dilatazione del ventricolo sinistro causata o da miocardiopatia dilatativa o da shock cardiogeno a seguito di un infarto miocardico esteso 25. L’incapacità di contrarsi normalmente e pompare sangue determina l’inadeguatezza della gittata cardiaca e la conseguente ritenzione idrosalina 26. Il paziente lamenta debolezza, affaticamento, ridotta tolleranza allo sforzo e altri sintomi da ipoperfusione 22.
- il diastolico, indotto da una miocardiopatia restrittiva o ipertrofica, presenta ipertrofia concentrica ventricolare che altera il normale rilasciamento. Il quadro clinico è correlato all’innalzamento della pressione di riempimento del ventricolo sinistro e/o destro con conseguente congestione polmonare o venosa periferica 26.
- Nella maggioranza dei pazienti coesistono anomalie sia della contrazione che del rilasciamento.
Come sostenuto da Hunt et al. nell’ultimo aggiornamento delle linee guida 1, l’insufficienza cardiaca è una sindrome caratterizzata da una storia clinica costellata da sintomi specifici (dispnea e astenia) e da segni evidenti
(edemi e rantoli). Non esiste un test diagnostico per lo scompenso cardiaco perché la sua diagnosi è principalmente clinica e si basa su un attento studio anamnestico e un accurato esame obiettivo. La presenza di almeno due criteri maggiori di Framingham (Tab. 3), o di uno maggiore e due minori, permette di fare diagnosi.
Tabella 3. 26
Criteri di Framingham per l'insufficienza cardiaca
Criteri maggiori
Dispnea parossistica notturna Distensione delle vene del collo Rantoli
Cardiomegalia radiografica Edema polmonare acuto 53 galoppo
Aumento della pressione venosa centrale >16 cm H,O Tempo di circolo ~25 sec
Reflusso epatogiugulare
Edema polmonare, congestione viscerale o cardiomegalia all' autopsia Perdita di peso ~4,5 kg in 5 giorni in risposta al trattamento dell'insufficienza cardiaca
Criteri minori
Edema bilaterale delle caviglie Tosse notturna
Dispnea per attività ordinarie Epatomegalia
Versamento pleurico
Diminuzione della capacità vitale di un terzo dal valore massimo registrato Tachicardia (frequenza ~120 battiti/min)
Da Ho KL, Pinsky IL, Kannel WB, Levy D: The epidemiology of heart failure: The Framingham Study. J Am Coll Cardiol 22(Suppl A):6A, 1993.
13
3.
Fisiopatologia
dello
scompenso
cardiaco
congestizio
Si definisce edema un turgore palpabile provocato dall’espansione del liquido interstiziale. Sono due i passaggi fondamentali nella formazione dell’edema :
1. l’alterazione dell’emodinamica capillare che favorisce il movimento dei liquidi dallo spazio vascolare all’interstizio;
2. la ritenzione renale di acqua e di sodio.
La permeabilità capillare è regolata dalla Legge di Starling, relazione che stabilisce lo spostamento dei liquidi sulla base della differenza di pressione idrostatica e oncotica tra due compartimenti. Perché si sviluppi edema è necessaria l’alterazione di una o di più forze di Starling, in modo da favorire la filtrazione netta dai vasi all’interstizio. Questo si verifica nello scompenso cardiaco in seguito alla ritenzione renale di acqua e sodio che comporta un incremento della pressione idrostatica capillare 27.
La ritenzione di liquidi da parte del rene negli stati edematosi è il risultato o di una sua incapacità a eliminare sodio e acqua introdotti con la dieta (a causa di una patologia renale) o, più spesso, di una risposta compensatoria appropriata alla deplezione di volume 27.
Per volume circolante effettivo s’intende quella parte del liquido extracellulare che è presente nel sistema arterioso e che effettivamente perfonde i tessuti. Più correttamente, dal punto di vista fisiologico, è definito come la pressione di perfusione dei barocettori arteriosi del seno carotideo e dell’arteriola glomerulare afferente 28. Nella maggioranza dei casi, è direttamente proporzionale alla gittata cardiaca, quindi quando questa si
riduce, come accade nello scompenso cardiaco, il rene tenta di riportare alla norma la volemia trattenendo sodio e acqua 27.
L’edema negli scompensi cardiaci a diversa eziologia è sempre legato a un aumento della pressione venosa che provoca un incremento parallelo della pressione idrostatica capillare. Nonostante la patogenesi sia simile, le zone di accumulo di liquido edematoso sono variabili e dipendono dalla natura della cardiopatia.
1. Le coronaropatie o l’ipertensione arteriosa preferibilmente provocano una diminuzione della funzionalità ventricolare sinistra. Ne risulta che i pazienti affetti da queste patologie presentano tipicamente edema polmonare, ma non periferico.
2. Il cuore polmonare, invece, è inizialmente associato a scompenso cardiaco destro puro e questo provoca edemi marcati agli arti inferiori e, a volte, ascite.
3. Le cardiomiopatie che tendono a coinvolgere in eguale misura sia il ventricolo destro che il sinistro, spesso provocano l’insorgenza simultanea di edema polmonare e periferico.
Nell’edema polmonare acuto dovuto a infarto miocardico o a ischemia, la disfunzione del ventricolo sinistro provoca un aumento della pressione telediastolica ventricolare e atriale sinistra che viene trasmesso per via retrograda, attraverso le vene polmonari, ai capillari polmonari (ipotesi dello
scompenso cardiaco retrogrado). Qui la pressione deve superare i 18-20
mmHg (valori normali 5-12 mmHg) prima che si verifichi uno stravaso di liquidi nell’interstizio e quindi la formazione di edema polmonare 27.
Nello scompenso cardiaco cronico la patogenesi della formazione dell’edema differisce dalla precedente. L’aumento della pressione capillare in questo caso è il risultato dell’espansione del volume plasmatico e non
15
l’effetto diretto delle alterazioni patologiche dell’organo cardiaco. Questa teoria, detta ipotesi anterograda dello scompenso cardiaco, individua nella riduzione della gittata cardiaca l’evento primario 27.
