1
Premessa
Oggetto di questa tesi di laurea è la sintesi e la valutazione funzionale di composti a nucleo pirazolo[3,4-d]pirimidinico, che agiscono come inibitori multi-target di tirosin-chinasi, risultando così attivi nel carcinoma tiroideo, in particolare nel carcinoma papillare tiroideo dedifferenziato (DePTC). Recenti evidenze dimostrano infatti che le tirosin-chinasi, in qualità di importanti mediatori nei processi di trasduzione del segnale, giocano un ruolo chiave nella patofisiologia del cancro.
Esse rappresentano una famiglia di enzimi ad attività chinasica, capaci, in risposta a stimoli esterni ed interni alla cellula, di fosforilare residui di tirosina in proteine bersaglio, usando ATP; questa modifica covalente post-traduzionale è essenziale per la comunicazione cellulare ed il mantenimento dell’omeostasi. In condizioni normali la loro attività è strettamente regolata, cosicché possano mediare diversi processi biologici, come la crescita e la differenziazione cellulare, il metabolismo e l’apoptosi.
Risulta quindi chiaro che segnali aberranti provenienti dalle tirosin-chinasi conferiscano a questi enzimi uno status di oncoproteine, che indirizzano la cellula verso una progressione neoplastica. Per questa ragione gli inibitori tirosin-chinasici hanno significativamente cambiato le prospettive riguardo l’attuale terapia contro il cancro.
Nel laboratorio presso il quale ho svolto la mia tesi sperimentale, sono stati
precedentemente sintetizzati due composti contenenti il nucleo
pirazolopirimidinico (CLM3 e CLM29), di cui è stata dimostrata l’attività antitumorale in vitro e in vivo nel DePTC. Si tratta di composti ATP-mimetici, che si distinguono come agenti multi-target visto l’ampio spettro di attività; risultano infatti efficaci nell’inibire VEGFR-2 (vascular endothelial growth factor receptor 2), la chinasi RET (REarranged during Trasfection), EGFR (epidermal growth factor
2
antineoplastica risulta quindi dalla combinazione di un effetto antiproliferativo, associato ad un incremento dell’apoptosi e di uno inibitorio della neovascolarizzazione tumorale, soprattutto per quel che riguarda il CLM3.
I composti da me sintetizzati nascono dall’esigenza di approfondire le relazioni struttura-attività di questa classe di inibitori, così da ottimizzare l’attività nei confronti della RET chinasi, pur preservando quella anti-angiogenica.
3
Abstract:
The purpose of my thesis work is the synthesis and the biological evaluation of pyrazolo[3,4-d]pyrimidine derivates as multi-target tyrosine kinase inhibitors with antitumoral activity in thyroid cancer treatment, especially in papillary dedifferentiated thyroid cancer (DePTC). Recent advances have shown that tyrosine kinases are important mediators of the signaling cascade, playing a key role in the pathophysiology of cancer.
Tyrosine kinases are a family of enzymes with kinase activity, which selectively phosphorylate tyrosine residues in different substrates, using ATP, in response to external and internal stimuli; this post-translational covalent modification is a pivotal component of normal cellular communication and maintenance of homeostasis. In physiological conditions tyrosine kinases’ activity is tightly regulated, so that they can mediate diverse biological processes such as growth and cell differentiation, metabolism and apoptosis.
It is clear that aberrant signals from tyrosine kinase enzymes conferred them the status of oncoproteins, which could direct cells to a neoplastic progression. For this reason, the tyrosine kinase inhibitors have significantly changed the outlook about the current cancer therapy.
In the laboratory where I did my thesis, two compounds containing the pyrazolopyrimidine core (namely CLM3 and CLM29) have been previously synthesized and their antitumoral activity in vitro and in vivo in DePTC has been demonstrated.
They are ATP-mimetic agents that stand out as multi-target, given the wide spectrum of activities; indeed, they can inhibit VEGFR-2 (Vascular Endothelial Growth Factor-type 2), RET (Rearranged during Transfection), EGFR (Endothelial Growth Factor Receptor) and the related pathway signaling. Their antitumoral activity is the result of an antiproliferative effect associated with the
4
increase of apoptosis and the inhibition of the neoplastic neovascularization, especially for CLM3.
All the compounds I synthesized were developed from the need to deepen structure-activity relationships of this class of inhibitors, so as to optimize the activity against RET kinase, while preserving the anti-angiogenic one.
5
Capitolo 1
-
La tiroide e il tumore tiroideo
1.1 La tiroide: sito, morfologia esterna e struttura
La tiroide è la ghiandola endocrina più voluminosa, situata nella regione anteriore
del collo. E’ formata da:
- due lobi laterali, destro e sinistro, che sono in rapporto con la cartilagine tiroidea e i primi cinque anelli tracheali;
- un ponte mediano, detto istmo, che collega tra loro i lobi tiroidei;
- un prolungamento che origina dall’istmo e si spinge in alto, raggiungendo così il margine inferiore dell’osso ioide.
La tiroide presenta una struttura follicolare, con parenchima compatto rivestito da una capsula fibrosa, che invia dei tralci in profondità; questi dividono la ghiandola in lobuli. Ciascun lobulo è formato da un numero elevato di follicoli: vescicole di forma sferica od ovoidale, la cui parete è costituita dall’epitelio follicolare monostratificato, formato dai tireociti [1], che producono gli ormoni triiodotironina (T3) e tetraiodotironina (T4), in risposta allo stimolo da parte
dell’ormone tireotropo (TSH), secreto dall’adenoipofisi.
In intimo rapporto con i tireociti e con i capillari sanguiferi, sono presenti cellule argentofile, dette cellule C o parafollicolari, appartenenti al sistema neuroendocrino diffuso e secernenti la calcitonina, un ormone polipeptidico ad azione ipocalcemizzante in risposta a condizioni di ipercalcemia. Il polipeptide infatti, in sinergia ad altri ormoni, prevalentemente sessuali, promuove il deposito di calcio nelle ossa ed inibisce l’escrezione renale di calcio.
6
1.2 Il tumore tiroideo
Il cancro della tiroide rappresenta la neoplasia endocrina maligna più diffusa e costituisce approssimativamente l’1% di tutti i tumori maligni. Il tasso di incidenza è aumentato notevolmente a partire dalla metà del 1900, diventando così il tipo di tumore a più rapida crescita, sia negli uomini che nelle donne, nei Paesi Occidentali. Questo è presumibilmente dovuto ad un potenziamento delle tecniche diagnostiche dei tumori micropapillari. Ad oggi, in accordo con le previsioni dell’American Cancer Society, nel 2013 verranno diagnosticati 60.000 nuovi casi negli USA e più di 2.000 morti, a sottolineare la marcata accelerazione nel tasso di incidenza nell’ultima decade, pari al 5,5% e al 6,5% negli uomini e nelle donne, rispettivamente [2-4]. La prognosi è generalmente molto buona, anche se dipende dal tipo di tumore, dallo stadio di avanzamento e da altri fattori.
1.2.1 Carcinoma tiroideo differenziato
La maggior parte delle neoplasie della tiroide (circa il 90%) origina dalle cellule tiroidee epiteliali ed è solitamente indicata con l’espressione generica di “carcinoma tiroideo differenziato” (DTC). Il tumore papillare tiroideo rappresenta approssimativamente l’85% di tutti i DTC, mentre quello follicolare circa il 10%; le restanti sono forme tumorali a carico delle cellule di Hürthle o delle cellule ossifile delle paratiroidi.
Il trattamento dei DTC prevede tre differenti strategie, a seconda dell’estensione del tumore: quella di prima scelta è l’intervento chirurgico, che consiste preferenzialmente in una tiroidectomia totale. A seconda del livello di rischio del paziente, segue poi una terapia radiometabolica, ovvero la somministrazione di iodio radioattivo (131I) a dosi ablative, per distruggere il tessuto tiroideo residuo. In
ogni caso viene seguita una terapia TSH-soppressiva con levotiroxina, per ridurre lo stimolo sulla tiroide e quindi l’eventuale proliferazione di foci neoplastici occulti.
7
Ulteriori possibilità di trattamento per il tumore allo stadio avanzato sono rappresentate dalla radioterapia a fasci esterni e dalla chemioterapia, principalmente basata sulla somministrazione di doxorubicina; in realtà nessuno dei due approcci ha dimostrato una sostanziale efficacia.
La sopravvivenza complessiva (OS- Overall Survival) è molto elevata: oltre il 90% dei pazienti sopravvive a cinque anni dalla diagnosi. I pazienti refrattari al trattamento con radioiodio vedono invece drasticamente diminuita al 3% la probabilità di sopravvivenza a dieci anni.
1.2.2 Carcinoma midollare tiroideo (MTC)
Si tratta di un sottotipo istologico non comune, che rappresenta il 5-8% di tutte le neoplasie tiroidee. Questo tumore neuroendocrino origina dalle cellule C della tiroide secernenti calcitonina e mostra una lenta crescita, accompagnata da un decorso clinico indolente.
