INDICE
CAPITOLO 1 – INTRODUZIONE
……… pag 3CAPITOLO 2 – INIBITORI DEL FVIII E FIX NELL’EMOFILIA
CONGENITA ED ACQUISITA
2.1 FVIII e FIX: Caratteristiche biochimiche……….. pag 8
2.2 Inibitori anti-FVIII e anti-FIX………... pag 9
CAPITOLO 3 – EMOFILIA CONGENITA ED EMOFILIA
ACQUISITA: ASPETTI CLINICO-DIAGNOSTICI
3.1 Emofilia Congenita ……… pag 30
3.2 Emofilia Acquisita ………. pag 34
3.3 Esplorazione funzionale del sistema coagulativo ……….. pag 38
3.4 Diagnosi di Inibitori ……….. pag 41
CAPITOLO 4 – STRATEGIE TERAPEUTICHE NELL’EMOFILIA
CONGENITA CON INIBITORI
4.1 Terapia del sanguinamento acuto ……….. pag 43
4.2 Terapia di induzione dell’immunotolleranza (ITI) nel paziente
emofilico con inibitori………. pag 46
4.3 Terapia del sanguinamento acuto e ITI nei pazienti con emofilia B e
CAPITOLO 5 – STRATEGIE TERAPEUTICHE NELL’EMOFILIA
ACQUISITA
5.1 Terapia del sanguinamento acuto ………... pag 55
5.2 Terapia immunosoppressiva ……….. pag 65
CAPITOLO 6 – DUE CASI DI EMOFILIA ACQUISITA: SUCCESSO
TERAPEUTICO CON FARMACI
IMMUNOSOPPRESSORI E ANTITROMBOTICI
..pag 70CAPITOLO 7 – CONCLUSIONI
……….…pag 781. INTRODUZIONE
Il sistema emostatico è un meccanismo di difesa che preserva l’integrità del sistema circolatorio. Coinvolge la parete dei vasi, rivestita da cellule endoteliali, e necessita di proteine plasmatiche solubili, alcune delle quali coinvolte direttamente nel processo emostatico e alcune come regolatori della coagulazione (vedi Tab.1). Le componenti cellulari del sangue comprendono i globuli rossi, le piastrine e i leucociti, compresi i granulociti, monociti e linfociti. Anche microparticelle, derivate da leucociti e piastrine, circolano nel sangue, sebbene le conoscenze relative al loro ruolo funzionale siano limitate. Queste componenti rivestono un ruolo chiave in varie forme di difesa, ma possono partecipare direttamente nel processo emostatico. In condizioni normali circolano nel sangue come costituenti inattive e inerti. In seguito ad un danno tissutale, il sistema è attivato. La lesione endoteliale determina l’esposizione della matrice subendoteliale. Alternativamente, le cellule endoteliali sono attivate con l’espressione di recettori sulla membrana plasmatica, l’esocitosi dei corpi di Weibel-Palade e l’attivazione di processi biochimici intracellulari. Cambiamenti nella membrana plasmatica alterano la composizione fosfolipidica presentata al flusso sanguigno. Con l’espressione del fattore tissutale (Tissue Factor, TF), i proenzimi (zimogeni) dei fattori della coagulazione del sangue sono convertiti sequenzialmente ad enzimi, e rapidamente viene prodotta trombina. Viene così iniziata la formazione del trombo[1].
Per quanto la cascata emocoagulativa abbia risentito di un’eccessiva semplificazione, per come è stata descritta essere costituita da due vie funzionalmente distinte, via intrinseca e via estrinseca, di fatto queste due vie sono funzionalmente connesse. In particolare, l’evento biochimico che descrive la generazione di trombina per attivazione del fattore XII (via intrinseca) non ha rilevanza dal punto di vista emostatico. Di fatto l’attivazione della cascata emocoagulativa in vivo è iniziata dal complesso TF-FVII in grado di promuovere quantità nanomolari di trombina (IIa), per via dell’attivazione del fattore X, ma soprattutto, in maniera e in modo estremamente più efficace dal punto di vista
della cinetica enzimatica, di attivare il fattore IX (FIX). Il FIX, in presenza di fattore VIII attivato (FVIIIa), rappresenta un evento enzimatico straordinariamente efficace e funzionalmente adeguato all’attivazione del FX, nell’ordine di migliaia di volte superiore di quanto rilevabile sperimentalmente mediante l’attivazione diretta del FX da parte del FVIIa (Fig. 1). Questo evento, identificabile come attività by-passante del FVIII, dà ragione del funzionamento attuale della cascata emocoagulativa e della sua straordinaria efficacia. E’ evidente che la carenza di FVIII, di FIX o la presenza di inibitori nei confronti di questi due fattori abbia un ruolo così rilevante nella determinazione di eventi emorragici.
Le malattie emorragiche dovute alla carenza di FVIII e FIX sono rispettivamente l’emofilia A e l’emofilia B.
Esiste poi una patologia associata alla carenza acquisita del FVIII e FIX, definita emofilia acquisita, che nella descrizione di due eventi clinici caratterizzati da manifestazioni emorragiche è l’oggetto di questa tesi.