3.1 - Adattamento cardiaco
L’evento scatenante acuto (infarto transmurale del miocardio) o cronico (ipertensione arteriosa o valvulopatie), determina una perdita massiva di miociti associata alla sostituzione fibrotica del tessuto perso 29. Per garantire un’adeguata frazione d’eiezione, si attivano numerosi meccanismi di compenso:
– l’adattamento a breve termine, vede il ventricolo insufficiente mantenere una normale funzionalità attraverso il meccanismo di
Frank-Starling. La legge di Frank-Starling afferma che l’aumento della
forza contrattile (contrattilità) è proporzionale all’allungamento dei sarcomeri (fino a 2,2 mm). Poiché la contrattilità ventricolare dipende dalla distensione delle fibre (provocata dal volume sanguigno che passa dall’atrio al ventricolo) e dal loro successivo accorciamento, un aumento del volume di carico (cioè del precarico) permetterà di ottenere l’effetto cercato. Lo smisurato aumento della pressione di riempimento pone un limite all’efficacia a lungo termine di questo meccanismo 23;
– l’adattamento a lungo termine, è rappresentato dal rimodellamento
del miocardio 30. I ventricoli reagiscono a un sovraccarico emodinamico cronico mediante lo sviluppo d’ipertrofia. In presenza d’insufficienza valvolare, ad esempio, il ventricolo è chiamato a pompare una gittata cardiaca elevata per periodi prolungati e sviluppa
trattata, determinano invece un sovraccarico di pressione e un’ipertrofia concentrica. In entrambi i casi, la tensione parietale è inizialmente mantenuta a livelli normali e la funzione cardiaca può rimanere stabile per molti anni. Tuttavia, alla fine, la funzione miocardica può deteriorarsi, con conseguente riduzione della frazione d’eiezione e comparsa del quadro clinico dell’insufficienza cardiaca. A questo punto il ventricolo va incontro a rimodellamento, si dilata, diventa sferico e incrementa lo stiramento su ciascuna unità del miocardio. Ciò comporta un’ulteriore dilatazione e un aumentato stress emodinamico sulla parete, instaurando un circuito perverso che si autoalimenta 22.
3.2 – Adattamenti extra-cardiaci
3.2.1 - Risposte emodinamiche alla riduzione della gittata cardiaca. Una caduta della gittata cardiaca determina una riduzione della pressione sistemica in base alla relazione esistente tra pressione arteriosa media, gittata cardiaca e resistenze vascolari sistemiche:
Pressione arteriosa media = gittata cardiaca x resistenze vascolari periferiche
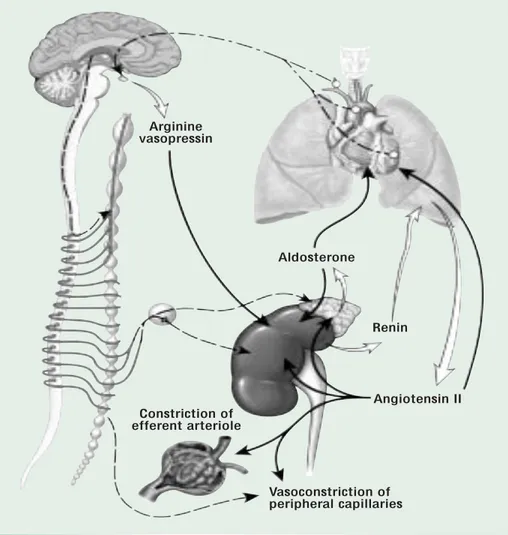
I barocettori del seno carotideo e dall’arco aortico percepiscono la modificazione emodinamica e attivano il sistema nervoso simpatico che determina vasocostrizione arteriosa nei distretti periferici (cute, circolazione splacnica e renale) e mantiene la perfusione cerebrale e coronarica. Il ripristino della pressione arteriosa è ottenuto grazie all’aumento del ritorno venoso (mediato in parte dalla vasocostrizione attiva), della contrattilità e della frequenza cardiaca (variazioni che nell’insieme contribuiscono ad aumentare la gittata cardiaca) e all’aumento delle resistenze vascolari, dovuto sia all’azione diretta del simpatico che all’incremento della secrezione di renina da parte del rene 31. L’arteriola afferente di ogni
17
glomerulo renale possiede cellule specializzate, chiamate cellule juxtaglomerulari, in grado di sintetizzare la pro-renina, precursore della renina, convertita successivamente nell’enzima proteolitico attivo. L’ipoperfusione renale e l’aumento dell’attività nervosa simpatica sono gli stimoli fisiologici principali che inducono la secrezione di renina. L’enzima converte l’angiotensinogeno in angiotensina I, trasformata in seguito in angiotensina II ad opera dell’enzima di conversione ACE (Angiotensin Converting Enzym) presente nei polmoni, nella membrana luminale delle cellule endoteliali dei vasi e nel glomerulo stesso. L’angiotensina II ha essenzialmente due effetti: la ritenzione di sodio e acqua e la vasocostrizione.
La vasocostrizione sistemica è ottenuta grazie all’azione diretta del neurormone sulla muscolatura liscia dei vasi e si va a sommare all’effetto del sistema simpatico.
La ritenzione di sodio e acqua a livello renale avviene attraverso due meccanismi: uno stimolo diretto dell’angiotensina II, che promuove il riassorbimento di sodio nel tubulo prossimale e l’aumento della secrezione surrenalica di aldosterone che incrementa il trasporto di sodio nel tubulo collettore corticale 32. Con il progredire della malattia cardiaca il riassorbimento prossimale di sodio aumenta per l’influenza dell’angiotensina II e per la concomitante azione del sistema simpatico. Un elevato riassorbimento al livello dell’ansa di Henle, è di solito un evento più tardivo che compare con la riduzione della pressione arteriosa sistemica. Inoltre, la riduzione del flusso ematico papillare, dovuto all’azione del neurormone, favorisce indirettamente il riassorbimento passivo di sodio nella branca ascendente sottile, aumentando la tonicità della midollare 27. In parallelo all’attivazione del sistema simpatico e del sistema
renina-angiotensina, si ha una maggiore liberazione, da parte dell’ipofisi posteriore, di arginina-vasopressina (AVP o ormone antidiuretico ADH) 33. L’effetto principale dell’ADH al livello renale è l’aumento della permeabilità all’acqua dei tubuli collettori della corticale e della midollare renale. Gli effetti antidiuretici sono mediati dal recettore V2 mentre, attraverso il V1 l’ormone aumenta le resistenze vascolari 34. Il rilascio dell’arginina-vasopressina è notevolmente incrementato dalla deplezione del volume circolante effettivo e dall’aumento dell’osmolarità del liquido extracellulare (via osmotica) 33.
