analisi della significatività statistica
In questo capitolo verranno analizzate alcune tecniche utilizzate per individuare quali geni risultano differenzialmente espressi in un esperimento di microarray.
La tecnica più semplice fa uso di una soglia empirica sull’istogramma del “fold change”, cioè del rapporto fra le intensità dei due canali, per stabilire l’insieme dei geni che presentano espressione differenziale.
Un metodo maggiormente adattativo è quello della distanza dalla media, che pone una soglia dimensionata in base alla varianza della distribuzione dei rapporti delle intensità.
Il criterio della soglia non lineare cerca, invece, di stabilire il livello di abbattimento del rumore sui dati in modo da poter modellare una soglia che tenga conto di questa informazione aggiuntiva.
Nessuno di questi metodi produce una valutazione statistica degli errori che possono essere commessi nell’affermare che un gene è differenzialmente espresso. Un miglioramento in questo senso viene realizzato con l’analisi della significatività statistica dei microarray o SAM (Significance Analysis of Microarray), che propone il False Discovery Rate (FDR) come parametro statistico di confidenza nella risposta ottenuta.
3.1 “Fold change”
I dati di espressione genica ottenuti dal processo di normalizzazione possono essere rappresentati sottoforma di matrice; ogni riga della tabella individua un gene, o una sua sequenza specifica, mentre ogni colonna contiene, generalmente, valori quali la media e/o la mediana sia del segnale che del background, la mediana del segnale con il background sottratto, i punteggi assegnati ad ogni spot dal controllo di qualità, le percentuali di pixel saturati in ciascun canale per ogni spot, le posizioni degli spot sul microarray, i nomi dei geni e i loro codici identificativi nelle banche dati ed altro ancora.
Di particolare importanza è il valore relativo di espressione genica, anche detto “fold change” , definito come il rapporto fra le intensità di segnale misurate in entrambi i canali.
fold change = R/G
dove R è l’intensità del segnale generato dal campione marcato con il fluorocromo Cy5 e G è quella prodotta dal campione marcato con il Cy3 per ogni spot. Il “fold change” è, dunque, il parametro che, in prima istanza, può essere utilizzato per dichiarare se un gene è differenzialmente espresso in uno dei due canali.
Il valore di questa grandezza, mai negativo in quanto rapporto di quantità positive o nulle, è compreso nell’intervallo [0,+∞): i geni sovraespressi sono identificati da valori del “fold change” compresi in (1,+∞), mentre i geni sottoespressi presentano valori in [0,1).
E’ evidente che valori del “fold change” più grandi o più piccoli di 1 indicano una maggiore o minore espressione del gene in uno dei due
canali, mentre valori prossimi ad 1 indicano un’espressione simile in entrambi i canali. 0 log2 = G R 0 +∞ = G R 2 log −∞ = G R 2 log 1 = G R +∞ = G R
Figura 3.1: Rappresentazione del fold change in scala lineare e in scala
logaritmica
Per ovviare alla differente ampiezza dei due intervalli di valori di espressione viene solitamente applicata una trasformazione logaritmica ai dati in modo tale che non sia più il semplice rapporto delle intensità il valore di riferimento ma la sua trasformata logaritmica, generalmente in base 2.
In conseguenza di ciò gli intervalli di variazione assumono dimensioni uguali, rispettivamente ]0, +∞] per i geni sovraespressi e [-∞,0[ per i geni sottoespressi, e un gene viene considerato differenzialmente espresso se il logaritmo del suo fold change non cade in un intorno “sufficientemente” largo dello zero.
Figura 3.2: Trasformazione logaritmica sui dati di espressione differenziale
3.2 Metodo del valore di soglia
Un gene viene considerato differenzialmente espresso se il suo livello di espressione si discosta in modo “significativo” da quello della condizione di controllo. Il termine “significativo” è strettamente correlato alla teoria statistica della verifica delle ipotesi, che valuta se gli scostamenti osservati da una determinata condizione siano attribuibili o meno al caso.
Il metodo più semplice, e forse anche attualmente più usato, è il metodo del valore di soglia; con questo criterio si determina sui valori del “fold change” un intervallo simmetrico rispetto allo zero e i geni che hanno livelli di espressione maggiori del suo limite superiore vengono considerati
sovraespressi, mentre quelli con livello minore del limite inferiore vengono considerati sottoespressi.
Per visualizzare meglio questo metodo è possibile considerare l’istogramma dei rapporti R/G, come in figura 3.3.