Circa il 65-75% degli MTC sono sporadici, ovvero non legati a cause genetiche, mentre il restante 25-35% viene trasmesso su base autosomica dominante ed è dovuto a mutazioni puntiformi del proto-oncogene RET. Tali forme ereditarie comprendono l’MTC familiare (FMTC) e la neoplasia endocrina multipla (MEN) di
tipo 2A e 2B, caratterizzate entrambe dalla compresenza di feocromocitoma.
Il trattamento si limita all’intervento chirurgico, che ha scopo curativo nei casi di tumore manifesto e profilattico nei bambini, qualora sia riconosciuta una predisposizione genetica. Il trattamento sintomatico dei disturbi dovuti all’azione sistemica della calcitonina, che includono vampate di calore, diarrea e riduzione del peso corporeo, prevede la somministrazione dell’octreotide, un analogo della somatostatina, ma di limitata efficacia.
Anche in questo caso, come nel precedente, la sopravvivenza complessiva è elevata, nonostante essa dipenda dallo stadio di avanzamento della malattia: la sopravvivenza a dieci anni per i pazienti con tumore localizzato e metastatico è rispettivamente del 96% e 40%.
8
1.2.3 Carcinoma tiroideo anaplastico (ATC)
Rappresenta la forma più rara e al tempo stesso più aggressiva di cancro alla tiroide, con un’incidenza approssimativamente del 2%. Origina anch’esso dalle cellule epiteliali della tiroide, sviluppandosi de novo oppure evolvendosi a partire dal DTC già manifesto.
Il trattamento attualmente disponibile è considerato quasi sempre inefficace e la prognosi è infausta; il tasso di sopravvivenza media è infatti di 3-4 mesi e leggermente migliore per i pazienti con tumore localizzato [5].
1.3 Analisi molecolare del tumore tiroideo
Negli ultimi decenni sono state descritte diverse alterazioni genetiche alla base del carcinoma tiroideo, che dimostrano chiaramente come le tirosin-chinasi siano fondamentali nel sostenere lo sviluppo e la progressione del tumore. In particolare sono quattro le mutazioni individuate: queste includono mutazioni puntiformi
BRAF e RAS e riarrangiamenti RET/PTC e PAX8/PPARγ e rappresentano
importanti marcatori di utilità diagnostica e prognostica nei pazienti colpiti da carcinoma papillare o follicolare.
I tumori scarsamente differenziati e il carcinoma anaplastico possono recare invece mutazioni principalmente a carico della via PI3K/AKT, che risultano rare nei tumori ben differenziati.
Queste mutazioni generano chinasi attivate in maniera costitutiva o inattivano soppressori tumorali, che possono cooperare nel trasformare la cellula tiroidea in una meno differenziata, aumentando così la propensione ad una proliferazione indipendente dalla tireotropina.
In aggiunta a tali alterazioni genetiche, è stato scoperto che altri meccanismi sono implicati nella carcinogenesi tiroidea; si tratta essenzialmente di modificazioni epigenetiche, in grado di alterare la funzione di un gene, senza modificarne la sequenza nucleotidica. Tra di esse la più comune nel tumore della tiroide è la
9
deacetilazione degli istoni, che ha fornito il fondamento logico per lo studio del Vorinostat, un inibitore dell’enzima istone-deacetilasi, nel trattamento di questa neoplasia [6,7].
Figura 2. Frequenza delle mutazioni nei carcinomi papillare e follicolare [6]
1.3.1 BRAF
La proteina BRAF è una serina-treonina chinasi, appartenente alla famiglia delle proteine RAF, famiglia che include effettori intracellulari della via del segnale delle MAP (mitogen-activated protein) chinasi che, fosforilando proteine che si legano al DNA, regolano il processo di trascrizione genica.
Le mutazioni puntiformi a carico di questa proteina rappresentano la più comune alterazione molecolare nel carcinoma papillare tiroideo e generalmente coinvolgono il nucleotide 1799, determinando sulla proteina la sostituzione di una valina con un glutammato, a carico del residuo 600 (V600E). Tale mutazione comporta l’attivazione costitutiva della BRAF chinasi e si traduce in una stimolazione prolungata della via delle MAPK, che causa in ultimo tumorigenesi a carico della tiroide [6].
1.3.2 RET/PTC
La proteina RET è stata una delle prime tirosin-chinasi recettoriali ad essere stata associata a neoplasie maligne. Viene codificata dal proto-oncogene RET sul cromosoma 10 (10q11.2) ed è espressa ad alte concentrazioni durante
10
l’embriogenesi, a livello del sistema nervoso periferico e di quello urogenitale. Nell’adulto si ritrova invece nelle cellule tiroidee, paratiroidee, nella midollare del surrene, nei gangli enterici e a livello del sistema nervoso autonomo. [8] Negli anni ’90 è stato dimostrato che il proto-oncogene RET è associato al carcinoma papillare tiroideo, attraverso un riarrangiamento cromosomico RET/PTC, che è un chiaro esempio di mutazione con guadagno di funzione; consente infatti alla tirosin-chinasi di attivare la via del segnale delle MAPK, sopra menzionata. Inoltre mutazioni puntiformi a carico del proto-oncogene RET sono state scoperte in oltre il 50% dei casi di MTC sporadico e nelle forme ereditarie.
Degli undici riarrangiamenti RET/PTC descritti, quelli maggiormente rappresentativi nei carcinomi tiroidei sono il RET/PTC1 e il RET/PTC3, il primo associato alla forma istologica papillare classica e l’altro più comune nelle varianti solide [6].
Il proto-oncogene RET produce cinque trascritti primari che codificano due differenti isoforme della proteina, RET9 e RET51, generate da uno splicing alternativo a livello della regione 3’-carbossiterminale [9]. Queste isoforme hanno ruoli evolutivi distinti e i diversi profili di espressione genica, ottenuti mediante analisi su microarray, suggeriscono possibili differenze nella regolazione a valle delle interazioni cellula-cellula.
11
La proteina-chinasi RET è costituita da tre domini:
- un dominio extracellulare: contiene una ripetizione in tandem di quattro domini simili alle caderine (CLD-catdherin like domains) e una regione altamente conservata, ricca di residui di cisteina (CRD), essenziale per la struttura terziaria e per la dimerizzazione del recettore, attraverso la formazione di ponti disolfuro; - un dominio trans-membrana, che attraversa il doppio strato fosfolipidico; - un dominio citoplasmatico tirosin-chinasico.
I ligandi del recettore RET sono stati identificati per la prima volta nel 1996 e sono fattori di crescita appartenenti alla famiglia dei GDNF (glial cell line-derived
neurotrophic factor), che include il GDNF, la neurturina, l’artemina e la persefina.
Perché il recettore possa interagire con i propri ligandi, è necessario l’intervento di co-recettori, appartenenti alla famiglia dei GFRα (growth factor receptor alfa): si
12
tratta di recettori privi di un dominio trans-membrana o di uno intracellulare, che si ancorano alla membrana attraverso una molecola di glicosilfosfatidilinositolo. Essi si associano al ligando, portando alla dimerizzazione della RET-chinasi, attraverso la formazione di un complesso eteroesamerico -GFL(2)-GFRα(2)-RET(2)- che innesca la sua attività chinasica intrinseca.
Anche i raft lipidici, microdomini planari della membrana plasmatica, sono essenziali per la trasduzione del segnale RET-mediata; sono stati proposti due differenti meccanismi di interazione con il complesso di attivazione. I recettori GFRα sono tipicamente localizzati nei raft lipidici e, a seguito del legame con il ligando, reclutano il recettore RET all’interno degli stessi raft, dove la chinasi dimerizza ed interagisce con specifiche proteine per la trasduzione del segnale. In alternativa il recettore GFRα può essere legato alla membrana plasmatica e interagire col ligando prima dell’associazione col recettore, che è poi reclutato dai raft lipidici [9].
1.3.3 RAS
Le mutazioni RAS compaiono in tutti i tumori epiteliali maligni della tiroide: sono state identificate nel 40-50% dei carcinomi follicolari e nel 10-20% dei carcinomi papillari.
La famiglia delle proteine RAS include piccole GTP-asi, codificate dai tre geni
HRAS, KRAS e NRAS e coinvolte nei processi di trasduzione del segnale a partire
dai recettori tirosin-chinasici e metabotropici lungo le vie delle MAPK e PI3K/AKT.
Le mutazioni puntiformi maggiormente riscontrate nei tumori tiroidei sono a carico del codone 60 dei geni NRAS e HRAS [6] e portano alla espressione di proteine permanentemente attive, anche in assenza di segnali in ingresso.