Le variazioni neurormonali che s’instaurano prima che compaia insufficienza cardiaca conclamata hanno inizialmente effetti positivi, perché cercano di normalizzare la gittata cardiaca e la pressione arteriosa (Fig.1)27. L’espansione del volume intravascolare è la conseguenza dell’ipersecrezione di ormoni pressori e antidiuretici (noradrenalina, angiotensina II, vasopressina, aldosterone) per compensare la ridotta funzione ventricolare 35,36,37.
I meccanismi di contro-regolazione che l’organismo mette in atto per bilanciare l’eccessiva e persistente vasocostrizione e la ritenzione idrosalina sono:
- i peptidi natriuretici, sostanze vasoattive prodotte e liberate dalle cellule muscolari degli atri, dei ventricoli e della parete dei vasi, in risposta a uno stiramento della parete delle cavità cardiache o a un aumento dello stress longitudinale esercitato sui vasi 33.
19
Fig.1 38
C L E V E L A N D C L I N I C J O U R N A L O F M E D I C I N E V O L U M E 7 3 • S U P P L E M E N T 2 J U N E 2 0 0 6 S3 water by the kidney in heart failure has been
debated for years. An intrinsically normal kid-ney continues to retain sodium and water, despite expansion of extracellular fluid volume in heart failure, which implies that it must be responding to “inadequate” signals from the volume regulatory system. This suggests that some sensor in the vascular tree is “underfilled” or that some process for detecting body fluid appropriateness fails to perceive the elevated circulating volume. This arterial underfilling is picked up on by baroreceptors in the left ven-tricle, the aortic arch, the carotid sinus, and the renal afferent arterioles. Decreased activation of these receptors during the evolution of arte-rial underfilling leads to compensatory neuro-humoral responses, which include stimulation of the sympathetic nervous system, activation of the RAAS, and nonosmotic release of vaso-pressin. These compensatory responses pre-serve circulatory integrity by increasing periph-eral and renal vascular resistance and by foster-ing renal sodium and water retention.4
Importance of GFR
The level of renal function is an important determinant of sodium and water excretion. The basis for sodium and water retention when heart failure first manifests relates to ele-ments other than a reduced glomerular filtra-tion rate (GFR). Over time, however, a gradu-ally falling GFR, either in association with heart failure progression or relating to medica-tion effects on the level of renal funcmedica-tion, becomes more critical in sodium and water retention. Although serum creatinine values have often been offered as a good gauge of renal function, in most cases “true” renal func-tion is appreciably lower than the “eyeball” estimate derived from a specific serum creati-nine value.5 In the heart failure patient with
progressive renal disease, diuretics generally become less effective in that the filtered load of sodium drops in parallel with a falling GFR.6
In other instances, transient changes in GFR, provoked by hemodynamic change, can attenuate the natriuretic response to a loop diuretic.7,8For example, the reduction in blood
pressure that occurs with ACE inhibitor ther-apy can reduce renal perfusion pressure (and GFR) to such a degree that diuretic action is significantly weakened.7 Also, diuretic
infu-sions may diminish the early, volume-inde-pendent activation of the RAAS triggered by the rapid increase in plasma loop diuretic con-centration after bolus loop diuretic therapy.
Interestingly, activation of the RAAS by loop diuretic therapy is accompanied by dele-terious hemodynamic effects.8 Under these
conditions, cardiac output, renal blood flow, and GFR can decrease, diminishing tubular delivery of the diuretic in the process.
! RENIN-ANGIOTENSIN-ALDOSTERONE AXIS
The kidney contains all elements of the RAAS and is functionally independent in its
genera-FIGURE 1. Efferent pathways in the sympathetic nervous system are activated in heart failure. Sympathetic nervous system activity contributes to peripheral and renal vasoconstriction and to sodium and water retention. Activation of renal sympathetic nerves leads to angiotensin II release, stimulating the
renin-angiotensin-aldosterone system. Sympathetic stimulation also prompts release of arginine vasopressin, excess levels of which lead to water retention and hypona-tremia. Angiotensin II acts as a potent vasoconstrictor, stimulates aldosterone release from the adrenal gland, and promotes renal tubule sodium reabsorption. Aldosterone increases reabsorption of sodium in the collecting duct.
Arginine vasopressin Aldosterone Renin Angiotensin II Vasoconstriction of peripheral capillaries Constriction of efferent arteriole
La loro azione è la vasodilatazione diretta, che riduce la pressione arteriosa sistemica, e l’aumento dell’escrezione urinaria di sodio e di acqua. Questi ormoni riducono la secrezione basale di renina, contrastano gli aumenti del riassorbimento di sodio nel tubulo prossimale e il rilascio di aldosterone, indotti dall’angiotensina II, e diminuiscono la risposta del tubulo collettore all’ADH 34.
- le prostaglandine, che derivano dall’acido arachidonico e sono prodotte in diversi siti al livello renale.
La loro sintesi è incrementata dalla presenza di angiotensina II, noradrenalina, vasopressina ed endotelina e hanno un’azione vasodilatante che minimizza l’ischemia renale indotta dalla vasocostrizione e dall’ipovolemia 34.
A lungo termine la risposta neurormonale è chiaramente dannosa 27. Mentre in acuto la vasocostrizione è utile per la ridistribuzione del flusso ematico agli organi nobili (cuore e cervello), alla lunga peggiora lo scompenso con un’ulteriore riduzione della portata cardiaca e la consegue necessità di una maggiore vasocostrizione per la ridistribuzione del flusso insufficiente. Anche la ritenzione di sodio e acqua operata dal rene rappresenta un meccanismo di compenso e consente un’espansione del compartimento extracellulare dell’organismo aumentando il volume ematico e interstiziale. L’aumento del volume sanguigno migliora il riempimento ventricolare e la prestazione cardiaca, fino al raggiungimento del massimo stiramento efficace delle fibre miocardiche (2,2 µm). Oltre questo limite, raggiunto rapidamente nei pazienti scompensati a causa del rimodellamento cardiaco, il meccanismo di Frank-Starling non è più efficace e un’ulteriore espansione del volume ematico risulta dannosa33, provocando l’insorgenza di edema nel polmone e nei muscoli scheletrici e
21
contribuendo alla dispnea, all’intolleranza all’esercizio e all’astenia 35,36,37. 3.2.2 - Risposta endoteliale.