Fig.1
Tabella 1. I fattori della coagulazione [2]. Denominazione
del Comitato
Nome e sinonimi Sito di produzione Funzioni principali VIIa TF X Xa Kcat/Km = 1.5 x 109 IX IXa VIIIa X Xa Kcat/Km = 7.9 x 109 Va II IIa
internazionale
Fattore I Fibrinogeno Fegato Si trasforma in
fibrina
Fattore II Protrombina Fegato,
dipendente dalla vitamina K
Viene convertita in trombina Fattore III Fattore tissutale
(TF) o tromboplastina tissutale Cellule danneggiate Forma complessi con il FVIIa, in presenza di ioni Ca2+, per attivare il FX
Fattore IV Ioni Ca2+ Dalla dieta o riserva ossea Favoriscono l’interazione tra enzimi, cofattori e fosfolipidi Fattore V Proaccelerina o acceleratore della protrombina fattore labile Fegato Agisce da cofattore non enzimatico nel formare complessi con il FXa e con i fosfolipidi
piastrinici per attivare la protrombina
Fattore VI Non più in uso -- --
Fattore VII Proconvertina o acceleratore della conversione della protrombina sierica o autoprotrombina I o fattore stabile Fegato, dipendente dalla vitamina K Forma complessi con il TF, in presenza di ioni Ca2+, per attivare il FX
Fattore VIII Fattore antiemofilico (AHF) o fattore antiemofilico A Fegato La porzione a basso peso molecolare agisce da cofattore non enzimatico nel complesso tenasico per attivare il FX. Fattore IX Fattore di Christmas o fattore antiemofilico B; componente della Fegato, dipendente dalla vitamina K Agisce con il complesso tenasico per attivare il FX; è, a propria volta,
tromboplastina plasmatica o autoprotrombina II attivato dal complesso
TF/FVIIa della via estrinseca (cross-over tra via estrinseca e via intrinseca) Fattore X Fattore di
Stuart-Prower o autoprotrombina III Fegato, dipendente dalla vitamina K E’ l’enzima direttamente responsabile della formazione della trombina dalla protrombina Fattore XI Antecedente della
tromboplastina plasmatica o fattore
antiemofilico C
Fegato Attiva il FIX
Fattore XII Fattore di
Hageman (HF) o fattore da contatto Fegato Legandosi al sottoendotelio innesca la via intrinseca della coagulazione Fattore XIII Fattore di
Laki-Lorand o fattore stabilizzante la fibrina o transglutaminasi plasmatica Fegato Causa la polimerizzazione della fibrina solubile in fibrina insolubile Precallicreina (PK)
Fattore di Fletcher Fegato Attivata dal FXIIa, con il contributo di HMWK, promuove a sua volta l’attivazione del FXII (amplificazione dell’attivazione) Chininogeno ad alto peso molecolare (HMWK) Fattore di Fitzgerald Fegato Contribuisce all’attivazione del sistema plasmatico attivabile da contatto convertendo la precallicreina in
callicreina e fornendo l’attacco per il FXI
2. INIBITORI DEL FVIII E FIX NELL’EMOFILIA CONGENITA E ACQUISITA
2.1 FVIII e FIX: Caratteristiche biochimiche
Il fattore VIII (FVIII) è una glicoproteina a singola catena, di P.M. di circa 330 kD. Il gene del FVIII mappa sul braccio lungo del cromosoma X in posizione Xq28. Il FVIII è presente in circolo legato non covalentemente al fattore di von Willebrand. La sua molecola è composta da sei domini così suddivisi e disposti: A1-a1-A2-a2-B-a3-A3-C1-C2. La sua attivazione, da parte della trombina o del FXa, richiede una rapida idrolisi di tre legami in corrispondenza dell’Arg372-Ser373, Arg740-Ser741 e Arg1689-Ser1690. Inoltre la trombina agisce, in maniera più lenta, anche in posizione Arg336-Ser337. Il FVIII attivato, separato dal vWF, è formato da una catena pesante di circa 93 kD, composta dai domini A1-A2, e da una catena leggera di circa 77 kD, comprendente i domini A3-C1-C2. Il FVIIIa, in forma eterotrimerica, entra a far parte del complesso tenasico, dove svolge la funzione di cofattore per il FIX. Il FVIII è inattivato dal clivaggio operato dalla proteina C attivata o dalla spontanea dissociazione del suo dominio A2 [3,4]. Il vWF è una grande glicoproteina multimerica, sintetizzata nelle cellule endoteliali e nei megacariociti. Il gene codificante per il vWF è localizzato sul cromosoma 12 e contiene 52 esoni. Il vWF maturo è costituito da 11 domini (D’-D3-A1-A2-A3-D4-B1-B2-B3-C1-C2). Oltre a mediare l’adesione e l’aggregazione piastrinica alla parete vasale danneggiata, il vWF agisce da trasportatore del FVIII. Quest’ultimo presenta 2 siti di legame per il vWF, entrambi localizzati sulla catena corta, nel dominio a3 e C2. L’interazione tra vWF e FVIII protegge il FVIII dal legame alla superficie piastrinica e alle cellule endoteliali e dall’attacco proteolitico di serin-proteasi, compresa la proteina C attivata. Il vWF trasporta e rilascia il FVIII al sito in cui è richiesta la coagulazione. Inoltre facilita il clivaggio e l’attivazione del FVIII da parte della trombina. L’emivita del FVIII è di 12-16 ore. E’ eliminato dal circolo attraverso specifiche interazioni con recettori endofitici presenti sulla superficie di cellule scavenger. Il vWF previene
una precoce eliminazione del FVIII impedendo il legame ai recettori catabolici. Modificazioni dei livelli plasmatici di vWF sono frequentemente associati a concordanti modificazioni dei livelli di FVIII [4].
Il fattore IX (FIX) è una glicoproteina a singola catena di P.M. di circa 57kD, contenente nella sua porzione NH2-terminale 10 residui di acido γ-carbossiglutammico (Gla), seguita da due moduli epidermal grogwth factor (EGF)-like, un peptide di attivazione e un dominio serin-proteasi [5]. E’ uno dei fattori epatici vitamina K-dipendente. L’attivazione implica un’idrolisi di due legami peptidici con rilascio di un cosiddetto peptide di attivazione del P.M. di 11 kD e la formazione di due catene tenute insieme da un legame disolfuro. La catena pesante contiene il sito attivo mentre la catena leggera contiene i residui di Gla. Il gene codificante per il FIX mappa sul braccio lungo del cromosoma X in posizione Xq27.1-q27.2. Il FIX ha un’emivita di circa 24 ore [3].
2.2 Inibitori anti-FVIII e anti-FIX
Il problema della formazione degli inibitori del FVIII e FIX sta catturando l’attenzione della comunità scientifica che si occupa di emofilia da più di cinquanta anni.
La presenza di inibitori è difatti associata ad un peggior controllo degli eventi emorragici, un più grave quadro della patologia articolare, una peggiore qualità della vita e maggiori costi terapeutici [6]. I pazienti con inibitori non hanno una maggiore frequenza e/o più gravi sanguinamenti ma quando questi si presentano sono più difficili da gestire [7].
Superato il problema della trasmissione di agenti infettivi con la terapia sostituiva dei fattori della coagulazione, il principale problema che si impone nella gestione terapeutica del paziente emofilico è rappresentato dalla formazione di anticorpi diretti contro il FVIII o FIX, denominati inibitori.