13
1.3.4 PAX8/PPARγ
Il riarrangiamento PAX8/PPARγ è il risultato di una traslocazione, che porta alla fusione del gene PAX8 con quello codificante per il recettore PPARγ (recettore gamma attivato dalla proliferazione dei perossisomi). I carcinomi follicolari positivi per questa mutazione compaiono in giovane età, sono di dimensioni inferiori e spesso associati ad invasione vascolare [6], parametro quest’ultimo che indica la presenza di cellule tumorali isolate o di aggregati nei vasi sanguigni e linfatici circostanti il tumore.
E’ stata a lungo dibattuta l’ipotesi che le mutazioni RET/PTC, BRAF e RAS possano sovrapporsi all’interno di un unico caso di PTC; ad oggi è stato dimostrato che non è un evento raro che tumori recanti un riarrangiamento
RET/PTC includano anche una mutazione V600EBRAF. Tuttavia non è possibile
discriminare se entrambe le mutazioni siano presenti all’interno di una stessa cellula o in cellule differenti della stessa massa tumorale. I risultati di uno studio condotto su 14 casi di PTC, recanti una mutazione duale, dimostrano che le suddette mutazioni non coesistono nella stessa cellula, ma ciò nonostante possono cooperare nel modulare le interazioni intercellulari e il processo di angiogenesi. Le due mutazioni inoltre sono associate a diversi fattori eziologici; i riarrangiamenti RET/PTC insorgono frequentemente dopo esposizione a radiazioni ionizzanti, ma non rappresentano un fattore di rischio per lo sviluppo di una neoplasia più aggressiva. Quelle V600EBRAF sono invece correlate a fattori
14
Capitolo 2
-
Le tirosin
-
chinasi: nuovo target della terapia
anti
-
tumorale
Le chinasi (o fosfotransferasi) sono enzimi che catalizzano il trasferimento di un gruppo fosfato da una molecola donatrice ad alta energia, quale ad esempio l’ATP, a specifiche proteine substrato, determinandone cambiamenti funzionali.
Figura 4. Rappresentazione schematica del meccanismo d'azione delle tirosin-chinasi [13] Tipicamente, le proteine chinasi sono suddivise in due classi: chinasi che fosforilano residui di serina e/o treonina e chinasi che fosforilano invece residui tirosinici; sebbene esse differiscano nell’amminoacido cui trasferiscono il gruppo fosfato, i loro domini catalitici sembrano avere molte caratteristiche comuni e una modalità d’azione sostanzialmente identica [10].
Delle 518 proteine chinasi che costituisco il kinoma umano, 90 appartengono al gruppo delle tirosin-chinasi. La fosforilazione selettiva di residui di tirosina in proteine target rappresenta una modifica covalente post-traduzionale, essenziale per la comunicazione cellulare [11]. Essa è determinante nello stimolare l’attività catalitica delle tirosin-chinasi stesse e nel generare siti di attacco per il reclutamento di proteine substrato [12].
2.1 Classificazione
Le tirosin-chinasi appartengono approssimativamente a 30 famiglie, quali la famiglia del VEGFR e quella dell’FGFR (fibroblast growth factor receptor). Oltre alla classificazione in famiglie, le tirosin-chinasi possono anche essere distinte in:
15
recettori tirosin-chinasici (RTK), come ad esempio EGFR, PDGFR (platelet-derived
growth factor receptor) e FGFR (fibroblast growth factor receptor) e tirosin-chinasi non
recettoriali (NRTK), tra cui Src, Abl, Fak e Janus [13].
I recettori tirosin-chinasici sono essenziali per la trasduzione di segnali extracellulari all’interno della cellula, mentre le proteine non recettoriali permettono la comunicazione intracellulare [11].
2.2 Struttura RTK
Un recettore tirosin-chinasico è costituito da un dominio extracellulare N-terminale che lega il ligando, da un dominio transmembrana e da uno intracellulare C-terminale ad attività chinasica. Il dominio chinasico presenta una struttura bi-lobare, con una fessura legante l’ATP, localizzata tra i due lobi N- e C-terminali.
Nella figura a fianco è riportato l’esempio del recettore per l’insulina, complessato ad un analogo dell’ATP e al peptide substrato; come si può notare il lobo N-terminale comprende un foglietto β (mostrato in giallo) a cinque filamenti e un’α-elica (rappresentata in rosso), mentre il lobo C-terminale, più ampio, è principalmente un’α-elica [12].
Figura 5. Struttura di un recettore tirosin-chinasico [11]
Figura 6. Architettura del dominio chinasico [16]
16
Il lobo C-terminale contiene inoltre un loop di attivazione, indicato come “motivo DFG”, perché contraddistinto da una specifica sequenza amminoacidica: acido aspartico-fenilalanina-glicina. Esso può assumere diverse conformazioni; nella conformazione “aperta” crea una tasca idrofobica, nelle vicinanze della fessura legante l’ATP, essenziale per un sottogruppo di inibitori tirosin-chinasici [11], di seguito descritti.
Il sito di legame dell’ATP è altamente conservato tra le tirosin-chinasi, poiché necessario nel garantire la loro attività e può essere suddiviso in cinque regioni:
a) Regione dell’adenina: contiene due legami idrogeno, formati per interazione degli atomi N-1 e N-6 del nucleo purinico dell’adenina con i gruppi –NH e carbonilico della cosiddetta “regione a cerniera” della proteina chinasi; molti potenti inibitori utilizzano almeno uno di questi due legami idrogeno. Sebbene non siano sfruttati dall’ATP, alcuni gruppi carbonilici della “regione a cerniera” possono essere utilizzati come accettori di legami idrogeno per il legame dell’inibitore;
b) Regione dello zucchero: è idrofilica nella maggior parte delle proteine chinasi; fa eccezione EGFR, particolarità questa che è sfruttata nella progettazione di inibitori potenti e selettivi per lo stesso EGFR;
17
c) Tasca idrofobica: non vi si lega l’ATP, ma è utilizzata dalla maggior parte degli inibitori ed è essenziale al fine di ottenere un’inibizione selettiva;
d) Canale idrofobico: rappresenta un’apertura accessibile al solvente e, poiché non è utilizzata dall’ATP, può essere sfruttata per ottimizzare l’affinità di legame;
e) Regione legante i gruppi fosfato: è la meno importante in termini di affinità di legame, ma può essere utile per migliorare la selettività. Il gruppo tri-fosfato dell’ATP è limitato da un loop ricco di glicina ed è vincolato da una matrice altamente conservata di amminoacidi basici, che assieme al residuo D (acido aspartico) del motivo “DFG” che deprotona il gruppo –OH fosfoaccettore, è coinvolta nel processo catalitico [13].
Secondo questo modello farmacoforico, risulta alquanto complessa l’individuazione di una farmaco che sia selettivo verso una singola chinasi; l’attenzione è quindi rivolta a composti aventi un profilo inibitorio sufficientemente selettivo, tale da limitare il blocco di altre tirosin-chinasi e conseguentemente gli effetti collaterali.
18
2.3 Meccanismo d’azione
I recettori tirosin-chinasici sono attivati dal legame del ligando al loro dominio extracellulare; tale legame porta alla dimerizzazione del recettore (eccezion fatta per il recettore dell’insulina), cui fa seguito una rapida autofosforilazione intramolecolare di residui tirosinici del dominio citoplasmatico, che vede così innescata la sua attività chinasica.
I ligandi sono molecole segnale extracellulari, che possono adottare differenti strategie per raggiungere la conformazione dimerica più stabile; nel caso ad esempio dell’ormone della crescita e del relativo recettore, il ligando lega due molecole del recettore per formare un complesso 1:2. Il più semplice meccanismo di dimerizzazione è però quello che vede due ligandi legarsi simultaneamente a due recettori, a dare un complesso 2:2; è questo il caso del VEGF (vascular endothelial
growth factor) e del VEGFR [14].
19
I residui tirosin-chinasici autofosforilati servono come siti di legame ad alta affinità per altre molecole segnale; si tratta di proteine adattatrici, come quelle contenenti il dominio SH2 (Src homology 2). L’associazione tra il recettore fosforilato e una di queste proteine comporta la fosforilazione e l’attivazione di questa stessa proteina effettrice ed è funzionale anche a fornire un sito di attacco per altre molecole segnale. [11] Il risultato è l’attivazione di una cascata di segnali biochimici intracellulari, che portano all’attivazione o alla repressione della trascrizione di determinati geni, definendo così la risposta biologica a segnali extracellulari.
Durante questi processi i recettori migrano all’interno della membrana plasmatica e sono internalizzati attraverso delle invaginazioni rivestite di clatrina, che infine sigillano a formare delle vescicole endocitotiche; queste ultime si fondono con i lisosomi, i cui enzimi provvedono alla degradazione del recettore e del proprio ligando. Nel corso dell’intero processo di internalizzazione il complesso ligando-recettore viene dissociato e questo si traduce in una terminazione del segnale. In alcuni casi i recettori possono anche essere riciclati [14].