Le cellule endoteliali hanno come sistema regolatore locale l’endotelina, potente vasocostrittore che si trova in alte concentrazioni nei pazienti scompensati 39,40,41. Il suo scopo è mantenere elevati i valori pressori in risposta a un’ipoperfusione 33,30.
3.2.3 - Citochine infiammatorie.
I livelli circolanti di TNF-α e di IL-6 aumentano nei pazienti con insufficienza cardiaca 42,43,44,45. Il miocardio insufficiente, di per sé, può essere una fonte di citochine infiammatorie, presenti a concentrazioni localmente elevate 46. Nelle cellule miocardiche in cultura è stato dimostrato che il TNF-α e l’IL-1β stimolano l’ipertrofia e l’apoptosi, mediata in parte dall’azione dell’ossido nitrico (NO) 30.
3.2.4 - NO e Stress Ossidativo.
In corso di scompenso cardiaco le citochine infiammatorie stimolano la forma inducibile della Ossido Nitrico Sintetasi (NOS) in grado di produrre una grande quantità di ossido nitrico (NO) a livello cardiaco. L’NO sembra essere citotossico e pro-apoptotico per il cuore 23. L’aumento delle citochine infiammatorie sistemiche 43, l’azione dei neurormoni e la tensione meccanica sul miocardio, indotta dal sistema simpatico e dall’endotelina, inducono la produzione di radicali liberi dell’ossigeno, sostanze in grado di provocare ipertrofia miocardica e apoptosi 30.
4. Terapia
4.1 – Terapia Standard dello Scompenso Cardiaco
Le più recenti linee guida raccomandano l’applicazione di un trattamento farmacologico atto a ridurre sia la sintomatologia dello scompenso cardiaco che la morbidità e la mortalità 47. Il trattamento standard è centrato sull’inibizione dei meccanismi di compenso extracardiaci che nell’immediato possono portare a un beneficio, ma a lungo termine sono solo dannosi determinando la progressione della patologia. Gli ACE inibitori o gli
antagonisti del recettore dell’angiotensina II e i β-bloccanti, rappresentano i
capisaldi nella gestione di tutti gli stadi dello scompenso cardiaco. A questi si affiancano gli antagonisti dell’aldosterone, la digitale, i nitrati e i diuretici, considerati fondamentali per la gestione della ritenzione idrosalina.
La figura 2 riassume gli approcci terapeutici nei diversi stadi della patologia. 4.1.1 - Inibitori del Sistema Renina-Angiotensina-Aldosterone 1
Questa categoria di farmaci è composta da tre diversi tipi di molecole in grado di agire in maniera differente:
- gli ACE Inibitori disattivano l’enzima che trasforma l’angiotensina I in angiotensina II. Sono indicati in tutte le fasi di questa patologia in associazione ai β-bloccanti;
- gli ARBs (antagonisti del recettore dell’angiotensina) hanno un’azione sinergica agli Ace inibitori, neutralizzando l’azione dell’angiotensina circolante prodotta da altre vie enzimatiche, e mantengono il loro effetto benefico riducendone gli effetti avversi. Si somministrano in associazione o in sostituzione degli ACE inibitori quando questi non sono tollerati dai pazienti;
23
dell’Aldosterone. Sopprime l’aldosterone non eliminato dagli inibitori del
sistema renina-angiotensina, riducendo gli effetti lesivi che l’ormone esercita sul cuore 1. Lo studio RALES (Randomised Aldactone Evaluation) ha scoperto che questo farmaco, se associato ad ACE inibitori, riduce i sintomi d’insufficienza cardiaca, le ospedalizzazioni e la mortalità dei pazienti affetti da scompenso cardiaco moderato-severo e blocca l’evolversi dell’insufficienza renale 48,49,50.
4.1.2 - β-bloccanti 1
Inibiscono gli effetti deleteri del sistema nervoso simpatico bloccando i recettori β1, β2 e α1. Il Bisoprololo, il Metoprololo e il Carvedilolo, riducono il rischio di morte nei pazienti con insufficienza cardiaca cronica e sono somministrati soprattutto nei pazienti appartenenti allo Stadio C dell’insufficienza cardiaca.
4.1.3 - Digitale 1
La Digossina, il digitalico maggiormente utilizzato nella pratica clinica, è un farmaco inotropo positivo che agisce anche a livello renale. Aumentando la concentrazione del sodio nei tubuli distali attraverso il suo mancato riassorbimento nei tubuli prossimali, riduce lo stimolo alla secrezione di renina dell’apparato juxtaglomerulare. Nello scompenso lieve-moderato migliora la sintomatologia, la qualità della vita e la tolleranza all’esercizio. 4.1.4 - Idralazine e Isosorbide Denitrato 1
Sono vasodilatatori arteriolari e venosi. I risultati ottenuti dagli studi su questi farmaci sono contrastanti e non permettono di individuare una chiara linea di utilizzo. L’associazione di questa terapia all’approccio standard con ACE inibitori e/o β-bloccanti si è rivelato utile solamente nella popolazione americana di discendenza africana. In questo gruppo si consiglia quindi la suddetta combinazione se, nonostante la terapia farmacologia ottimale,
continuano a essere presenti i sintomi. 4.1.5 - Diuretici
I diuretici sono i farmaci di prima linea utilizzati per la gestione della ritenzione di liquidi. Inibiscono il riassorbimento del sodio o del cloro in specifici punti del tubulo renale e proprio sulla base del sito d’azione si distinguono in:
- diuretici dell’ansa, Bumetanide, Furosemide e Torsemide, che agiscono sull’Ansa di Henle e incrementano l’escrezione del sodio filtrato del 20-25%, aumentano la clearance dell’acqua libera e mantengono la loro efficacia fino a che il rene non è funzionalmente compromesso;
- tiazidici (Metolazone) e diuretici risparmiatori di potassio (Spironolattone), che agiscono sulla porzione distale del tubulo. I primi aumentano l’escrezione frazionale del sodio solo del 5-10%, riducono la clearance dell’acqua libera e perdono la loro efficacia con la progressiva riduzione della funzione renale. Lo Spironolattone è l’antagonista del recettore dell’aldosterone.