E’ riportata un’incidenza tra i pazienti affetti da emofilia A severa fino al 50%, del 3-13% in quelli con la forma lieve/moderata [8].
Il riscontro di inibitori in soggetti non emofilici, senza precedenti di sanguinamento, definisce l’emofilia acquisita [9].
Quanto verrà di seguito detto per l’emofilia A vale, ipoteticamente, anche per l’emofilia B, pur essendo molto più scarsi i dati a disposizione. Le più evidenti differenze nelle due condizioni sono la diversa incidenza di inibitori, in meno del 5% dei pazienti con emofilia B, e il minor successo della terapia ITI (induzione di immuno-tolleranza), solo il 15% dei pazienti trattati [9,10]. Inoltre nei pazienti con emofilia B la presenza di inibitori è associata a complicanze quali manifestazioni allergiche fino ad una reazione anafilattica, un fenomeno raramente incontrato nell’emofilia A [11].
Ad oggi molto si sa dei meccanismi immunologici che sono alla base dello sviluppo di inibitori ma ancora molte sono le domande senza risposta.
Mentre nel primo caso, paziente emofilico, gli inibitori sono il risultato di una risposta immunitaria verso antigeni non-self (risposta alloimmune), nel secondo caso, emofilia acquisita, essi sono l’espressione di una risposta immunitaria di tipo autoimmune [9].
Ad un diverso, sotto certi aspetti, meccanismo immunologico corrisponde, però, un quadro clinico simile.
Tra le numerose variabili che entrano in gioco nello sviluppo degli inibitori nei pazienti emofilici troviamo severità della malattia, tipo di difetto genico del FVIII, HLA, numero e modalità di esposizione ai concentrati di FVIII, tipo di prodotto usato, polimorfismi del gene codificante per IL-10, probabilmente molte altre non sono state ancora individuate [9,12].
Ben poco si sa invece circa i fattori individuali che possono condizionare lo sviluppo di inibitori nell’emofilia acquisita.
La maggior parte degli inibitori acquisiti sono diretti contro siti localizzati sui domini del FVIII A2 e A3, dove interferiscono con il legame al FX e al FIXa, e C2, situato all’estremità C-terminale, dove alterano il legame ai fosfolipidi e al vWF. Non sono differenti i bersagli degli inibitori dell’emofilia congenita ma non si possono escludere differenze trattandosi di due diverse tipologie di risposta immunitaria (alloimmune e autoimmune) [5,13,14,].
I bersagli degli inibitori sul FIX sono localizzati nella regione NH2-terminale con i residui di Gla e nel dominio serin-proteasi. Alcuni pazienti sviluppano inibitori verso entrambe le regioni ma non sono stati trovati inibitori verso i moduli Epidermal Growth Factor-like (EGF-like) [5].
Gli anticorpi inibitori neutralizzano la funzione di fondamentale cofattore, nella cascata coagulativa, del FVIII interferendo nel modo suddetto [13,14].
L’interferenza dell’anticorpo diretto verso il dominio cui si lega il vWF, accelera il catabolismo del FVIII. In aggiunta alcuni anticorpi sarebbero in grado di degradare direttamente il FVIII attraverso proteolisi diretta [15].
L’effetto inibitorio degli anticorpi anti-FIX comprende l’inibizione dell’attivazione del FX dipendente dal FVIII, del legame del dominio Gla ai fosfolipi e del legame del FVIII [5].
Risulta ormai accertata la presenza di anticorpi non inibitori che sarebbero diretti contro epitopi non coagulanti del FVIII. Il significato clinico di tali anticorpi non inibitori non è chiaro, sebbene sia stata suggerita una maggiore clearance del FVIII nei pazienti che li possiedono. Circa il 20% dei soggetti normali produce anticorpi che sembrano non neutralizzanti. Questi potrebbero spiegare il riscontro in alcuni pazienti di un titolo BU (Bethesda Unit) negativo ma concentrazione ed emivita del FVIII ridotte. Altre tecniche per la determinazione di anticorpi anti-FVIII, come l’ELISA (enzyme-linked immunosorbent assay), rappresentano un metodo migliore del test di Bethesda per l’identificazione di anticorpi non neutralizzanti. La relazione tra il significato clinico degli anticorpi neutralizzanti e non neutralizzanti resta attualmente difficile da predire [16].
Aspetti caratteristici degli inibitori acquisiti (autoanticorpi)
Sono autoanticorpi di classe IgG non fissanti il complemento, leganti il FVIII in modo tempo e temperatura dipendente. Mentre gli inibitori che compaiono nell’emofilia congenita seguono una cinetica di primo ordine, quelli acquisiti mostrano un pattern di inattivazione del FVIII non lineare (tipo 2 o cinetica di secondo ordine).
Il titolo esatto degli inibitori può avere un minore significato biologico nell’emofilia acquisita rispetto a quella congenita, ma risulta comunque importante quale misura dell’efficacia della terapia impostata [14].
I principali aspetti differenziali tra lo sviluppo di inibitori nell’emofilia acquisita e nella forma congenita sono riportati, schematicamente nella seguente tabella (Tab.2).
Tab.2 Aspetti differenziali relativi allo sviluppo di inibitori INIBITORI-EMOFILIA ACQUISITA INIBITORI-EMOFILIA CONGENITA Tipo di risposta immunitaria
Autoimmune Allo immune
Fattori individuali
condizionanti lo sviluppo di inibitori
Per lo più non noti In gran parte noti
Metodi di determinazione Test mixing-plasma Test di Bethesda/Nijmegen Test mixing-plasma Test di Bethesda/Nijmegen Cinetica degli anticorpi Tipo 2 o secondo ordine Tipo 1 o primo ordine Quadro Clinico Sanguinamenti di
difficile controllo
Sanguinamenti di difficile controllo
Aspetti immunologici dello sviluppo degli inibitori
Il background immunologico dell’emofilia acquisita non è ancora noto a causa della sua bassa incidenza. Con molta probabilità si sviluppa come conseguenza di complesse interazioni tra suscettibilità genetica e altri fattori ambientali e non, che portano alla rottura della regolazione della risposta immunitaria con conseguente formazione di autoanticorpi verso il FVIII endogeno [17].