Partendo dall’ipotesi che molti geni non saranno differenzialmente espressi, si dovrà trovare una distribuzione gaussiana del “fold change” centrata sullo zero; per selezionare i geni differenzialmente espressi si deve collocare una soglia bilaterale sulla distribuzione e considerare tutti quei geni che sono posizionati all’esterno dell’intervallo così ottenuto.
Se, per esempio, si reputa che il valore di soglia sul “fold change” possa essere fissato a ±2, allora i geni differenzialmente espressi saranno quelli con rapporto logaritmico maggiore di +1 e minore di –1.
Figura 3.3: Istogramma del fold change con apposizione della soglia empirica bilaterale
per la selezione dell’espressione differenziale.
Dal punto di vista grafico, se su uno “scatterplot” del logaritmo delle intensità del controllo verso il logaritmo di quelle del trattato si vogliono visualizzare i geni differenzialmente espressi, basta tracciare due linee parallele alla bisettrice del quadrante la cui distanza da quest’ultima
corrisponde al valore di soglia, e selezionare come differenzialmente espressi i geni fuori dall’area delimitata dalle due rette.
Figura 3.4: Scatterplot del campione di controllo contro quello di trattato con
apposizione della soglia empirica bilaterale.
E’ possibile osservare l’effetto dell’apposizione della soglia anche in un altro tipo di “scatterplot” dei dati, detto RI-plot, che ha in ascissa il prodotto delle intensità e in ordinata il loro rapporto: anche in questo caso la soglia bilaterale individua un’area al di fuori della quale i geni vengono considerati differenzialmente espressi.
Figura 3.5: RI-plot dei dati di intensità con posizionamento della soglia bilaterale
Questo metodo di selezione pur avendo il vantaggio di essere molto intuitivo ha, per contro, la totale mancanza di una teoria statistica alla base della scelta dei valori di soglia. Se, per esempio, il trattamento non provoca un’espressione differenziale dell’entità della soglia fissata, non vengono rilevati geni sovra o sotto regolati anche se è molto probabile che queste condizioni si siano verificate.
In termini statistici, come specificato nell’appendice A, questa caratteristica si traduce nell’incapacità di rilevare i veri positivi, cioè la metodica manca di sensibilità.
Analogamente, quando il trattamento è tale da provocare una marcata espressione differenziale e la soglia selezionata è troppo bassa, si evidenzia l’inadeguatezza del metodo nel rilevare i veri negativi, cioè la metodica è poco specifica.
3.3 Metodo della distanza dalla media
Un approccio alternativo basato sul concetto di soglia, utilizzato per individuare i geni differenzialmente espressi, consiste nel collocare la soglia ad una distanza prefissata, prendendo come punto di riferimento la media della distribuzione del rapporto dei due canali.
Ogni esperimento produce una diversa distribuzione ed essa è generalmente di tipo gaussiano, per cui è possibile descriverla attraverso il suo valore medio e la sua varianza σ2.
Questo metodo posiziona una soglia di selezione sulla base della deviazione standard σ della distribuzione; tipicamente questa soglia viene fissata a ±2σ.
Si può facilmente intuire che, rispetto al metodo di soglia empirica, questo sistema consente di “adattare” la larghezza dell’intervallo alla variazione di espressione in relazione al trattamento applicato, ossia si adatta alla distribuzione dei dati.
Dal punto di vista grafico, per effettuare una scelta sulla base della deviazione standard è necessario fare una trasformazione dei dati di “fold change” in modo da sottrarre la media e dividere per la deviazione standard, questo corrisponde a fare una trasformata z dei dati:
σ μ
− = X
Z
dove X sono i dati da trasformare, μ è la media della distribuzione e σ è la sua deviazione standard.
La nuova distribuzione dei dati avrà media nulla e varianza unitaria, quindi sul suo istogramma, che ha in ascissa la deviazione standard e in ordinata il valore di z, si posizionerà una soglia su ±2 come nel caso del valore empirico.
Z
σ
Figura 3.6: Distribuzione dei logaritmi dei rapporti con apposizione della soglia bilaterale
a ±2σ
Posizionare la soglia a ±2σ coincide con il considerare il 5% di geni come differenzialmente espressi e questo perché se si sommano la probabilità che i nuovi dati trasformati siano <-2 e la probabilità che invece siano >+2 si raggiunge questa percentuale. Da questo semplice calcolo si può osservare il limite più evidente del metodo: questa percentuale resta fissa anche se in realtà non ci sono geni differenzialmente espressi, o se ve ne sono di più di quanto ipotizzato.