Proprio per il loro effetto prolungato, i diuretici dell’ansa sono i composti maggiormente utilizzati nella terapia dello scompenso cardiaco e dovrebbero essere prescritti in tutti quei pazienti in cui c’è evidenza di ritenzione idrosalina, associati agli ACE inibitori e ai β- bloccanti 1. Un uso inappropriato di questi agenti può portare a riduzione del volume effettivo circolante, con il maggior rischio che i farmaci associati sviluppino i propri effetti collaterali (ipotensione e insufficienza renale), e con il conseguente potenziamento proprio di quei processi fisiopatologici che l’approccio terapeutico vorrebbe controllare, rendendo inefficace la terapia e ingestibile la patologia.
25 Fig.2 1
noncardiac disorders or behaviors that might cause or accelerate the development or progression of HF. (Level of Evidence: C) 2. A careful history of current and past use of alcohol, illicit drugs,
current or past standard or “alternative therapies,” and chemo-therapy drugs should be obtained from patients presenting with HF. (Level of Evidence: C)
3. In patients presenting with HF, initial assessment should be made of the patient’s ability to perform routine and desired activities of daily living. (Level of Evidence: C)
4. Initial examination of patients presenting with HF should include assessment of the patient’s volume status, orthostatic blood pressure changes, measurement of weight and height, and calculation of body mass index. (Level of Evidence: C) 5. Initial laboratory evaluation of patients presenting with HF
should include complete blood count, urinalysis, serum electro-lytes (including calcium and magnesium), blood urea nitrogen, serum creatinine, fasting blood glucose (glycohemoglobin), lipid profile, liver function tests, and thyroid-stimulating hormone. (Level
of Evidence: C)
6. Twelve-lead electrocardiogram and chest radiograph (posterior-anterior and lateral) should be performed initially in all patients presenting with HF. (Level of Evidence: C)
7. Two-dimensional echocardiography with Doppler should be per-formed during initial evaluation of patients presenting with HF to assess LVEF, left ventricular size, wall thickness, and valve function. Radionuclide ventriculography can be performed to assess LVEF and volumes. (Level of Evidence: C)
8. Coronary arteriography should be performed in patients present-ing with HF who have angina or significant ischemia unless the patient is not eligible for revascularization of any kind (15–19). (Level of Evidence: B)
CLASS IIa
1. Coronary arteriography is reasonable for patients presenting with HF who have chest pain that may or may not be of cardiac origin who have not had evaluation of their coronary anatomy and who have no contraindications to coronary revascularization. (Level of
Evidence: C)
2. Coronary arteriography is reasonable for patients presenting with HF who have known or suspected coronary artery disease but who do not have angina unless the patient is not eligible for revascularization of any kind. (Level of Evidence: C)
3. Noninvasive imaging to detect myocardial ischemia and via-bility is reasonable in patients presenting with HF who have known coronary artery disease and no angina unless the patient is not eligible for revascularization of any kind (20). (Level
of Evidence: B)
4. Maximal exercise testing with or without measurement of respi-ratory gas exchange and/or blood oxygen saturation is reason-able in patients presenting with HF to help determine whether HF is the cause of exercise limitation when the contribution of HF is uncertain. (Level of Evidence: C)
5. Maximal exercise testing with measurement of respiratory gas exchange is reasonable to identify high-risk patients presenting Figure 1. Stages in the Development of Heart Failure/Recommended Therapy by Stage
ACEI indicates angiotensin-converting enzyme inhibitor; ARB, angiotensin II receptor blocker; EF, ejection fraction; FHx CM, family history of cardiomyopathy; HF, heart failure; LV, left ventricular; LVH, left ventricular hypertrophy; and MI, myocardial infarction.
e9
JACC Vol. 53, No. 15, 2009 Huntet al.
5. Effetti collaterali della terapia diuretica
Sebbene i diuretici dell’ansa agiscano direttamente sull’eliminazione dei liquidi in eccesso, questi farmaci non riportano alla normalità la funzione cardiaca e si associano a numerosi effetti avversi. I meccanismi attivati dai diuretici dell’ansa, responsabili del peggioramento della prognosi nei pazienti affetti da scompenso cardiaco, includono l’ipovolemia, la riduzione della funzionalità renale, gli squilibri elettrolitici e le alterazioni dell’equilibrio acido-base. Tutti questi effetti sfavorevoli possono essere ingigantiti dallo sviluppo di resistenza ai diuretici 10,51,52.
5.1 – Deplezione di volume.
Se la ritenzione di sodio e di acqua messa in atto dal rene durante lo scompenso cardiaco rappresenta un sistema appropriato di compenso, perché riporta a livelli normali il volume ematico, una sua rimozione, messa in atto dalla somministrazione di diuretici, avrà l’effetto opposto, cioè diminuirà il volume circolante effettivo. L’edema deriva dall’eccesso di liquidi trattenuti dal rene, a seguito della caduta della gittata cardiaca, che si distribuiscono nell’interstizio. Alla somministrazione dei diuretici segue un’iniziale perdita di liquido di origine plasmatica. Ciò provoca una riduzione della pressione venosa e, di conseguenza, della pressione idrostatica capillare, favorendo il ripristino del volume plasmatico con il movimento del liquido edematoso verso lo spazio vascolare. Una volta rimosso l’edema, la perdita di liquidi con le urine continua ad avvenire a discapito del volume plasmatico, con una diminuzione del ritorno venoso al cuore e delle pressioni cardiache di riempimento 27 e con conseguente caduta della gittata cardiaca e della perfusione tissutale nei pazienti sottoposti a dosi saturanti o inappropriate di diuretico 53,54,55. A conferma di questo, Feigenbaum et al., hanno dimostrato che una terapia a lungo termine con
27
diuretici, anche se associata ad ACE Inibitori, determina una diuresi eccessiva e provoca ipovolemia 56. Bayliss et al., hanno studiato pazienti, mai precedentemente trattati con furosemide, che lamentavano dispnea da sforzo. L’introduzione del farmaco induceva contrazione del volume intravascolare e attivazione del sistema renina-angiotensina-aldosterone (RAAS) a riposo e durante l’esercizio. Il tutto si normalizzava con la sospensione della terapia 4.