2.4 Le tirosin-chinasi: target della terapia anti-tumorale
In condizioni fisiologiche il livello di fosforilazione delle tirosin-chinasi è finemente regolato dall’’effetto antagonista di tirosin-chinasi e tirosin-fosfatasi [14]; tuttavia, come già accennato nel capitolo precedente, attraverso diversi meccanismi le tirosin-chinasi possono acquisire funzioni trasformanti, che le rendono, da un lato capaci di innescare la proliferazione e la differenziazione cellulare e dall’altro insensibili ai meccanismi di retroinibizione. L’attivazione oncogenica è generalmente il risultato di una mutazione; diversi casi di glioblastoma multiforme, ad esempio, sono da ricondurre alla delezione di un amminoacido nel dominio extracellulare dell’EGFR, che dà luogo ad un recettore, detto mutante EGFRv III (EGFR class III variant), in grado di promuovere la crescita cellulare anche in assenza del ligando. Analogamente, mutazioni puntiformi a carico del dominio extracellulare dell’FGFR-3 producono un residuo
20
di cisteina spaiato, che conduce ad una dimerizzazione abnorme del recettore, attraverso legami disolfuro intermolecolari.
Anche il circuito autocrino-paracrino rappresenta un importante meccanismo per l’attivazione costitutiva dei recettori tirosin-chinasici ed è frequentemente associato ad un’ampia varietà di tumori; questo loop si innesca ogni qualvolta il recettore è espresso in maniera abnorme rispetto al ligando associato oppure quando il ligando è overespresso in presenza del recettore affine. A titolo d’esempio, è stato osservato che la produzione congiunta del PDGFR e del suo ligando PDGF-A e PDGF-B negli astrociti è alla base di molti tumori cerebrali e gliomi [12].
Tali eventi favoriscono la progressione delle cellule sane in maligne, portando in ultimo a carcinogenesi.
Visto dunque il coinvolgimento delle tirosin-chinasi nell’eziopatogenesi del cancro, negli Stati Uniti la Food and Drug Administration ha approvato diversi farmaci per la terapia anti-tumorale e di malattie legate all’attivazione oncogenica di tirosin-chinasi. Questi appartengono a due differenti categorie: anticorpi monoclonali e inibitori tirosin-chinasici, che si legano al sito di legame dell’ATP, nel dominio tirosin-chinasico intracellulare [15].
2.4.1 Anticorpi monoclonali
Rappresentano una classe di prodotti biotecnologici, in grado di legarsi al dominio extracellulare dei recettori tirosin-chinasici.
Il primo anticorpo monoclonale ad essere stato approvato dall’FDA nel 1998 è stato il Trastuzumab (®Herceptin), per il trattamento del carcinoma mammario. Si
tratta di un anticorpo monoclonale umanizzato, in grado di legarsi al recettore HER-2 per il fattore di crescita epidermico (EGF), che risulta spesso overespresso nel tumore al seno. Il risultato finale dell’azione del farmaco consiste nell’internalizzazione del recettore e nella conseguente inibizione della progressione del tumore, sostenuta da una divisione cellulare incontrollata [14].
21
2.4.2 Inibitori tirosin-chinasici
Sono molecole a basso peso molecolare che, a differenza degli anticorpi monoclonali, attraversano la membrana plasmatica in virtù delle loro caratteristiche idrofobiche, potendo così facilmente interagire col dominio intracellulare dei recettori tirosin-chinasici o con molecole segnale intracellulari [11].
Per la maggior parte sono composti ATP-mimetici, appartenenti a tre differenti categorie:
- Inibitori tirosin-chinasici di tipo I: riconoscono la conformazione attiva del recettore, legandosi al sito di legame dell’ATP, attraverso uno dei legami idrogeno normalmente formati dall’ATP stesso;
- Inibitori tirosin-chinasici di tipo II: sono inibitori allosterici dell’attività chinasica recettoriale, in quanto competono indirettamente con l’ATP, legandosi alla tasca idrofobica adiacente al sito di legame dell’ATP. Questa tasca, come già precedentemente descritto, è creata dalla conformazione aperta del “motivo DFG” dell’ansa di attivazione, conformazione conosciuta anche come sito allosterico; - Inibitori tirosin-chinasici di tipo III: noti anche come inibitori “covalenti”. Essi si legano irreversibilmente a residui di cisteina, localizzati a livello di siti specifici delle chinasi, attraverso la condivisione di elettroni; il legame si stabilisce infatti tra l’atomo di S elettron-ricco della cisteina e un gruppo elettrofilo dell’inibitore [11]. Tra gli inibitori tirosin-chinasici assumono particolare rilevanza gli inibitori dell’angiogenesi tumorale che, come descritto da Hanahan e Weinberg, costituisce una delle sei principali alterazioni nella fisiologia cellulare, alla base della carcinogenesi. Tra le altre rientrano: un’autosufficienza nei segnali di crescita, una mancata sensibilità nei riguardi di fattori inibenti la crescita, l’evasione dalla morte cellulare programmata (apoptosi), un potenziale replicativo illimitato e l’evasione tissutale con conseguente formazione di metastasi [16].
22
2.5 Angiogenesi tumorale
Con il termine angiogenesi o neovascolarizzazione si intende la crescita di nuovi vasi sanguigni a partire da altri pre-esistenti.
Nell’adulto è promossa fisiologicamente durante il ciclo mestruale e la gravidanza e in condizioni patologiche nei processi di riparazione e rigenerazione tissutali [11]. Durante l’embriogenesi si affianca invece alla vasculogenesi nella formazione dei costituenti dei vasi sanguigni: cellule endoteliali, periciti e membrana basale. Le cellule endoteliali costituiscono lo strato più interno della parete vascolare e sono in diretto contatto con i periciti, cellule connettivali cui si attribuisce una funzione contrattile, mentre la membrana basale è uno strato sottile ed uniforme che riveste quasi l’intera lunghezza delle cellule endoteliali [17].
L’angiogenesi rappresenta anche, come già accennato, un processo critico nella progressione neoplastica; per supportare infatti la crescita della massa tumorale oltre gli 1-2 mm3 è richiesta la formazione di nuovi vasi sanguigni, in grado di
garantire l’apporto di ossigeno e nutrienti e di permettere la diffusione metastatica. Ne deriva che, mentre in condizioni normali l’angiogenesi è controllata dall’azione antagonista di fattori pro- e anti-angiogenici, nel cancro questo equilibrio risulta alterato, determinando così quello che viene definito “switch angiogenico” [11]. E’ comunque necessario sottolineare che, a confronto con i normali vasi sanguigni, i componenti e la struttura del sistema vascolare tumorale risultano differenti: i vasi sanguigni vedono infatti persa la loro organizzazione gerarchica in arteriole, capillari e venule, non sono più in intimo contatto con i periciti e sono soltanto debolmente connessi alla membrana basale [17].
Vista quindi la secrezione da parte delle cellule tumorali di fattori pro-angiogenici che, favorendo la proliferazione e la migrazione delle cellule endoteliali, vascolarizzano la massa tumorale, sono stati sviluppati diversi inibitori tirosin-chinasici anti-angiogenici. Essi agiscono bloccando la formazione di nuovi vasi sanguigni oppure distruggendo la rete capillare pre-esistente, limitando così l’apporto di nutrienti [12].
23
Dal momento che tra gli attivatori dell’angiogenesi, il fattore di crescita dell’endotelio vascolare (VEGF) è uno dei principali elementi regolatori in condizioni sia fisiologiche che patologiche, sono stati sintetizzati diversi composti anti-VEGF che, se associati a chemioterapia e/o radioterapia, permettono di ottenere risultati migliori rispetto a quelli attesi con una monoterapia.
Possiedono importanti proprietà angiogeniche anche il fattore di crescita basico dei fibroblasti (bFGF) e il fattore di crescita derivato dalle piastrine (PDGF), che facilita il reclutamento dei periciti e delle cellule muscolari lisce, oltre ad essere essenziale per la maturazione e la stabilità vascolare [11].
2.5.1 Funzioni biologiche del VEGF e trasduzione del segnale VEGFR-
mediata
La famiglia del fattore di crescita dell’endotelio vascolare è costituita da cinque membri, che agiscono tutti come importanti stimolatori della proliferazione e della migrazione delle cellule endoteliali: VEGFA, VEGFB, VEGFC, VEGFD e il fattore di crescita placentare (PIGF).
Gli effetti dei differenti VEGF sono mediati dal recettore tirosin-chinasico
VEGFR, la cui famiglia comprende tre recettori: VEGFR-1, -2 e -3 [11].