Nel primo caso il problema è dovuto essenzialmente al fatto che i dati provenienti da microarray sono sempre affetti da rumore. Si supponga per esempio di fare un esperimento nel quale lo stesso materiale viene marcato con entrambi i fluorocromi e poi ibridizzato su un microarray. Idealmente a tutti i geni dovrebbe corrispondere lo stesso valore di espressione nei due canali, ma ciò non si verifica per la presenza di rumore nella misurazione.
Se la condizione ideale venisse rispettata la distribuzione dei rapporti dovrebbe essere tanto stretta da poter essere approssimata con un delta di Dirac centrato sullo zero e la deviazione standard dovrebbe essere nulla. A causa della presenza del rumore, invece, la distribuzione presenta una forma a campana e la deviazione è diversa da zero, per questo motivo viene evidenziata un’espressione differenziale anche quando
essa è assente. In termini statistici è stata rifiutata l’ipotesi nulla quando essa è vera, per cui è stato commesso un errore di tipo I.
Nel secondo caso, la percentuale di geni differenzialmente espressi rimane fissa anche quando ve ne è una quantità maggiore, evidenziando l’incapacità del metodo di rilevare falsi negativi: ciò corrisponde a commettere un errore di tipo II.
3.4 Metodo della soglia non lineare
Il rumore che presentano i dati di espressione genica non si abbatte uniformemente su di essi, ma, poiché presenta una distribuzione gaussiana, ha un maggior effetto sui dati a bassa intensità piuttosto che su quelli ad alta intensità.
Figura 3.7: Distribuzione del rumore e banda di abbattimento sui dati
Per sfruttare questa informazione dal punto di vista della selezione di geni differenzialmente espressi si può pensare di generare una soglia non-lineare modulabile con il livello di rumore che viene riscontrato. L’idea
è quella di dare una caratterizzazione statistica al rumore e confrontare la sua distribuzione con quella dei logaritmi del fold change in modo da determinarne la “percentuale” di abbattimento sul dato a tutte le intensità.
Da questo confronto si può ricavare una misura dell’affidabilità dei dati non in maniera globale, ma puntualmente su ognuno di essi e in modo da poter generare una curva non-lineare che funzioni da soglia per la selezione dei geni differenzialmente espressi.
Figura 3.8: Distribuzione dei dati con apposizione della soglia lineare (in
arancione) e della soglia non lineare (in giallo)
3.5 Analisi della significatività sui microarray
I metodi precedentemente illustrati non sono realizzati su base statistica; nessuno di loro, infatti, esprime un livello di confidenza sui geni selezionati come differenzialmente espressi oppure quantifica il numero di errori di tipo I per caratterizzare l’affidabilità del risultato.
Sebbene i metodi a soglia siano estremamente intuitivi, la presenza di rumore e di numerosi fattori di variabilità dei dati, non sempre ben quantificabili ed eliminabili, rende necessaria l’adozione di approcci statistici per la selezione dei geni differenzialmente espressi.
In questo contesto, il metodo che va sotto il nome di analisi della significatività statistica (Tusher et al., 2001) conferisce una solida base statistica ad un criterio di selezione a soglia.
Nell’analisi della significatività statistica (SAM) viene assegnato un punteggio ad ogni gene effettuando misure virtuali ripetute della sua espressione e considerando i cambiamenti relativi, rispetto alla deviazione standard, di ogni livello di espressione.
Capita spesso che un metodo di analisi statistica utilizzi “bootstrap” o permutazioni, cioè un campionamento dei dati con o senza sostituzione delle osservazioni, per creare degli insiemi di dati surrogati a partire dai quali effettuare le necessarie speculazioni statistiche; questi approcci hanno tanto più valore quanto minore è il numero delle informazioni o dei dati disponibili.
SAM si basa su un test statistico, noto come t-test, in cui vengono messe a confronto le medie delle due distribuzioni di dati che si vogliono paragonare, attraverso la verifica di due ipotesi: l’ipotesi nulla, secondo cui i due campioni di dati provengono dalla stessa popolazione, e l’ipotesi alternativa, che afferma che i dati appartengono a due popolazioni distinte.