La somministrazione di diuretici dell’ansa determina anche un aumento della pressione di riempimento del ventricolo sinistro e delle resistenze periferiche 57 secondario a un incremento della secrezione dei tre ormoni controregolatori: renina, noradrenalina, ADH 58,59. Francis et al., hanno dimostrato che l’attività reninica plasmatica è maggiore nei pazienti sintomatici con disfunzione ventricolare sinistra sottoposti a terapia diuretica, rispetto a pazienti asintomatici non sottoposti al medesimo trattamento. L’attivazione del RAAS nei pazienti con scompenso cardiaco moderato si verifica più in risposta alla terapia diuretica protratta e aggressiva, che alla progressione della malattia 36,4,60,61. Inoltre Tsutsui et al., hanno misurato la concentrazione di renina attivata e di noradrenalina in pazienti sottoposti per lungo periodo a furosemide, rilevando livelli più alti rispetto a una popolazione similare trattata con un altro diuretico. Da qui si deduce come la furosemide, causando una fluttuazione giornaliera marcata nel volume di sangue circolante, possa stimolare il rilascio di neurormoni e catecolamine 62. Come dimostrato da Francis et al. in un altro studio, la contrazione del volume indotto dalla diuresi, rappresenta uno dei meccanismi alla base del rilascio di renina 57.
Ci sono evidenze di un’associazione tra gli elevati livelli di aldosterone e un maggior rischio cardiovascolare per i pazienti affetti da scompenso sistolico
63,64: la furosemide e altri diuretici dell’ansa aumentano i livelli plasmatici di questo ormone 65,61. Inoltre, come dimostrato da numerosi studi 66,67, l’aldosterone gioca un importante ruolo nella fibrosi miocardica, con rimodellamento e disfunzione ventricolare. Il RAAS favorisce il rimodellamento strutturale e influisce sull’aumento della mortalità per progressione dell’insufficienza cardiaca 68.
Rialzi significativi di azotemia e della concentrazione plasmatica di creatinina indicano una deplezione di volume legata a un’eccessiva perdita di liquidi con la diuresi. L’incremento dell’azotemia è significativamente superiore rispetto all’aumento della creatinina: il maggior recupero di sodio e acqua conseguente all’ipovolemia determina il riassorbimento passivo di urea nel tubulo prossimale. Il tutto dipende dall’azione dello scambiatore Na+-H+ e urato- - OH- a livello della membrana luminale. Il riassorbimento di urati varia in modo diretto rispetto al trasporto del sodio nel tubulo prossimale. Nei pazienti con deplezione di volume indotta dal diuretico diminuisce sia l’escrezione di sodio che quella di urato. L’angiotensina II, liberata in risposta all’ipovolemia, favorisce l’attività del fattore di scambio Na+-H+ che conseguentemente determina un aumento parallelo dello scambio urato--OH-. Inoltre, un aumento del riassorbimento prossimale di acqua incrementa la concentrazione di urato nel liquido tubulare, facilitandone l’ingresso nelle cellule renali 27.
5.2 – Ridotta funzionalità renale
Oggi si da’ maggiore importanza al ruolo che il rene svolge nello scompenso cardiaco: un peggioramento della sua funzione, anche lieve, è segno di prognosi infausta 69,8,70,71,72,73,74,75,76,77,78,79. Soman et al. e Wase et al., hanno dimostrato che l’insufficienza renale correla a un maggior rischio di morte per aritmia e a una prognosi infausta legata a un’aumentata attività
29
del sistema nervoso simpatico in questi pazienti 80,81. Alla base dell’iperattivazione noradrenergica sembra esserci l’ischemia intrarenale, spesso provocata da un’ipoperfusione dell’organo 82. Un aumento dei livelli di creatinina sierica nei pazienti ricoverati, anche modesto (>26.5 µmol/L o 0,3 mg/dL), è stato associato a un aumento del tasso di mortalità e a risultati peggiori 83. Dai dati di Sun et al., si evince che un dosaggio di diuretico superiore a 114 mg/dl e una durata della terapia più lunga di 7.4 giorni si associano allo sviluppo di insufficienza renale acuta 71. Le cause di insufficienza renale nello scompenso cardiaco cronico potrebbero essere legate alla cronica ipoperfusione renale, all’aumento della pressione venosa renale, ad una sottostante infiammazione e alla disfunzione endoteliale 84. L’impatto dei diuretici sul Filtrato Glomerulare dipende dagli effetti diretti sul nefrone e, indirettamente, dalla conseguente deplezione di volume 73. Si pensa che i diuretici siano in grado di attivare direttamente il sistema simpatico e renina-angiotensina-aldosterone contribuendo allo sbilanciamento tra fattori vasocostrittori e vasodilatatori 71. Studi effettuati su animali 85,86,87 e su esseri umani 88, hanno dimostrato che la diuresi indotta dalla furosemide nei pazienti con insufficienza cardiaca cronica provoca, come effetto indiretto, la riduzione del Filtrato Glomerulare attraverso diversi meccanismi. La deplezione di liquido intravascolare determina l’attivazione neurormonale, con riduzione della pressione di perfusione attraverso la contrazione dell’arteriola afferente, la dilatazione dell’efferente o effetti sul flusso plasmatico renale 37,16,60. L’attivazione dell’innervazione simpatica renale, fisiologica e indotta dalla deplezione di volume, ha un effetto antidiuretico e antinatriuretico 89,90,91,92 attraverso un aumento delle resistenze vascolari renali 93,94,95,96. L’attivazione dei nervi renali e l’ipovolemia indotta dai diuretici stimolano il rilascio di renina e la
generazione di angiotensina II (potente vasocostrittore renale che agisce in sinergia al simpatico), limitando la risposta natriuretica del rene e riducendo ulteriormente il GFR 97,98. Precedenti studi dimostrano che la somministrazione di alte dosi di furosemide attivano l’angiotensina II e l’aldosterone 55. L’aumentato trasporto nel tubulo distale del sodio, conseguente all’uso di diuretici dell’ansa, attiva il meccanismo di feedback tubulo glomerulare, con ulteriore vasocostrizione dell’arteriola afferente e riduzione del flusso plasmatico renale 72,99,55.
Galve et al., hanno dimostrato che la sospensione della terapia diuretica determina un miglioramento della funzione renale nei pazienti con scompenso sistolico stabile 21.
Hillege et al., hanno scoperto che anche basse dosi di diuretici dell’ansa possono ridurre il Filtrato Glomerulare 74.