24
In accordo al modello precedentemente descritto, essi sono costituiti da una porzione extracellulare,che presenta sette domini simili alle immunoglobuline, da una singola regione transmembrana e da un dominio chinasico intracellulare, interrotto da una regione non catalitica, indicata in figura come KI (Kinase Insert). Il dominio extracellulare del VEGFR-1 è anche espresso indipendentemente come proteina solubile. VEGFR-1 e VEGFR-2 sono localizzati a livello dell’endotelio vascolare e VEGFR-1 anche nei monociti, mentre VEGFR-3 è preferenzialmente espresso a livello linfatico.
VEGFR-1 e VEGFR-2 legano entrambi il VEGFA, meglio noto come VEGF e
principale mediatore dell’angiogenesi; sembra tuttavia che VEGFR-1 agisca prevalentemente come modulatore negativo del VEGFR-2, attraverso la forma solubile del suo dominio extracellulare. Il VEGFR-3 è membro della stessa famiglia di recettori tirosin-chinasici, ma non è recettore del VEGF; lega infatti VEGFC e VEGFD [18].
Il VEGF interagisce inoltre con una famiglia di co-recettori, le neurolipine ed in particolare con la neurolipina 1 (NP-1): una proteina transmembrana non tirosin-chinasica, inizialmente identificata come recettore per i membri della famiglia delle semaforine, implicate nella crescita assonale [19].
Il VEGF è espresso in condizioni di ipossia, quali quelle che caratterizzano diverse condizioni patologiche, stimolato dal fattore di trascrizione HIF-1 (hypoxia-inducible
factor 1), che si lega all’elemento di risposta all’ipossia, all’interno del promoter del
gene codificante il VEGF.
Tale gene è costituito da otto esoni separati da sette introni; lo splicing alternativo, attraverso un arrangiamento degli esoni, produce quattro isoforme: VEGF121,
VEGF165, VEGF189 e VEGF206, differentemente nominate sulla base degli
amminoacidi costituenti. VEGF165, privo dei residui codificati dall’esone 6,
rappresenta l’isoforma predominante; è una glicoproteina omodimerica di 45 kDa legante l’eparina e che viene secreta, sebbene una frazione significativa rimanga legata alla superficie cellulare e alla matrice extracellulare [19].
25
Il VEGF è stato inizialmente identificato come fattore di permeabilità vascolare, grazie alla sua capacità di indurre, supportato presumibilmente da fattori vasodilatatori come NO e la prostaciclina PGI2, la formazione di fenestrazioni
endoteliali, ovvero di regioni specializzate della membrana plasmatica, altamente permeabili a piccoli soluti. Tali aperture nella parete vascolare sono tipiche dei glomeruli renali, del tratto gastrointestinale, delle ghiandole endocrine e di particolari aree cerebrali, mentre sono assenti nel resto del cervello, nel muscolo scheletrico, nella pelle e nel fegato. Anche la neovascolarizzazione indotta dal VEGF nel glioblastoma è altamente fenestrata [18] e l’aumento di permeabilità dei vasi sanguigni tumorali comporta un incremento della pressione interstiziale, che inverte i gradienti pressori normalmente presenti nel tessuto, impedendo la penetrazione delle molecole, inclusi i farmaci chemioterapici.
Un importante meccanismo cellulare attraverso il quale il VEGF promuove la formazione di nuovi vasi sanguigni, preservandone l’integrità, è rappresentato dall’attivazione di fattori anti-apoptotici e di vie del segnale, che mediano la sopravvivenza cellulare. Quest’ultima è infatti garantita, in parte dall’attivazione PI3K-mediata della chinasi anti-apoptotica Akt/PKB, in parte da effetti a lungo termine, legati ad una upregulation dei componenti che costituiscono il macchinario cellulare anti-apoptotico [18].
In particolare, a trasdurre gli effeti pro-angiogenici del VEGF è il VEGFR-2, il cui residuo tirosinico fosforilato crea un sito di legame per proteine segnale, quali la fosfolipasi C-γ (PLCγ) e le molecole adattatrici Grb2 (growth factor receptor-bound
26
Come schematizzato nella fig.10A, la PLCγ fosforila la proteina chinasi C (PKC), che a sua volta fosforila una serie di mediatori molecolari, quali le chinasi MEK ed ERK (extracellular-signal-regulated kinase), che attivano la via delle MAPK, in grado di promuovere la trascrizione genica di fattori che sono noti regolare la proliferazione cellulare. Un’altra molecola segnale coinvolta nella cascata delle MAPK è la proteina Grb2, che contiene domini SH2 e SH3 ed è in grado di attivare la proteina G Ras, che, attraverso il legame e la fosforilazione della proteina Raf, attiva la via MEK/MAPK.
Il legame della proteina adattatrice Shb al VEGFR-2 attiva invece la via del fosfatidilinositolo-3-chinasi (PI3K) che sembra tra l’altro essere coinvolta nella migrazione cellulare [11] e che risulta geneticamente alterata in un’ampia gamma di tumori, tra cui il carcinoma tiroideo. Ciò che viene attivata è la subunità catalitica del PI3K, che fosforila il fosfatidilinositolo-4,5-bifosfato a produrre il fosfatidilinositolo-3,4,5-trifosfato (PIP3), la cui funzione consiste nel traslocare sulla membrana cellulare la proteina Akt [20] : una serina-treonina chinasi ad azione anti-apoptotica, che stimola la proliferazione e la sopravvivenza cellulare, attraverso l’attivazione o l’inibizione di un’ampia varietà di substrati. Come schematicamente rappresentato in fig.10B, la proteina chinasi B fosforila ed inibisce la proteina pro-apoptotica BAD (Bcl-2 associated death promoter), così come
27
la GSK3 (glycogen synthase kinase 3). Akt è inoltre in grado di attivare la proteina effettrice mTOR (mammalian target of rapamicina) [11], che è stato dimostrato avere un ruolo ben definito nella carcinogenesi [19] e la NO sintasi endoteliale (eNOS) a livello della serina 1177, coinvolta nel mediare la migrazione delle cellule endoteliali, attraverso un processo di degradazione della membrana basale, che assicura l’angiogenesi in condizioni fisiopatologiche.
La migrazione cellulare indotta dal VEGF è favorita inoltre dalla fosforilazione della proteina tirosin-chinasica non recettoriale FAK (focal adhesion kinase) e da una via indipendente, che vede attivata la p38 MAP chinasi. Essenziale al riguardo è anche la produzione di NO da parte del VEGF stesso; l’ossido nitrico è infatti implicato nel regolare il movimento scalare non-chemiotattico delle cellule endoteliali e la fosforilazione del residuo tirosinico della proteina FAK [18].
Visto quindi il coinvolgimento della neovascolarizzazione nel sostenere la crescita tumorale, negli ultimi decenni è stata sviluppata una nuova strategia di trattamento anti-neoplastica, basata sull’inibizione dell’angiogenesi. In effetti le terapie anti-angiogeniche hanno dimostrato un’efficacia clinica in diversi tipi di cancro, sebbene siano suscettibili allo sviluppo di tossicità e farmaco-resistenza, considerato il coinvolgimento di più vie del segnale ed elementi regolatori [11].
2.5.2 Inibitori tirosin-chinasici anti-angiogenici
Molti di essi sono denominati inibitori multi-target, poiché attivi nei riguardi di differenti chinasi; ne deriva che questi agenti possiedono uno spettro di attività più ampio rispetto ad inibitori a singolo target [11].
La FDA negli Stati Uniti ha approvato tre farmaci anti-VEGF, che agiscono bloccando il ligando oppure inibendo la funzione del suo recettore VEGFR: bevacizumab, sunitinib malato e sorafenib.
Il bevacizumab è stato il primo inibitore anti-VEGF ad essere stato impiegato per uso sistemico nel cancro. Si tratta di un anticorpo monoclonale umanizzato che lega selettivamente il VEGF; è attualmente approvato in associazione al
5-28
fluorouracile per via e.v. nel trattamento di prima o seconda scelta del carcinoma metastatico del colon o del retto e in associazione al paclitaxel e al carboplatino, per il trattamento di prima scelta del carcinoma polmonare non a piccole cellule. Mostra anche attività nel tumore mammario e in quello del fegato.
Il sunitinib e il sorafenib agiscono entrambi come inibitori multipli di tirosin-chinasi implicate nella crescita tumorale, nell’angiogenesi e nella progressione metastatica [20]. Il primo rientra nella classe di inibitori tirosin-chinasici di tipo I, l’altro invece compete indirettamente con l’ATP, occupando la tasca idrofobica del dominio chinasico (inibitore tirosin-chinasico di tipo II) [11].
Il sunitinib è approvato per il trattamento del carcinoma renale avanzato e dei tumori stromali gastrointestinali, in caso di progressione della malattia o di intolleranza all’imatinib mesilato. Il sorafenib è anch’esso approvato nel tumore renale avanzato.
D’altro canto, come precedentemente accennato, questi agenti non sono privi di tossicità; il bevacizumab è generalmente ben tollerato, ma può essere accompagnato da effetti collaterali, intensificati dalla co-somministrazione di agenti chemioterapici, che includono: ipertensione, proteinuria, infezioni delle alte vie respiratorie, epistassi, dispnea, stanchezza e dermatiti esfoliative.