Dal punto di vista dell’espressione differenziale dei geni queste ipotesi si possono definire come:
Ipotesi nulla: i due valori di espressione che si stanno
confrontando indicano che il gene non è differenzialmente espresso;
Ipotesi alternativa: i due valori di espressione indicano che il
Calcolate le medie XA e XB delle misure relative ad ogni gene
nelle due condizioni, nell’assunzione che i dati siano normalmente distribuiti, esse vengono confrontate attraverso una statistica, detta t, la cui forma analitica è:
(
)
(
XA XB)
B Ae
X
X
t
−−
−
≡
0
dove al numeratore vi è la differenza fra le medie delle misure associate ad ogni gene e la media nulla della distribuzione della statistica, mentre al denominatore vi è l’errore standard relativo allo stesso gene nelle due condizioni.
Questa statistica è una variabile aleatoria che segue una distribuzione, nota come t-Student, il cui andamento è caratterizzato dai gradi di libertà forniti dalle osservazioni disponibili. I gradi di libertà vengono definiti come il numero di osservazioni indipendenti necessarie per ottenere la misura della statistica rispetto al numero di osservazioni totali.
Per affermare che l’ipotesi nulla è vera (XA =XB ), o che bisogna
rigettarla (XA≠XB ), occorre stabilire un livello di confidenza α,
generalmente posto uguale a 0.01 o 0.05 nella statistica biomedica, e ricavare dalla distribuzione t-Student, per la quale si sono fissati i gradi di libertà, il valore della variabile t, detto tα, cui corrisponde quel livello di
confidenza..
Dal confronto fra la variabile tα e la t ricavata dalle misure si
stabilisce che:
|t| < |tα|-> ipotesi nulla vera;
La soluzione di fare tanti t-test accoppiati per quanti sono i geni è molto semplice ed intuitiva, tuttavia, come spiegato nell’appendice A, ciò fa aumentare la probabilità di commettere almeno un errore di tipo I.
Per ovviare a questo problema, il metodo di analisi della significatività concepisce una nuova statistica, molto simile alla statistica t, con l’intento di generare un punteggio per il test che si sta effettuando; questo punteggio viene poi utilizzato per verificare le ipotesi di test sia sui dati originali che su quelli surrogati generati attraverso le permutazioni.
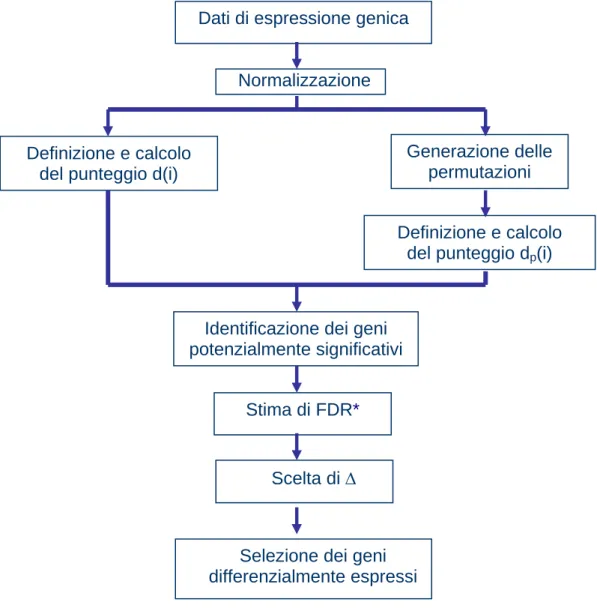
Per visualizzare meglio il procedimento, di seguito è riportato il diagramma delle operazioni eseguite dall’analisi della significatività:
Dati di espressione genica
Normalizzazione
Definizione e calcolo del punteggio d(i)
Generazione delle permutazioni
Identificazione dei geni potenzialmente significativi
Stima di FDR*
Definizione e calcolo del punteggio d
Figura 3.9: Diagramma delle operazioni effettuate nell’analisi della significatività Scelta di Δ
(i)
p
Selezione dei geni differenzialmente espressi
Partendo dall’ipotesi che le fluttuazioni del rumore sul dato sono gene specifiche, il primo passo del trattamento consiste nella normalizzazione con i metodi usuali e nella visualizzazione dei dati attraverso “scatterplot” o “cube root plot”, che è il grafico della radice cubica delle due intensità e permette di evidenziare il comportamento dei geni a basse intensità di fluorescenza e di identificare i geni differenzialmente espressi a queste intensità, come mostrato nella figura 3.10.