Francis et al., hanno individuato la relazione esistente tra la terapia diuretica cronica e la riduzione della clearance della creatinina/ridotto GFR, nei pazienti con una funzione renale già compromessa 52.
Secondo Waldum et al., una riduzione clinicamente significativa del GFR al di sotto di 60mL/min è un fattore predittivo indipendente per tutte le cause di morte. Il tempo di sopravvivenza media nei pazienti con una moderata riduzione del GFR è di 50 mesi circa. Con un Filtrato Glomerulare al di sotto dei 30 mL/min, la sopravvivenza si riduce a 32 mesi. Solo l’età dei pazienti, la classe NYHA e il GFR rappresentano nelle analisi multivariate un fattore predittivo indipendente della mortalità totale. Preservare la funzione renale è di massima importanza nei pazienti con scompenso di cuore 8.
Sia Sun et al. che Weinfeld et al., hanno dimostrato che l’insufficienza renale indotta dai diuretici nei pazienti affetti da scompenso cardiaco cronico, trova nell’età un importante fattore predittivo. Nei soggetti anziani
31
c’è una fisiologica e graduale riduzione del GFR, della riserva funzionale renale e del flusso plasmatico renale che non deriva solamente da un aumento delle resistenze vascolari conseguente all’aterosclerosi delle arteriole renali (soprattutto l’arteriola afferente), ma anche dalle alterazioni funzionali nella vascolarizzazione renale. A questi cambiamenti provocati dall’invecchiamento si sommano:
- le alterazioni neurormonali associate allo scompenso cardiaco, che impediscono l’aumento della frazione di filtrazione normalmente osservato in soggetti di più giovane età;
- l’ulteriore aumento della vasocostrizione pre-glomerulare indotto dalla furosemide 100.
5.3 – Squilibri elettrolitici
5.3.1 - Iposodiemia.
L’iposodiemia si verifica comunemente nel corso di insufficienza cardiaca, ma è anche conseguenza della terapia diuretica 38. L’iponatriemia (concentrazione di sodio nel sangue <134 mmol/L), predice risultati negativi nei pazienti con scompenso cardiaco 101,102,103. Lee et al., hanno dimostrato che la concentrazione sierica di sodio è uno dei migliori predittori di mortalità cardiovascolare, con pazienti iponatriemici che mostrano una sopravvivenza sostanzialmente più corta rispetto a soggetti con una normale concentrazione di sodio plasmatica 104.
I diuretici dell’ansa sono considerati i responsabili del 94% dei casi di iponatriemia severa in 129 pazienti affetti da questo disturbo elettrolitico 105. Studi europei hanno evidenziato l’alta incidenza dell’uso dei diuretici nei pazienti iponatriemici 106. I meccanismi alla base di questo effetto collaterale sono:
- la riduzione della GFR per la deplezione di volume;
- l’aumentata secrezione di ADH a causa dell’ipovolemia 98. - l’attivazione dell’angiotensina II che stimola la sete;
- il ridotto trasporto del sodio nei segmenti distali del nefrone ad opera dei neurormoni e quindi il danneggiamento della capacità di eliminare l’acqua libera 71.
I diuretici dell’ansa di per sé raramente provocano iponatriemia, ma è necessario tenere presente il quadro generale. La fisiopatologia dello scompenso cardiaco si associa all’attivazione di tutti i meccanismi antidiuretici e antinatriuretici per sopperire alla deplezione del volume circolante effettivo. Alla somministrazione dei diuretici fa seguito un progressivo rilascio di AVP e un peggioramento dell’ipovolemia conseguente alla diuresi, con maggior rischio di sviluppare iponatriemia 107. Molti studiosi hanno osservato livelli plasmatici più alti di renina-angiotensina, catecolamine e vasopressina in individui con insufficienza cardiaca cronica e iponatriemia 108,109,110,111,104,112,113. In uno scenario tipico, il paziente è ricoverato per un aggravamento dello scompenso cardiaco e lieve iponatriemia. Per trattare i sintomi di congestione vengono somministrati diuretici endovena e all’aumento della diuresi l’iponatriemia peggiora. Questo porta all’insorgenza di gravi sintomi conseguenti al danno inflitto alle cellule del SNC (confusione e coma) 107.
Sun et al., hanno dimostrato come una bassa concentrazione sierica di sodio all’ammissione si correli a un rischio significativo di insufficienza renale acuta indotta dai diuretici 71.
33
5.3.2 - Ipopotassiemia.
I diuretici dell’ansa incrementano le perdite urinarie di potassio e spesso determinano ipopotassiemia (concentrazione plasmatica di K+ compresa tra 3.0-3.5 mEq/l). I fattori responsabili sono:
- l’aumento del flusso di Na+ e H2O nel tratto distale, conseguente all’inibizione del riassorbimento nei segmenti più prossimali;
- l’aumento della secrezione di aldosterone, dovuto sia alla patologia sottostante, che alla concomitante deplezione di volume.
Non è comunque molto frequente l’insorgenza di una grave ipopotassiemia, poiché la furosemide riduce il riassorbimento passivo di calcio nell’ansa di Henle e l’aumento del carico di ioni positivi nei tubuli distali riduce l’escrezione di K+.
Il significato clinico di una modesta ipopotassiemia resta controverso 114. Cohen et al., hanno dimostrato che si associa a una maggiore incidenza di aritmie ventricolari 115.
I diuretici dell’ansa provocano una profonda ipovolemia con riduzione della secrezione renale di bicarbonato di sodio e conseguente alcalosi metabolica e sviluppo di ipopotassiemia per l’aumentato scambio cellulare tra K+ e H+ 116.
5.3.3 - Ipomagnesemia.