Poiché le conoscenze relative allo sviluppo di inibitori nell’emofilia congenita sono sicuramente maggiori, le informazioni di seguito riportate sono per lo più riferite a quest’ultima, anche se gran parte dei meccanismi descritti, facendo anch’essi parte della risposta immunitaria, possono essere estesi all’emofilia acquisita.
Gli inibitori sono prodotti da cellule B istruite a differenziarsi in plasmacellule. E’ noto che il midollo osseo di qualunque individuo sano produce milioni di nuove cellule B ogni giorno. Tali cellule B acquistano specificità attraverso riarrangiamenti casuali delle loro catene variabili; ciò significa che centinaia di cellule B con la capacità di riconoscere il FVIII sono prodotte quotidianamente. Alcuni meccanismi che regolano tale risposta immunitaria sono: l’eliminazione della maggior parte delle cellule B reattive entro il midollo osseo, la possibilità di un riarrangiamento dei loro recettori in quelle a cui è stato permesso di sfuggire a tale delezione, l’induzione dell’anergia in periferia, la delezione o l’apoptosi a causa dell’assenza di stimolazione [15].
Sebbene la specificità degli inibitori non apporti differenze dal punto di vista del quadro clinico, le cellule B che producono tali anticorpi sono qualitativamente eterogenee. Gli inibitori acquisiti che appaiono in un contesto di sistemica autoimmunità sono parte di una tendenza generale a produrre autoanticorpi. Quelli osservati dopo interventi chirurgici o gravidanza, sono prodotti nel contesto di circostanze specifiche e temporanee. Al contrario, gli inibitori osservati come risposta alla terapia sostitutiva hanno tutte le ragioni per subire ogni passaggio maturativo verso una produzione a lungo termine di inibitori ad alta affinità [15].
Ruolo dei linfociti T-CD4+
Il ruolo dei linfociti T-CD4+ è suggerito da varie osservazioni come la scomparsa degli inibitori in pazienti HIV+ con riduzione della conta dei CD4+ e la mancata formazione degli stessi in un modello murino emofilico in cui venivano bloccati i segnali costimolatori indispensabili per i CD4+ [9].
Il FVIII o FIX fagocitato dalle cellule che esprimevano l’antigene (APC) veniva sottoposto a digestione proteolitica con clivaggio in particolari siti in relazione alle caratteristiche strutturali.
I frammenti così ottenuti vengono presentati sulla superficie delle APC, nel contesto del complesso maggiore di istocompatibilità di tipo II (MHC-II), il cui fenotipo condiziona quali peptidi saranno legati [10,18].
Si avrà quindi il riconoscimento da parte del recettore FVIII specifico presente sui linfociti T-CD4+ e, solo in presenza di adeguati segnali costimolatori, una risposta immunitaria efficace [9,10].
I linfociti T-CD4+ con recettori in grado di riconoscere sequenze del FVIII o FIX sono presenti nei pazienti emofilici perché, non essendo in questi prodotti il fattore VIII o IX, non si ha la delezione in utero dei linfociti auto-reattivi, che è l’evento fondamentale alla base della tolleranza verso gli antigeni self [18].
I segnali costimolatori sono rappresentati dal legame di B7.1 (CD80) e B7.2 (CD86) dell’APC con CD28 dei linfociti T, CD40 dei linfociti B con il CD40L dei linfociti T [5,10,18].
Il CD28 compete per il legame a B7 con l’antigene A dei linfociti T citotossici (CTLA4), il quale ha un’affinità di legame venti volte maggiore ed è espresso solo dalle cellule T attivate e ad un livello più basso rispetto al CD28. L’effetto del legame B7-CTLA4 è di tipo inibitorio, giocando un ruolo cruciale nella regolazione della stimolazione delle cellule T; recenti studi confermano, infatti, che il CTLA4 è coinvolto nella modulazione della risposta immunitaria e nel mantenimento della tolleranza periferica [10,17,18].
L’importanza della via CD28/B7/CTLA4 è suggerita da alcuni studi nei quali si è visto che topi affetti da emofilia con deficit di B7.2 non sviluppano anticorpi contro il FVIII e la creazione di una proteina, data dalla fusione di CTLA4 con la catena pesante di IgG1 in grado di bloccare l’interazione B7-CTLA4, sopprime la formazione di anticorpi anti-FVIII [18].
Proprio in relazione al ruolo del CTLA4, un recente studio ha evidenziato la presenza di associazione tra polimorfismi del gene CTLA4 e l’emofilia acquisita. Il gene del CTLA4 è localizzato sul cromosoma 2q33, in una regione immunologicamente inportante per la presenza nelle sue vicinanze di altri due geni codificanti per molecole coinvolte nella risposta immunitaria, per esempio il CD28-R. Sono stati analizzati diversi polimorfismi del gene CTLA4 e ci sono consistenti evidenze circa l’associazione con l’emofilia congenita con inibitori e malattie autoimmuni come la malattia di Graves, l’ipotiroidismo autoimmune, il diabete mellito di tipo 1, la sclerosi multipla e la celiachia. Rimane comunque non
chiaro con quale esatto meccanismo i polimorfismi del gene CTLA4 contribuiscano allo sviluppo dei suddetti quadri patologici. In relazione, in particolare, all’emofilia acquisita è stata osservata una maggiore frequenza del polimorfismo CTLA4 49 G rispetto ad una popolazione sana di controllo, tale dato però risulta statisticamente significativo se si confronta la popolazione femminile affetta da emofilia acquisita con il controllo, sempre di sesso femminile, mentre non è statisticamente significativo se si confrontano le due popolazioni maschili, rispettivamente emofilici acquisiti e controlli sani. Inoltre tale polimorfismo ricorre con una frequenza maggiore significativa, rispetto al gruppo di controllo, nei pazienti con emofilia acquisita idiopatica o con sottostante malattia autoimmune ma non in quelli con sottostante neoplasia o gravidanza. Osservazioni basate su studi condotti in vitro sostengono che soggetti con CTLA4 49 G/G rispetto ai CTLA4 49 A/A avrebbero un CTLA4 con minore effetto inibitorio sui linfociti T [17].
Anche la via CD40-CD40L potrebbe avere un ruolo cruciale ed è stato condotto uno studio sulla possibilità di impiego di un anticorpo monoclonale umanizzato anti-CD40L; non si hanno però dati definitivi in quanto la sperimentazione è stata interrotta per il rischio di effetti collaterali di natura trombo embolica [18,19].