Figura 3.10: Scatterplot e cube root plot dei dati
Il passo successivo calcola il punteggio d(i) per ogni gene i nelle due condizioni che si vogliono verificare
0 2 1
)
(
)
(
)
(
)
(
s
i
s
i
x
i
x
i
d
C C+
−
=
dove: al numeratore vi è la differenza fra le medie delle misure
relative alle due condizioni C1 e C2 per il gene i-esimo (possono
al denominatore vi è la somma fra la stima della deviazione
standard del numeratore e un valore additivo s0, detto “fudge
factor”.
La deviazione standard del numeratore può essere calcolata in base alla seguente formula di stima:
⎭
⎬
⎫
⎩
⎨
⎧
−
+
−
⎟⎟
⎠
⎞
⎜⎜
⎝
⎛
−
+
+
=
∑
∑
= = 2 2 1 1 2 1 1 2 1 2 2 1 1 1)]
(
)
(
[
)]
(
)
(
[
2
)
(
n k C k n h C h n ni
x
i
x
i
x
i
x
n
n
i
s
dove: le due sommatorie sono estese al numero di misure effettuate
nei due stati;
n1 è il numero di misure nello stato C1;
n2 è il numero di misure nello stato C2.
A bassi livelli di espressione la varianza di d(i) può essere alta a causa di piccoli valori di s(i). Per assicurare l’indipendenza della distribuzione dei d(i) dal livello di espressione del gene è necessario
aggiungere un fattore additivo so al denominatore del punteggio. Il valore
di s0 viene scelto in modo da minimizzare il coefficiente di variazione di d(i)
in funzione di s(i) attraverso un procedimento a finestre mobili sui dati. In generale si sceglie s0 in maniera che il coefficiente di variazione di
d(i) sia approssimativamente costante al variare di s(i).
L’acquisizione di una stima di confidenza (FDR) sui dati richiede la realizzazione di numerosi esperimenti al fine di ottenere un’informazione il più possibile completa sui livelli di espressione di tutti i geni. Poiché eseguire molti esperimenti è dispendioso sia in termini di tempo che economici, vengono effettuate una serie di permutazioni dei dati, ognuna
delle quali produce un nuovo valore del punteggio d(i); tali permutazioni devono essere bilanciate.
Una permutazione è bilanciata se per ogni gruppo di g esperimenti, con g pari al numero di campioni che sono stati ibridizzati, vi sono g/2 esperimenti per campione.
Si analizzi, per esempio, l’esperimento effettuato da Tusher su due linee umane linfoblastoidi “wild-type” (Tusher et al., 2001). In questo esperimento le cellule di ogni linea sono state fatte crescere in uno stato irradiato (I), sottoposte a 5 Gy di radiazione ionizzante per una durata di quattro ore, e in uno stato non irradiato (U). I campioni sono stati, poi, marcati e divisi in due aliquote uguali A e B; si hanno, quindi, otto campioni U1A, U1B, I1A, I1B, U2A, U2B, I2A, I2B. I campioni sono stati successivamente confrontati fra di loro in esperimenti con uguale linea cellulare e aliquota, ossia (U1A vs I1A), (U1B vs I1B), ecc.
Per ognuna delle due condizioni sperimentali (U e I), costituite da quattro campioni, due per linea cellulare, si possono ottenere
6 2 * 2 2 * 3 * 4 )! 2 4 ( ! 2 ! 4 2 4 = = − = ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛
raggruppamenti a due a due dei campioni stessi.
In questo caso, le permutazioni bilanciate sono date da tutti i possibili accoppiamenti fra le coppie di campioni delle due condizioni
sperimentali: si ottengono in totale 36 permutazioni bilanciate.
2 4 2 = ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛
Figura 3.11: Esempio di due permutazioni bilanciate con due linee cellulari (i colori
Per stimare l’ordine delle statistiche d(i), vengono calcolati per ogni
permutazione p i punteggi dp(i) da attribuire al gene i di ogni coppia di
esperimenti, secondo la definizione:
0 2 1
)
(
)
(
)
(
)
(
s
i
s
i
x
i
x
i
d
p G G+
−
=
dove con Gi si indicano i due gruppi della permutazione, ossia le due
condizioni sperimentali.
I punteggi così ottenuti sono ordinati in senso ascendente:
( )
k
d
d
d
d
p(
1
)
≥
p(
2
)
≥
p(
3
)
≥
...
≥
pdove k indica la posizione del punteggio all’interno dell’insieme ordinato dei dp(i) .