Effetti indesiderati simili sono propri anche del sorafenib e del sunitinib e sono essenzialmente correlati all’inibizione del VEGF; ad esempio, l’azione pro-ipertensiva di questi composti è da ascrivere ad una riduzione della produzione di NO. In condizioni normali infatti, il fattore di crescita dell’endotelio vascolare promuove la sintesi di NO, attraverso un’up-regulation dell’enzima NO-sintasi; l’inibizione del VEGF causa un decremento della concentrazione di questo agente vasodilatatore, promuovendo vasocostrizione e aumento della pressione sanguigna [21]. Oltre a ciò, i composti anti-angiogenici causano un’alterazione della funzionalità piastrinica, che si traduce in complicanze emorragiche e disturbi nei processi di guarigione delle ferite.
29
Un altro limite della terapia anti-angiogenica è dato dalla farmaco-resistenza; la maggior parte dei pazienti infatti trae un beneficio solo temporaneo dal trattamento in questione e il tumore ben presto comincia nuovamente a crescere e a metastatizzare. Inoltre una piccola percentuale di pazienti non riesce neanche a mostrare un iniziale beneficio clinico. Ciò potrebbe essere dovuto al fatto che, nonostante la via del segnale mediata dal VEGF sia predominante nello stimolare l’angiogenesi, tuttavia altre vie parallele sostengono la crescita tumorale.
In altri casi la resistenza agli inibitori tirosin-chinasici è il risultato di una mutazione a carico della proteina chinasi target, come accade per il gefitinib, un inibitore del fattore di crescita epidermico.
Per ridurre l’incidenza di questo fenomeno o comunque ritardarne l’insorgenza, è possibile intervenire associando gli inibitori tirosin-chinasici ad altri agenti anti-angiogenici, quali il bevacizumab, oppure alla convenzionale chemioterapia citotossica [11].
30
Capitolo 3
-
Trattamento farmacologico del carcinoma
tiroideo avanzato
La prognosi della maggior parte dei tumori tiroidei è generalmente buona, tuttavia i casi di carcinoma allo stadio avanzato sono di difficile controllo e i relativi trattamenti risultano deludenti [22]. Chirurgia, radioterapia a fasci esterni, chemioembolizzazione e bifosfonati non hanno infatti prodotto risposte durevoli né benefici in termini di sopravvivenza [23]. La situazione sta fortunatamente cambiando grazie alla disponibilità di inibitori tirosin-chinasici multi-target; la
Food and drug admistration ad oggi ha approvato due farmaci per il cancro tiroideo
avanzato o metastatico: vandetanib e cabozantinib. La loro efficacia terapeutica è stata valutata utilizzando, come endpoint primario, il parametro PFS
(progression-free survival), che risulta essere più che raddoppiato [24].
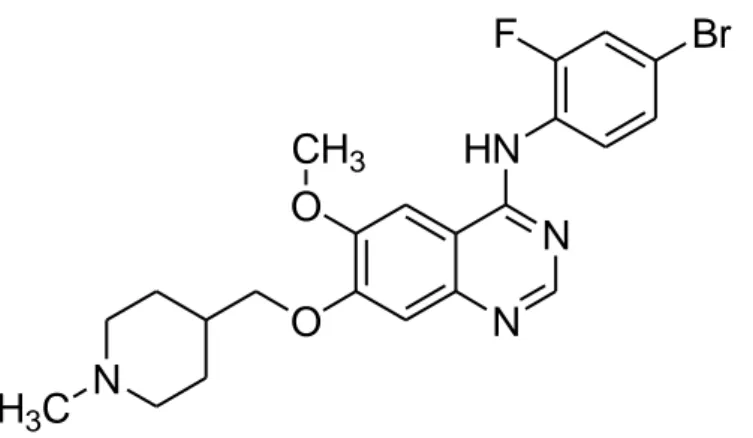
3.1 Vandetanib (ZD6474)
N O N N HN H3C F Br O CH3 Il vandetanib (N-(4-bromo-2-fluorofenil)-6-metossi-7-[(1-metil-4-piperidinil) metossil]-4-chinazolinammina) è approvato in Europa e negli Stati Uniti per il trattamento del carcinoma midollare tiroideo non asportabile, localmente avanzato (stadio III) o metastatico: si tratta di una molecola ATP-mimetica a basso peso31
molecolare, attiva per os, che agisce come potente inibitore in vitro di VEGFR-2 (IC50 40 nM), VEGFR-3 (IC50 110 nM), EGFR (IC50 500 nM) e della RET chinasi
(IC50 100 nM) [23]. In quest’ultimo caso l’inibizione si manifesta nei riguardi sia
della forma wild-type sia di diverse forme mutanti dell’enzima, eccezion fatta per le proteine recanti mutazioni a carico del residuo V804 (V804L e V804M): il valore di IC50 risulta infatti incrementato di circa 50 volte. Per questa ragione si ritiene
che le mutazioni V804, riscontrate nei casi di neoplasia endocrina multipla di tipo 2 e MTC sporadico, siano alla base della resistenza al vandetanib [23].
Per quel che concerne il profilo farmacodinamico, la dose raccomandata è di 300 mg una volta al giorno; dopo somministrazione orale, il vandetanib viene lentamente assorbito e la massima concentrazione plasmatica viene raggiunta dopo circa 6 ore. Si lega fortemente (92-94%) alle proteine plasmatiche ed è metabolizzato dall’isoforma 3A4 del CYP450 a N-desmetilvandetanib e al metabolita N-ossido dalle mono-ossigenasi contenenti flavina, FMO1 e FMO3, nel rene e nel fegato, rispettivamente. In termini di inibizione in vitro, la forma demetilata ha una potenza inibitoria nei riguardi di VEGFR-2 e dell’EGFR simile al vandetanib, mentre l’N-ossido è circa 50 volte meno attivo. Il farmaco viene escreto per via biliare e urinaria [23].
Studi in vivo hanno dimostrato la capacità del vandetanib di inibire, attraverso più vie del segnale intracellulari riassunte in fig.12, la neovascolarizzazione e la crescita tumorale in un’ampia gamma di modelli di xenotrapianti tumorali umani (seno, polmone, prostata, colon e ovaie), a dispetto della loro differente origine istologica [25].
32
Nel capitolo precedente è stato dato ampio spazio alla trattazione del ruolo del
VEGFR in condizioni fisiopatologiche; per quel che riguarda l’EGFR, occorre
invece sottolineare che la famiglia di questo recettore tirosin-chinasico include quattro membri: EGFR (ErbB-1), ErbB-2, ErbB-3 e ErbB-4, attivati a seguito del legame al loro dominio extracellulare di fattori di crescita peptidici.
La via mediata dall’EGFR gioca un ruolo chiave nel promuovere la crescita e la sopravvivenza di vari tipi di tumori solidi e l’utilizzo di agenti anti-EGFR è in grado di inibire la proliferazione e la migrazione delle cellule endoteliali [25].
Studi pre-clinici hanno suggerito che l’inibizione dei due differenti meccanismi che contribuiscono alla crescita tumorale, l’angiogenesi sostenuta dal VEGFR da un lato e la proliferazione EGFR-indotta dall’altro, permetta di ottenere risultati migliori, rispetto a quelli derivanti dal blocco di un’unica via, impedendo così alle cellule tumorali di sottrarsi all’attività di agenti citotossici e biologici [25].
L’efficacia del vandetanib per via orale, nel trattamento dei pazienti con MTC non resecabile, localmente avanzato o metastatico, è stata saggiata in tre
33
sperimentazioni cliniche; tra queste rientrano anzitutto due piccoli studi di fase II, cui sono stati sottoposti pazienti affetti dalla forma ereditaria e nei quali il vandetanib, alla dose di 100 o 300 mg una volta al giorno, ha dimostrato una significativa attività antitumorale, stabilizzando la malattia in circa il 53% dei casi. Ben più rilevante è la sperimentazione multi-nazionale, randomizzata, in doppio cieco, ZETA, che mette a confronto il vandetanib, alla dose di 300 mg/die, con il placebo in pazienti con MTC sporadico o ereditario; tale studio ha permesso di constatare come il vandetanib, durante un follow-up della durata media di due anni, sia in grado di migliorare notevolmente, rispetto al placebo, l’endpoint primario PFS, oltre ad un’ampia gamma di endpoint secondari, quali il tasso di controllo della malattia, il tempo di peggioramento del dolore e la valutazione di risposta biochimica (determinata attraverso cambiamenti della concentrazione plasmatica di calcitonina). E’ stato inoltre possibile dimostrare che il vandetanib presenta un beneficio clinico in termini di PFS, non solo nei pazienti recanti una mutazione a carico della chinasi RET, ma anche nel sottogruppo per il quale tale mutazione non compare.