Riferendosi al ruolo dei linfociti T-CD4+, sarebbe semplicistico parlare in generale di CD4+ in quanto la risposta alla stimolazione antigenica è molto più articolata.
Al contatto con l’antigene segue la possibilità dei CD4+ di differenziarsi in 3 diversi sottotipi: Th1, Th2 e Th3 [9,16].
Le cellule Th1 stimolate secernono le citochine proinfiammatorie IL-2 e IFN-γ, possono avere un’azione direttamente citotossica e contribuiscono alla produzione di anticorpi IgG1 fissanti il complemento e IgG2 [9,18].
Le cellule Th2 hanno una duplice funzione, da un lato attraverso la secrezione delle citochine antinfiammatorie IL-4 e IL-10, che hanno azione inibitoria sulle Th1 e sulle APC, e la stimolazione delle Th3, per mezzo dell’IL-4, che a loro volta producono TGF-β, potente immunomodulatore, esercitano un’azione di
down-regulation sulla risposta immunitaria; dall’altro esse sono in grado di indurre le cellule B a produrre IgG4 e quindi un’azione di up-regulation [9,18]. Andando a studiare la prevalenza di IgG1, IgG2 e IgG4 nei pazienti con emofilia A ed emofilia acquisita non si osservano differenze significative; si ottiene, però, un dato interessante dopo introduzione di una terapia ITI (induzione immuno-tolleranza). Nei pazienti nei quali si è avuto successo terapeutico si aveva prevalenza di IgG1 e IgG2 mentre nei casi di insuccesso prevalevano IgG4 [9]. Si deduce da ciò che sia Th1 che Th2 hanno un ruolo determinante nella risposta immunitaria ma la maggiore “aggressività” e quindi un titolo più alto di inibitori correla con l’attività delle cellule Th2, mentre il successo della terapia ITI correla con l’attività delle cellule Th1; quanto detto vale sia nell’emofilia A che nella forma acquisita [9].
Per meglio chiarire il ruolo regolatorio delle cellule Th CD4+, in uno studio sono stati esaminati i domini C2 e A3 del FVIII, sui quali si trovano i principali epitopi verso i quali sono diretti gli inibitori. E’ stato quindi valutato quali epitopi venivano riconosciuti dalle cellule CD4+.
Gli epitopi riconosciuti nei pazienti con emofilia A differivano da quelli dei pazienti con emofilia acquisita e ciò non sorprende visto che alla base vi è un diverso tipo di risposta immunitaria.
Il risultato più interessante di tale studio è che mentre alcuni epitopi sono riconosciuti da T-CD4+ sia di soggetti sani che emofilici con e senza inibitori, altri epitopi sono riconosciuti da T-CD4+ dei soli soggetti sani ed emofilici senza inibitori; ciò suggerisce che il mancato riconoscimento di tali epitopi si traduce nella mancata stimolazione di T-CD4+ con un ruolo regolatorio “protettivo” nella risposta immunitaria. In altri termini chi posside T-CD4+ in grado di riconoscere determinati epitopi non sviluppa inibitori [9,20].
Il concetto di cellule T regolatorie fu proposto già 40 anni fa ma solo di recente il loro ruolo nel mantenere lo stato di salute o nello sviluppare le malattie si sta definendo. Il concetto di cellule T ‘suppressor’ ha generato molto interesse negli anni ’70 ma studi pubblicati negli anni ’80, riportando dati discordanti, hanno creato molto scetticismo. Risalgono così agli anni ’90 le più dirette e convincenti
evidenze dell’esistenza di cellule T regolatorie. Oggi, ampiamente accettate, queste cellule sono comunemente indicate come cellule T regolatorie, TR, o Treg. Nel 2001 è stato identificato, prima in un modello murino e poi anche nell’uomo, un gene responsabile, qualora mutato, di un quadro patologico caratterizzato da gravi manifestazioni di autoimmunità e infiammazione; tale gene è Foxp3. Due anni dopo si è scoperto che tale gene codifica per una molecola chiave per lo sviluppo e la funzione delle cellule Treg. Le cellule Treg sono di due tipi: Treg adattative e Treg naturali. Le prime sono CD4+, CD25-, compaiono in periferia quando incontrano antigeni in un contesto ambientale di tolleranza e producono alti livelli di IL-10 e TGF-β, attraverso i quali mediano l’immunosoppressione. Le Treg naturali originano nel timo, sono CD4+, CD25+ ed esprimono il CTLA4, il recettore per TNF indotto da glucocorticoidi (GITR), il prodotto della trascrizione del gene Foxp3, e possono attivamente ridurre la risposta immunologica attraverso il contatto cellula-cellula. Sempre più evidenze ricavate da modelli umani e murini, sostengono che le cellule Treg giocano un ruolo importante nel modulare la risposta immunitaria verso il FVIII [20].
Un aspetto interessante è che non tutte le risposte immunitarie contro il FVIII sono patogene, cioè sono stati trovati anticorpi contro il FVIII non inibitori sia in soggetti affetti da emofilia A sia nel 15% dei donatori di sangue sani. In pratica è come se il sistema immunitario, oltre a fare una distinzione tra self e non-self, distinguesse anche tra antigeni patogeni e non e questo potrebbe dipendere dalle condizioni in cui viene presentato l’antigene cioè la presentazione in un contesto di stress favorirebbe una reazione immunitaria “patogena” (‘danger theory’ of tolerance) [9].
La risposta immunitaria che si osserva nei soggetti sani contro il FVIII coinvolge sempre i CD4+ ma è meno intensa e più transitoria rispetto a quella osservata nei pazienti emofilici [9].
Nei soggetti sani peraltro potrebbero essere coinvolti meccanismi regolatori diversi da quelli riscontrati nei pazienti emofilici, come la formazione di anticorpi anti-idiotipo [18].