La terapia diuretica induce deplezione di magnesio di modesta entità. Generalmente il magnesio filtrato è riassorbito nell’ansa di Henle, processo inibito direttamente dai diuretici dell’ansa 114. Anche l’aldosterone promuove l’escrezione di magnesio e di calcio attraverso il rene e l’intestino. La conseguente ipermagnesiuria e ipercalciuria, determina una perdita di minerali ossei con effetti di vasta portata sui tessuti sistemici 117. La deplezione di magnesio a carico delle scorte intracellulari è un evento
relativamente frequente: la concentrazione plasmatica spesso rimane entro i limiti della norma. La carenza della componente biologicamente attiva di questo importante catione bivalente, induce calcio e stress ossidativo nella cellula. Una riduzione del magnesio intracellulare e il conseguente carico di calcio nei mononucleati contribuisce a un’attivazione di queste cellule e alla creazione di un fenotipo pro-infiammatorio dei vasi sistemici e delle coronarie 118,119,120. Nel cuore, la risultante fibrosi interstiziale e perivascolare fa da substrato all’anormale reattività vasomotoria, alle aritmie e alla disfunzione ventricolare 117. E’ stato ipotizzato che la deplezione di magnesio predisponga ad aritmie, in particolare in presenza di una concomitante ipopotassiemia 114.
5.3.4 - Ipercalciuria.
L’escrezione urinaria di calcio indotta dalla furosemide porta a una riduzione del calcio ionizzato, che stimola le paratiroidi a produrre ormone paratiroideo (PTH). Il PTH determina la sintesi del metabolita attivo della Vitamina D. Insieme, questi ormoni cercano di preservare l’omeostasi plasmatica del calcio con maggior riassorbimento osseo e gastrointestinale dello ione e perdita di massa ossea. In pazienti con insufficienza cardiaca avanzata sintomatica in attesa di trapianto, con alle spalle una storia di prolungati trattamenti con furosemide, si riscontra spesso riduzione marcata (osteoporosi) o moderata (osteopenia) della densità minerale ossea e aumento del PTH 121,122.
La nefrocalcinosi frequentemente accompagna il trattamento con furosemide. Livelli di aldosterone plasmatico inappropriati e cronici aumentano l’escrezione urinaria di calcio e magnesio e la secrezione paratiroidea 123,124.
35
5.4 – Alterazione dell’equilibrio acido-base
L’ipopotassiemia indotta dai diuretici dell’ansa si associa di frequente ad alcalosi metabolica. I meccanismi sono:
- l’aumentata perdita urinaria di idrogenioni indotta dai diuretici e secondaria all’iperaldosteronismo. L’aldosterone stimola la pompa H+ -ATPasi dipendente distale e promuove il riassorbimento del catione Na+ creando un potenziale elettrico, con negatività verso il lume, che favorisce l’accumulo di H+ nel lume stesso rendendo minimo il grado di diffusione retrograda. Nella parte corticale della branca ascendente spessa di Henle il riassorbimento del sodio avviene attraverso il trasportatore Na+-K+-2Cl e lo scambiatore Na+-H+. I diuretici dell’ansa inibiscono il primo potenziando indirettamente l’attività del secondo, con perdita di idrogenioni;
- la contrazione del volume extracellulare senza grosse variazioni della quantità di HCO3- extracellulare, fenomeno chiamato “alcalosi da contrazione”.
5.5 – Ototossicità
L’ototossicità transitoria o permanente consegue ad alte dosi di furosemide, a un trattamento prolungato o a una somministrazione endovenosa 125. Se con una dose <50 mcg/ml non si riesce ad ottenere l’effetto diuretico desiderato, è allora consigliabile cambiare tipo di farmaco per minimizzare l’effetto ototossico 126.
5.6 – Resistenza ai diuretici
La resistenza ai diuretici nell’insufficienza cardiaca deriva dall’iterazione tra la fisiopatologica ritenzione di sodio e la risposta renale alla terapia diuretica 61. Tipicamente i pazienti con insufficienza cardiaca
lieve-moderata hanno una risposta al farmaco che è ⅓-¼ rispetto a quella osservata nei pazienti normali. Un aggravamento della patologia riduce ulteriormente l’effetto 38,127,128,57.
Un lento riassorbimento intestinale causa la resistenza alla somministrazione per os della furosemide. La minor perfusione intestinale, la scarsa motilità e forse l’edema della mucosa ritardano il riassorbimento intestinale 114.
La riduzione del Filtrato Glomerulare (conseguente all’ipoperfusione renale), associata a un eccessivo riassorbimento al livello del tubulo prossimale (in parte mediato dall’angiotensina II), determina un marcato calo dell’apporto di liquidi alle sedi sensibili al diuretico nei segmenti più distali del nefrone 38,114.
L’incremento distale del carico di sodio e l’azione dell’aldosterone sul tubulo collettore corticale aumenta il riassorbimento distale del sodio 114. L’adattamento renale segue una prolungata esposizione ai diuretici dell’ansa e si manifesta con l’ipertrofia e l’iperfunzione delle cellule tubulari distali, con il conseguente aumento del riassorbimento locale di sodio e con la secrezione di aldosterone 129,130,131.
La resistenza ai diuretici induce alla somministrazione di dosaggi sempre più alti per ottenere una risposta farmacologica adeguata 38,132,10,133 ed espone i pazienti agli effetti collaterali di una terapia medica aggressiva e inefficace 134,70.
37
6. SCOPO DELLA TESI
Lo scopo di questa tesi è analizzare gli effetti della terapia diuretica nell’ambito di una popolazione anziana affetta da scompenso cardiaco. Questo è volto a dimostrare che la somministrazione inappropriata di dosi saturanti di diuretici dell’ansa può provocare una riduzione del volume effettivo circolante e, per via dell’attivazione del sistema renina-angiotensina-aldosterone57,36,4, stimolare ulteriormente le forze sodio ritentive e la riduzione della perfusione renale.
La riduzione della sodiuria e della perfusione renale compete di fatto con la possibilità di ottenere un bilancio negativo di acqua e di favorire i cicli virtuosi che, riportando il sistema nella fase positiva della curva di Starling, consenta di ottenere una nuova fase di compenso cardiaco.
L’analisi eseguita esplora il cambiamento dei parametri clinici confrontando due popolazioni di riferimento: una nella quale la sospensione della terapia diuretica con Furosemide e della eventuale co-somministrazione di ACE inibitori e/o Antialdosteronici, era dovuta alla presenza di iperpotassiemia e di segni evidenti di ipoperfusione renale, l’altra nella quale il trattamento con la terapia diuretica non veniva sospeso in quanto consentito dallo stato clinico del paziente (es., disidratazione moderata correggibile con piccoli volumi infusionali).
Abbiamo valutato anche la possibilità che la relativa disidratazione e l’ipertono simpatico potessero essere associati a un’aumentata generazione di trombina e quindi a un fenotipo trombofilico.