Nella sperimentazione ZETA, quasi tutti i pazienti (99.6%) hanno manifestato almeno un effetto avverso:
34
alcuni tra i più comuni (diarrea, rash ed ipertensione) sono legati all’attività farmacologica del vandetanib, quale inibitore del VEGFR e dell’EGFR. La maggior parte di essi è comunque gestibile, attraverso una riduzione del dosaggio.
L’effetto collaterale che desta maggior preoccupazione è rappresentato invece da un prolungamento asintomatico dell’intervallo QT, alla luce della lunga emivita del farmaco [23]; tale effetto richiede particolare attenzione, perché correlato al rischio di sviluppare aritmie cardiache con torsioni di punta, che possono causare decessi improvvisi [25]. Per questa ragione, l’impiego del farmaco è limitato, attraverso l’utilizzo di una strategia di valutazione e riduzione del rischio (REMS).
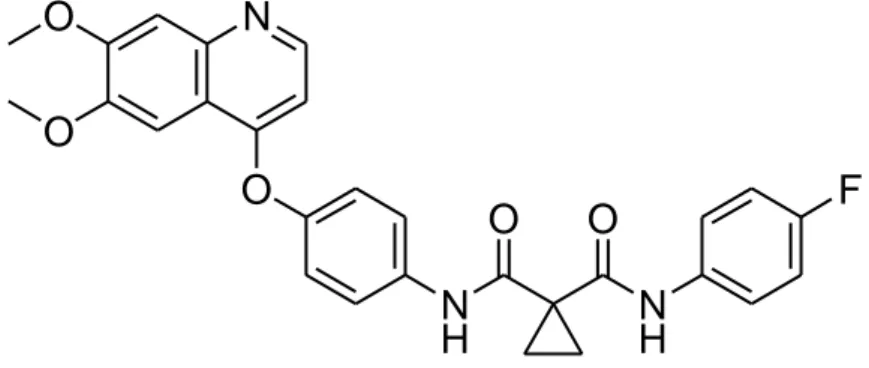
3.2 Cabozantinib (XL 184)
N O O O N H NH F O O Il cabozantinib (N–4-(6,7-dimetossichinolin-4ilossi)fenil)-N-(4-fluorofenil) ciclopropano-1,1-dicarbossammide) ha ricevuto l’approvazione dall’FDA per il trattamento del carcinoma midollare tiroideo nel novembre 2012. E’ un potente inibitore tirosin-chinasico, biodisponibile per os, in grado di bloccare più vie del segnale, per interazione con i recettori VEGFR-2 (IC50 0.035 nM), RET (5.2 nM) eMET-mesenchymal-epithelial transition factor- (IC50 1.3-14.6 nM).
MET è un proto-oncogene che codifica per il recettore del fattore di crescita degli epatociti, indicato come c-MET, un recettore tirosin-chinasico della superficie cellulare. Il fattore di crescita degli epatociti è prodotto in condizioni di ipossia e
35
promuove la neovascolarizzazione per ripristinare l’apporto di ossigeno ai tessuti; in condizioni fisiologiche, il recettore attivato innesca una cascata di segnali, mediando così la divisione e la motilità cellulare, in particolare per quel che riguarda le cellule endoteliali ed è di ausilio nei processi di angiogenesi e riparazione delle ferite.
Il proto-oncogene MET risulta frequentemente mutato, overespresso o amplificato in diversi tipi di tumore, tra cui il carcinoma midollare tiroideo; il risultato è l’attivazione costitutiva di vie del segnale a valle, quali quelle FAK, PI3K/Akt e Ras-Raf-Mek-MAPK, già menzionate, che promuovono la replicazione cellulare e una riduzione dell’apoptosi, oltre a favorire il processo di migrazione, responsabile della formazione di metastasi [26].
Studi pre-clinici, condotti su modelli di xenotrapianto, hanno dimostrato che la somministrazione orale del cabozantinib determina una rapida e significativa inibizione della crescita neoplastica, accompagnata da una regressione della vascolarizzazione tumorale e da una maggiore sopravvivenza [27].
Un trial clinico di fase I ha invece consentito di determinarne la farmacocinetica: il composto è rapidamente disponibile dopo somministrazione orale, raggiungendo un picco di concentrazione plasmatica dopo 5 ore. La dose raccomandata è di 175 mg una volta al giorno (corrispondente alla massima dose tollerata).
La risposta al cabozantinib, registrata nei casi di MTC mediante il suddetto trial, ha condotto ad una più ampia sperimentazione di fase III, nota come EXAM (Efficacy of XL184 in Advanced Medullary Thyroid Cancer); analogamente alla sperimentazione ZETA relativa al vandetanib, essa costituisce un trial internazionale, a doppio cieco, randomizzato, che compara il cabozantinib alla dose di 140 mg al giorno con il placebo, in pazienti con carcinoma midollare tiroideo avanzato o metastatico. I risultati ottenuti indicano che il farmaco ha una significativa attività in tali pazienti, inclusi quelli pre-trattati con terapie citotossiche; nel 47.3% dei soggetti sottoposti alla terapia, è stata registrata una
36
sopravvivenza senza progressione della malattia pari ad un anno, contro il 7.2% del placebo. L’endpoint secondario di tasso di risposta globale è stato del 28% per il gruppo trattato con cabozantinib, pari invece allo 0% per coloro cui era somministrato il placebo.
Tuttavia il farmaco non è privo di effetti collaterali, poiché ciascuna via che viene inibita ha un ruolo rilevante nelle normali funzioni cellulari e nel mantenimento dell’omeostasi. I più comuni effetti avversi includono diarrea e la sindrome mano piede, caratterizzata dall’arrossamento ed eventualmente dalla fessurazione del palmo delle mani e/o della pianta del piede, che impone una sospensione della chemioterapia.
Altri inibitori tirosin-chinasici hanno prodotto risultati promettenti nel trattamento del carcinoma midollare tiroideo; alcuni di essi sono già stati saggiati
in vitro e valutati attraverso trial clinici di fase II. In un’ampia percentuale di casi la
malattia può essere stabilizzata e alcuni pazienti mostrano anche una risposta parziale [28].
E’ questo il caso del sorafenib, ad attività inibente nei riguardi delle chinasi RAF,
RET e VEGFR; in uno studio di fase II condotto su 21 pazienti, trattati per un
periodo minimo di 16 settimane, si è dimostrato in grado sia di produrre una risposta nel 10% dei casi sia di stabilizzare la malattia (86%). Il farmaco inoltre si è
37
rivelato essere maggiormente efficace nei casi di carcinoma papillare, specialmente in quelli recanti una mutazione BRAF ed inefficace nel carcinoma follicolare e in quello anaplastico, oltre che nei casi di DTC scarsamente differenziato.
L’axitinib invece è in grado di inibire il solo VEGFR e il tasso di risposta registrato è la prova di un significativo effetto anti-angiogenico.
Il pazopanib agisce come inibitore del VEGFR e del PDGFR; in un trial di fase II, cui sono stati sottoposti 39 pazienti con carcinoma tiroideo differenziato a rapida progressione e refrattario alla terapia radiometabolica, il tasso di risposta è stato del 49% [23].
Il motesanib inibisce VEGFR, RET e PDGFR. La sperimentazione di fase II ha in questo caso dimostrato una risposta parziale pari al 2%; la malattia è inoltre stabilizzata nell’81% dei casi.
Ricordiamo infine l’imatinib e il gefitinib, che sono stati testati in pazienti affetti da MTC e/o DTC ma che non hanno mostrato alcun effetto antitumorale [21].
38
Capitolo 4
-
Introduzione alla parte sperimentale
Il laboratorio presso il quale ho svolto il mio lavoro di tesi è da tempo impegnato nello sviluppo di inibitori tirosin-chinasici, al fine di ottenere candidati farmaci per il trattamento del carcinoma tiroideo.