Ruolo delle cellule B memoria
Un punto cruciale nello sviluppo degli inibitori è se la risposta immunitaria delle cellule B evolverà o meno verso la formazione di cellule B memoria. Senza quest’ultimo passaggio le cellule B scompaiono rapidamente, dando luogo ai casi con i cosiddetti inibitori transitori, osservati per esempio quando ad un paziente viene somministrato FVIII per un breve periodo di tempo. Se, invece, si formano le cellule memoria è alta la possibilità che la produzione di inibitori si mantenga nel tempo. Il perché tale passaggio è importante è riconducibile a tre motivi:
1. L’attivazione delle cellule B memoria a plasmacellule vuol dire che le cellule producenti anticorpi si localizzano nel midollo osseo e nella milza, cioè in siti nei quali diventano meno accessibili alla terapia.
2. Una plasmacellula riduce il numero di recettori espressi sulla sua superficie, rendendosi meno suscettibile all’interazione con il suo ambiente. La plasmacellula vive così per settimane, se non mesi, soltanto producendo anticorpi.
3. Le cellule B memoria sono altamente attivabili, anche in assenza dell’antigene specifico. Ciò è dovuto alla presenza sulla loro superficie di recettori, denominati Toll-like receptors (TLR), che possono far virare la cellula verso l’attivazione per mezzo del legame con ligandi derivati da microrganismi. Quest’ultimo aspetto potrebbe spiegare perché, per esempio, alcuni pazienti emofilici continuano a produrre inibitori ad alto titolo nonostante non vengano esposti al FVIII, talvolta per anni [15]. Visto il ruolo cruciale delle cellule B memoria si può ben comprendere come esse rappresentino un ottimo target per l’induzione della tolleranza. La terapia convenzionale, basata sull’infusione di alte dosi di FVIII per prolungati periodi di tempo, ha un impatto significativo sulle cellule B memoria, inducendone l’apoptosi. Comunque la stessa procedura potrebbe rivelarsi un’arma a doppio taglio, in quanto l’esposizione al FVIII potrebbe determinare il reclutamento di nuove cellule B emergenti dal midollo osseo.
Una prospettiva terapeutica futura ipotizza di sfruttare alcune caratteristiche delle cellule B memoria. Quest’ultime offrono con i loro recettori (BCR) determinanti
unici, denominati idiotipi, e facilmente riconoscibili da anticorpi antiidiotipo. Partendo da anticorpi antiidiotipo ottenuti da pazienti emofilici con inibitori si è osservato che la risposta immunitaria verso il FVIII sembra essere molto meno diversificata di quanto si pensasse, suggerendo una possibile terapia per gli inibitori basata sulla specifica eliminazione delle cellule B memoria attraverso interazioni idiotipiche. Molto rimane comunque da comprendere, se si desidera sviluppare una tale strategia terapeutica, soprattutto è necessario avere più informazioni relative alle vie di segnale che sono attivate dall’interazione tra BCR e FVIII (o derivati) o anticorpi antiidiotipo (o derivati). Il segnale prodotto potrebbe portare la specifica cellula B verso diversi destini, inclusa la delezione attraverso apoptosi [15].
Fattori di rischio per lo sviluppo di inibitori
Sia i pazienti con emofilia severa che quelli con una forma lieve-moderata sviluppano inibitori dopo un numero relativamente piccolo di esposizioni al FVIII (9-12 ED negli emofilici severi).
Gli inibitori possono svilupparsi in qualunque momento della vita dei pazienti. In riferimento alla comparsa un primo picco d’incidenza si ha nella prima decade, con bassa incidenza nella seconda, terza e quarta decade e un secondo picco, più piccolo, nella sesta decade. Questi inibitori con comparsa tardiva spesso si presentano in pazienti che hanno accumulato centinaia di giorni di esposizione e spesso dopo un trattamento intensivo per un episodio di sanguinamento o per un intervento chirurgico. L’eziologia degli inibitori a precoce comparsa rispetto a quelli tardivi sembrerebbe diversa. I primi sarebbero l’espressione di una primitiva intolleranza verso un antigene non self, mentre i secondi sarebbero il risultato della rottura della tolleranza in un paziente precedentemente tollerante[12].
Tra i fattori di rischio per lo sviluppo di inibitori troviamo: - Fattori direttamente dipendenti dal paziente: 1. Tipo di mutazione del FVIII e FIX; 2. fenotipo MHC-I e MHC-II;
3. recettori per i linfociti T;
4. polimorfismi dei geni codificanti per le citochine;
5. polimorfismi dei geni codificanti per le molecole immunoregolatorie.
- Fattori indirettamente dipendenti dal paziente:
1. Età della prima somministrazione di fattore VIII/IX;
2. Stimoli per il sistema immunitario (vaccinazioni, infiammazioni, traumi, interventi chirurgici).
- Fattori dipendenti dalla terapia: 1. Tipo di prodotto;
2. Modalità di somministrazione.
In merito ai fattori di rischio direttamente dipendenti dal paziente, dati estremamente interessanti giungono da studi, almeno sei negli ultimi venti anni, condotti sulla popolazione di pazienti emofilici non precedentemente trattati con FVIII (PUP, pazienti precedentemente non trattati); essi rappresentano una popolazione naïve dal punto di vista immunologico ed altamente suscettibile [6]. Nei PUP l’incidenza di inibitori è circa dieci volte maggiore che nei pazienti precedentemente trattati (PTP) indipendentemente dal tipo di prodotto utilizzato [7].
Le mutazioni del FVIII in relazione allo sviluppo di inibitori possono essere distinte in mutazioni ad alto e a basso rischio.
Le mutazioni ad alto rischio sono ampie delezioni, mutazioni nonsense e inversione dell’introne 22; queste sono associate, generalmente, a bassissimi livelli di FVIII circolante e l’incidenza di inibitori riscontrata oscilla tra il 21% e 88% [6,18].
Le mutazioni a basso rischio, nelle quali l’incidenza di inibitori è inferiore al 10%, sono le mutazioni missenso, le piccole delezioni/inserzioni, le mutazioni del sito di splicing [18].