Si definisce la differenza relativa attesa sul numero di permutazioni come:
∑
==
p p p n p n k d Ek
d
1 ) ()
(
come nell’esempio indicato in figura 3.12.
k)
Per identificare i geni significativamente espressi si ordinano i
punteggi dei dati originali in senso ascendente e si indica con di(k) il
punteggio d(i) del gene che era in posizione i-esima e dopo l’ordinamento si trova in posizione k-esima.
Per mettere in relazione i punteggi di(k) con le differenze relative
attese dE(k) si fa uno scatterplot che prende il nome di “SAM plot”.
Figura 3.13: “SAM plot”
Dalla figura 3.13 si può osservare che per diversi geni si ha che
di(k)≅dE(k).
Una volta stabilita una soglia Δ si individuano il più piccolo d(i) positivo (t1) e il più grande d(i) negativo (t2) tali che:
Δ
≥
−
(
)
)
(
k
d
k
d
i Ee il gene i-esimo viene definito potenzialmente differenzialmente espresso se vale che di(k) ≥ t1 o di(k) ≤ t2
Figura 3.14: SAM plot con apposizione della soglia superiore t1 e della soglia
inferiore t2
Per dare una valutazione statistica dell’affidabilità con cui si è individuato l’insieme di geni differenzialmente espressi si stima il False Discovery Rate (FDR):
}
)
(
)
(
|
{
}
)
(
)
(
|
{
2 1 1 1 2 1t
i
d
t
i
d
i
card
t
i
d
t
i
d
i
card
FDR
p p n p p p n≤
∨
≥
≤
∨
≥
≈
∑
=dove, fissati i valori di soglia t1 e t2 , al numeratore si ha la media del
numero di geni individuati come differenzialmente espressi attraverso le permutazioni e al denominatore il numero di geni differenzialmente espressi ottenuti dall’analisi dei dati reali.
I geni differenzialmente espressi prodotti da una permutazione p sono detti “falsamente significativamente espressi” e sono individuati con la stessa procedura con cui si selezionano i geni significativamente espressi, ma sostituendo di(k) con dp(k), ossia:
Δ
<
−
(
)
)
(
k
d
k
d
p EOvviamente questa stima è differente a seconda della soglia impostata, per cui è possibile determinare il valore di Δ a seconda del FDR
he si desidera avere sui dati. Come si può osservare nella tabella 3.1, ad un m
oncetto intuitivo come quello del valore di soglia, irrobustito da una
aluta
i; queste ultime vengono catturate attraverso le permutazioni.
ad ave c
inore FDR corrispondono Δ maggiori e, come immaginabile, diminuisce il numero di geni differenzialmente espressi individuati.
Tabella 3.1: Lista dei geni differenzialmente espressi in funzione di FDR e Δ
Il metodo di analisi della significatività statistica utilizza, quindi, un c
v zione statistica del risultato ottenuto.
Rispetto alle metodiche illustrate nei precedenti paragrafi è bene evidenziare alcuni sostanziali vantaggi che il metodo qui discusso presenta.
Il criterio che individua i due valori di soglia nel SAM è di tipo iterativo, permettendo un maggiore controllo sui dati e sulle loro fluttuazion
Il processo di permutazione dei dati consente, infatti, di considerare i due insiemi di geni sovraespressi e sottoespressi come “indipendenti”, ossia è possibile ricavare per essi due valori di soglia che possono portare
re un intervallo non simmetrico sull’istogramma. Ciò è conseguenza del fatto che l’espressione differenziale non si manifesta necessariamente con la stessa intensità relativa sui geni sovraespressi e sottoespressi, ma può succedere che il “fold change” minimo per affermare la presenza di espressione differenziale non sia lo stesso.
L’uso di permutazioni, inoltre, ha il vantaggio di riuscire a migliorare la qualità dell’informazione in esperimenti che utilizzano un numero piccolo di campioni, generando insiemi di dati surrogati, coerenti con
icroarray, il carico computazionale diviene estrem
tima del livello di affidabilità dell’insieme di geni selezionati come differenzialmente espressi, evidenziando la l’esperimento realizzato.
Purtroppo, nel caso opposto, ossia con campioni molto numerosi o con molti geni per m
amente oneroso e può risultare ingestibile se non si dispone di un adeguato supporto hardware.
Infine, l’utilizzo di un parametro statistico come il False Discovery Rate permette un’immediata s