Uno degli scaffold privilegiati nel campo di questa classe di composti è rappresentato dal nucleo pirazolo[3,4-d]pirimidinico; ad oggi è stata testata l’efficacia su linee cellulari di carcinoma tiroideo umano di due derivati:
N N N NH2 PP1 PP2 N CH3 N N N NH2 N Cl 1-tert-butil-3-p-tolil-1H-pirazolo
[3,4-d]pirimidin-4-ammina 1-tertbutil-3-(4-clorofenil)-1H-pirazolo[3,4-d]pirimidin-4-ammina
Figura 15. Derivati pirazolo[3,4-d]pirimidinici attualmente studiati nel carcinoma tiroideo Questi composti hanno dimostrato potenti proprietà inibitorie nei confronti della RET chinasi e sono pertanto capaci di ridurre la proliferazione cellulare e, per quel che riguarda il PP1, il fenotipo invasivo di carcinoma tiroideo, sostenuto da riarrangiamenti RET/PTC1 (IC50≈30 µM) [29]. D’altra parte mentre la RET
chinasi è altamente sensibile al PP1, in virtù di un residuo di glicina, situato sul fondo della tasca idrofobica adiacente al sito di legame dell’ATP, in cui si inserisce la porzione fenilmetilica del composto [29], non vale altrettanto per il PP2; questo infatti non è selettivo per la RET, poiché in grado di inibire anche la proteina c-Src, un effettore essenziale a valle della via RET-mediata, e chinasi correlate. Questo non permette peraltro di escludere addizionali effetti indiretti in vivo [30]. Recentemente l’attenzione è stata focalizzata sulla messa a punto di una terapia mirata alla cura del carcinoma papillare tiroideo dedifferenziato (DePTC),
39
frequentemente associato a mutazioni V600EBRAF; come già menzionato nel
capitolo primo, il carcinoma papillare tiroideo è infatti solitamente curabile combinando chirurgia, ablazione con radioiodio e terapia soppressiva a base di levotiroxina; tuttavia in circa il 5% dei casi le cellule possono regredire ad uno stato di dedifferenziazione, di solito accompagnata da una crescita tumorale più aggressiva, dalla diffusione di metastasi e dalla perdita di capacità di uptake dello iodio, fattori questi che rendono il tumore refrattario ai tradizionali approcci terapeutici.
A tale riguardo, il gruppo di ricerca presso cui ho svolto la mia tesi sperimentale ha sviluppato due nuovi derivati a nucleo pirazolo[3,4-d]pirimidinico (CLM3 e CLM29): N N N N HN N N N N HN CLM3 CLM29 CH3
Figura 16. Nuovi derivati a nucleo pirazolo[3,4-d]pirimidinico
I due composti risultano attivi nei riguardi di diversi target, tra cui la RET chinasi, EGFR e VEGFR, in particolare VEGFR-2; la loro attività antitumorale, saggiata sia in vitro che in vivo, si esplica attraverso un’inibizione concentrazione- e tempo-dipendente sia della proliferazione cellulare, accompagnata da un incremento dell’apoptosi, sia della migrazione. Nella figura 17, sono riportati i risultati del saggio WST-1, che rappresenta un metodo sensibile ed accurato per la valutazione della proliferazione cellulare, basato sulla riduzione del sale di tetrazolio WST-1 da parte delle deidrogenasi cellulari:
40
Le concentrazioni di CLM3 e CLM29 utilizzate sono state di 1, 10, 30 e 50 µM; nelle cellule primarie di DePTC il CLM3 è in grado di inibire in maniera significativa, rispetto al controllo, la proliferazione cellulare (Fig. 17A) e risultati analoghi sono stati ottenuti con il CLM29 (Fig. 17B).
I risultati del saggio condotto su cellule tiroidee follicolari sane (Fig. 17C e Fig. 17D) mostrano invece una piccola seppur significativa riduzione della proliferazione rispetto al controllo.
Per il CLM3 è stata inoltre dimostrata una notevole capacità nell’inibire la crescita tumorale e la neovascolarizzazione neoplastica in assenza di un’apprezzabile tossicità; questi effetti sono stati saggiati in vivo attraverso l’iniezione sottocutanea in topi CD nu/nu della linea cellulare AL, ottenuta a partire da cellule primarie di DePTC e recante una mutazione V600EBRAF.
La massa tumorale è divenuta detectabile dopo 10 ore dallo xenotrapianto e negli animali di controllo ha mostrato un progressivo ma lento incremento delle proprie
Figura 17. Risultati del saggio WST-1 in cellule di DePTC o di controllo, trattate con CLM3 o
CLM29 per 24h [31]
41
dimensioni; al quarantesimo giorno gli animali di controllo e quelli trattati sono stati sacrificati.
I risultati ottenuti hanno rivelato che il CLM3 alla dose di 40 mg/kg die è in grado di inibire in maniera rilevante la crescita tumorale, con un effetto terapeutico divenuto significativo a partire dal diciannovesimo giorno dall’impianto cellulare (quattro giorni dopo l’inizio del trattamento). Il CLM3 inoltre non ha mostrato effetti tossici, come si evince dalla modesta perdita di peso degli animali trattati rispetto al controllo (Fig. 18B).
I meccanismi che sottendono invece all’azione anti-angiogenica del CLM3 sono presumibilmente correlati ad una up-regulation di uno dei principali inibitori endogeni dell’angiogenesi, il TSP-1 (trombospondilin-1). Il CLM3, come d’altro canto anche il CLM29, incrementa infatti nella linea cellulare AL l’espressione del TSP-1, che da un lato è in grado di promuovere l’apoptosi delle cellule endoteliali e dall’altro interagisce con molte proteine extracellulari, coinvolte nel processo di neovascolarizzazione, tra cui il VEGF [31].
Inoltre l’attività anti-proliferativa, saggiata attraverso un’inibizione dell’espressione del gene codificante per la ciclina D1 coinvolta nel ciclo cellulare e quella pro-apoptotica del CLM3 si manifestano non solo nei confronti delle cellule tumorali, ma anche di quelle endoteliali:
42
La linea cellulare HMVEC-d (human dermal microvascular endothelial cells) è particolarmente sensibile a basse concentrazioni di CLM3 (IC50 0.40±0.22 nM
dopo 72h di esposizione), mentre linee cellulari di carcinoma tiroideo, come quella dedifferenziata 8305C (IC50 9.20±5.06 µM dopo 72h di esposizione), che
esprime la RET endogena o la linea cellulare TT (IC50 26.93±7.60 µM dopo 72h
di esposizione), che esprime invece la forma mutata, richiedono concentrazioni più elevate affinché possa essere inibita la loro crescita.
Ne deriva che l’eccellente efficacia anti-angiogenica del CLM3 è accompagnata da un’attività inibitoria meno marcata nei riguardi della RET chinasi.
Nella figura sottostante sono invece riportati gli effetti pro-apoptotici del CLM3: Figura 19. Effetti del CLM3 in vitro sulla proliferazione cellulare dopo 72h di esposizione [32]
43
Dopo 72h di trattamento è stata riscontrata una significativa percentuale di apoptosi sia nelle cellule endoteliali HMVEC-d sia nelle linee cellulari 8305C e A549, seppure a più alte concentrazioni [32].
I risultati ottenuti sono estremamente incoraggianti ed hanno avviato la ricerca verso una ottimizzazione del CLM3, allo scopo di chiarire i requisiti strutturali, essenziali al raggiungimento di una piena e concomitante inibizione di VEGFR-2 e della RET chinasi, così da ottenere inibitori VEGFR-2/RET ad azione duale. Studi preliminari di docking, condotti sulle proteine target, hanno rivelato che il CLM3 è in grado di legarsi al sito di legame dell’ATP, in maniera del tutto analoga ad altri inibitori tirosin-chinasici; ciò è stato successivamente confermato saggiando il composto sulle proteine target ricombinanti umane.
Di seguito viene riportata la modalità di legame del lead in questione a VEGFR-2 (Fig. 21) e alla RET chinasi (Fig.22):
- L’atomo di azoto in posizione 2 dell’anello pirazolopirimidinico stabilisce un legame idrogeno (sopra indicato attraverso una linea nera tratteggiata) con il residuo di cisteina in posizione 919 dello scheletro principale;
- la porzione feniletilamminica è invece circondata dai residui amminoacidici delle catene laterali;

![Figura 3. Rappresentazione schematica della struttura della RET chinasi [9]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7633120.117535/11.892.279.645.128.605/figura-rappresentazione-schematica-struttura-ret-chinasi.webp)
![Figura 5. Struttura di un recettore tirosin-chinasico [11]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7633120.117535/15.892.286.763.582.818/figura-struttura-recettore-tirosin-chinasico.webp)
![Figura 7. Sito di legame dell'ATP delle tirosin-chinasi [14]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7633120.117535/16.892.268.689.433.762/figura-sito-legame-atp-tirosin-chinasi.webp)
![Figura 9. Ligandi VEGF e recettori VEGFR [18]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7633120.117535/23.892.222.764.790.1116/figura-ligandi-vegf-recettori-vegfr.webp)
![Figura 10. Via di trasduzione del segnale mediata dai recettori tirosin-chinasici nell'angiogenesi [11]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7633120.117535/26.892.162.805.163.440/figura-trasduzione-segnale-mediata-recettori-tirosin-chinasici-angiogenesi.webp)
![Figura 13. Tollerabilità del trattamento orale con vandetanib [23]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7633120.117535/33.892.205.791.803.1127/figura-tollerabilità-trattamento-orale-vandetanib.webp)
![Tabella 1. Inibitori TK già sottoposti a sperimentazione clinica [28]](https://thumb-eu.123doks.com/thumbv2/123dokorg/7633120.117535/36.892.174.805.737.904/tabella-inibitori-tk-già-sottoposti-sperimentazione-clinica.webp)