Le diverse mutazioni sono alla base della diversa incidenza di inibitori nella forma di emoflia A severa rispetto alla lieve e moderata, rispettivamente 20-30% e meno del 10-15% [18]. La maggior parte degli inibitori nei pazienti con emofilia lieve-moderata sono riscontrati in un numero relativamente piccolo di casi con mutazioni ad alto rischio nei domini A1-A2, che provocano un cambiamento conformazionale nel FVIII e sono associate ad un rischio di sviluppare inibitori fino al 50% [12]. Maggiore incidenza di inibitori si ritrova anche nel caso di mutazioni missenso, per definizione a basso rischio, situate nei domini A2 e C2, dove potrebbero determinare una modificazione dell’immunogenicità del FVIII. Questa regione risulta critica per il legame al vWF e ai fosfolipidi nel contesto del complesso tenasico; una sua modificazione nella conformazione tridimensioanale potrebbe spiegare la maggiore incidenza di inibitori. Si può desumere che oltre al tipo di mutazione riveste un ruolo, non sottovalutabile, anche la localizzazione della mutazione [10,21].
La bassa incidenza di inibitori nell’emofilia B (meno del 5%) sarebbe anch’essa attribuibile alla relativamente alta frequenza di mutazioni a basso rischio. Ciò non sembra comunque sufficiente a spiegare valori così bassi, è stato attribuito un possibile ruolo alla somiglianza dei fattori vitamina K dipendenti II, VII e X al FIX [10,18].
Esisterebbe anche una relazione tra tipo di mutazione e titolo di inibitori, maggiore nelle mutazione missenso rispetto alle ampie delezioni [10].
Inoltre, per ragioni non ancora note, è stata riscontrata un’incidenza doppia di inibitori nel caso di mutazioni della catena leggera rispetto a quella pesante del FVIII [12,18].
Prima di andare avanti, ulteriori aspetti relativi alla formazione di inibitori nell’emofilia lieve/moderata meritano di essere affrontati. I pazienti con emofilia lieve/moderata generalmente hanno un disordine emorragico di lieve entità e gli episodi vengono trattati con desmopressina, ma talvolta può essere necessaria l’infusione di FVIII. In questi pazienti si potrà avere lo sviluppo di inibitori e si troveranno non valori più bassi di FVIII, rispetto ai loro valori storici, ma titoli molto elevati di anticorpi inibitori diretti contro il FVIII esogeno, ma non contro
la proteina endogena di FVIII mutato. Gli inibitori diretti contro il FVIII esogeno legano particolari epitopi non espressi dal FVIII mutato; si realizza in pratica una distinzione tra FVIII non self e FVIII self seppur mutato. Inoltre gli inibitori nell’emofilia lieve/modareta sono sia di tipo 1 che di tipo 2 e, diversamente dall’emofilia severa, prevalgono questi ultimi [21,22,23].
Non è sufficiente il tipo di mutazione per giustificare la comparsa di inibitori: studi condotti su gemelli, che evidenziano una concordanza non del 100% e l’esistenza di un rischio maggiore di sviluppare inibitori nel caso di parenti emofilici di secondo e terzo grado con inibitori, suggeriscono l’esistenza di altri fattori costituzionali coinvolti [6,10,12,20]. Tali fattori potrebbero essere rappresentati da geni coinvolti nella risposta immunitaria. Questi ultimi potrebbero spiegare la diversa incidenza di inibitori nelle diverse etnie visto che i comuni genotipi dell’emofilia sono egualmente distribuiti in tutti i gruppi etnici. L’associazione con particolari aplotipi non è stata confermata da studi più recenti (MIBS, Malmö International Brother Study), che invece sosterrebbero il ruolo di geni codificanti per citochine. Alcuni polimorfismi dei geni per 1β, 4 e IL-10, che potrebbero essere coinvolti, si è già dimostrato essere associati a malattie autoimmuni e allo stato di atopia [12,18].
Non sono però state trovate associazioni tra incidenza di inibitori e polimorfismi dell’IL-1 e IL-4, mentre una forte correlazione c’è con un polimorfismo della regione promotore del gene dell’IL-10 [12,18,20].
L’Il-10 ha sia un’azione stimolatoria che inibitoria sul sistema immunitario, è prodotta dalle cellule Th2 e stimola le cellule B a differenziarsi, proliferare e produrre anticorpi. Il polimorfismo suddetto si tradurrebbe in una maggiore produzione di IL-10, il cui risultato sarebbe una maggiore proliferazione di linfociti B e quindi una maggiore produzione di anticorpi [10,18,20].
Tra le varie citochine, studi più recenti, sostengono il ruolo anche del TNFα, citochina con potenti funzioni proinfiammatorie e immunomodulatorie. In pazienti con il polimorfismo TNFα-308 A/A sono stati trovati inibitori con frequenza significativamente maggiore rispetto ai pazienti con TNFα-308 G/G e G/A, indicando che il polimorfismo suddetto può essere un utile marker ed un
potenziale modulatore della risposta immunitaria verso la terapia sostitutiva. E’ interessante notare, inoltre, che il genotipo A/A sembra essere prevalente nella popolazione Africana rispetto all’Europea e all’Asiatica [10,20].
L’aspetto interessante della possibile correlazione tra profilo citochinico di ciascun paziente e tipo di risposta inibitoria sviluppata è che la caratterizzazione di tale profilo nei pazienti potrebbe avere delle ripercussioni sulle scelte terapeutiche [18].
L’esistenza comunque anche di fattori ambientali, influenzanti il rischio di sviluppare inibitori, è suggerita soprattutto dal riscontro di discordanza in studi condotti su gemelli omozigoti [18].
Tra tali fattori diversi studi riportano l’età di inizio dei trattamenti sostitutivi, in realtà i dati sono discordanti. In due studi, uno spagnolo e uno olandese, veniva riportata una maggiore incidenza in chi aveva iniziato terapia prima dei sei mesi di età; ciò non è, però, stato confermato da altri studi nei quali invece si pone l’accento sulla maggiore incidenza di inibitori in chi è stato sottoposto a terapia al bisogno e non a trattamento profilattico [5,24]. Proprio in riferimento alla terapia profilattica Santagostino et al., confrontando pazienti sottoposti a profilassi e pazienti non sottoposti, mostra un’incidenza significativamente maggiore di inibitori in quelli non sottoposti a profilassi, anche quando i risultati venivano corretti sulla base di altre variabili quali l’età alla prima esposizione e fattori genetici. Inoltre, riporta una minore incidenza di inibitori in pazienti che avevano iniziato la profilassi prima dei 35 mesi di età rispetto a quelli che l’avevano iniziata più tardi [12,16]. Non è da sottovalutare, però, che la maggiore incidenza di inibitori in soggetti più giovani potrebbe essere ricondotta ad una forma più grave della malattia con più facili conseguenze emorragiche e quindi ricorso a terapia in condizioni di stress, cosa che potrebbe essere di stimolo per il sistema immunitario [18].
Riguardo quest’ultimo aspetto dati, non sempre confermati, suggeriscono che malattie infettive o vaccinazioni associate alla prima infusione di FVIII, nonché interventi chirurgici e traumi muscolari con emorragia e la conseguente massiva esposizione antigenica, sono tutti fattori potenzialmente in grado di esercitare una
potente azione di stimolo sul sistema immunitario, con conseguente produzione di inibitori [5,18].
I fattori che influenzano lo sviluppo di inibitori nei pazienti emofilici possono anche essere classificati, alla luce dei più recenti studi, in fattori certi e fattori potenziali.
Rientrano nel primo gruppo:
• Numero di giorni di esposizione al FVIII (ED, dose di esposizione), secondo alcuni studi il rischio maggiore di sviluppare inibitori si avrebbe nei primi cinquanta giorni [6];
• severità della malattia, che come si è detto correla al tipo di mutazione del FVIII;
• etnia, l’incidenza è doppia nella razza non caucasica (Afroamericani) rispetto alla caucasica [6,10,16,18];
• familiarità, più a rischio soggetti con una storia familiare di inibitori, riconducibile a fattori genetici ancora da individuare [6,12,18];
• fattori genetici non FVIII correlati come il polimorfismo genetico nella regione promotore dell’IL-10 [6,18].
Al secondo gruppo appartengono:
• Regime terapeutico: un trattamento profilattico potrebbe essere associato ad una minore incidenza di inibitori rispetto alla terapia al bisogno, come già sopra menzionato [6];
• età alla prima somministrazione del FVIII;
• modalità di somministrazione: soprattutto nei pazienti con limitati contatti con il FVIII l’infusione continua con l’esposizione ad elevate quantità di FVIII potrebbe essere particolarmente rischiosa per la formazione di inibitori [5,6];
• interventi chirurgici con il corredo di danno tissutale e infiammazione sarebbero di stimolo per una risposta immunitaria contro il FVIII esogeno[6];
• tipo di prodotto utilizzato: in alcuni studi si sostiene che i prodotti plasma derivati contenenti vWF (fattore di von Willebrand) sarebbero meno
immunogeni dei prodotti ricombinanti (rFVIII) ma altri studi non confermano tale dato, che non può perciò essere considerato conclusivo[6,16];
• cambio di tipologia di prodotto durante il decorso della malattia: anche in riferimento a ciò non ci sono dati conclusivi e si raccomanda una forte vigilanza [16];
• frequenza dei test per inibitori; l’esecuzione di tali test con bassa frequenza potrebbe non permettere il riconoscimento di forme di inibitori a basso titolo e transitori;
• vaccinazioni, di per sé rappresentano uno stimolo per il sistema immunitario potendo condurre con maggiore facilità ad una reazione immunitaria verso il FVIII esogeno [6].
Gli aspetti relativi al rapporto tra sviluppo di inibitori e terapia del paziente emofilico, per la notevole rilevanza che hanno nella pratica clinica, sono stati affrontati in numerosi studi e i principali risultati sono riportati di seguito; pur anticipando che al momento i dati sono estremamente discordanti e quindi non esistono raccomandazioni circa l’impiego di prodotti plasma derivati piuttosto che di prodotti ricombinanti o in relazione alla modalità di somministrazione, infusione continua piuttosto che in boli.
Relazione tra terapia dell’emofilia congenita e rischio di sviluppare inibitori
Da studi retrospettivi si evidenzierebbe un maggior rischio di sviluppare inibitori con l’impiego di rFVIII rispetto ai prodotti plasma derivati, con una incidenza di inibitori rispettivamente tra 29% e 32% e tra 0% e 12% [25,26].
Questi dati non possono però essere assolutamente considerati conclusivi in quanto le caratteristiche dei vari studi non li rendono confrontabili in modo statisticamente significativo.
Anche Peerlinck ed Hermans pur riportando incidenze di inibitori di 15-32% in pazienti precedentemente non trattati (PUPs) e 0.9-2.9% in pazienti precedentemente trattati (PTPs) con incidenza di inibitori ad altro titolo (>5 BU)
del 10-16% nel primo gruppo e 0-2.3% nel secondo, confermano l’impossibilità di confrontare in termini statisticamente significativi i diversi studi dai quali i dati sono stati desunti [7].
Molto interessanti a tal riguardo sono i risultati dello studio di coorte CANAL (Gouw et al.). L’obbiettivo di questo studio era stabilire l’esistenza o meno di differenze per quanto riguarda la formazione di inibitori nelle seguenti condizioni: - Utilizzo di rFVIII piuttosto che prodotti plasma derivati;
- utilizzo di preparati senza vWF in confronto a preparati con molto o poco vWF;
- impiego di diverse marche di rFVIII;
- cambiamento di marca di rFVIII durante la terapia [25].
Tutti i dati sono stati analizzati e quindi corretti alla luce delle variabili, già citate nel testo, influenzanti la formazione degli inibitori.
I risultati sono stati i seguenti:
1. Non c’è una netta riduzione dell’incidenza di inibitori con l’utilizzo di prodotti plasma derivati rispetto ai prodotti ricombinanti e ancora minore è la differenza nel caso di inibitori ad alto titolo;
2. il rischio di sviluppare inibitori era simile nel caso di rFVIII e di prodotti plasma derivati ad alto contenuto di vWF, era invece ridotto del 70% nel caso di prodotti plasma derivati con basso contenuto di vWF;
3. nei pazienti che hanno utilizzato FVIII ricombinante con il dominio B deleto (Refacto) avevano maggiore incidenza di inibitori rispetto agli utilizzatori di rFVIII completo (Kogenate) ma non in modo statisticamente significativo; risultati simili sono stati ottenuti con gli altri prodotti ricombinanti (Kogenate Bayer, Ricombinate);
4. non è stato osservato un aumento del rischio nei pazienti in cui si è dovuti passare dall’utilizzo di un determinato prodotto ricombinante ad un altro [25].
I risultati di questo studio riferiti alle differenze tra prodotti plasma derivati e prodotti ricombinanti sono in contrasto con quelli dello studio di Goudemand et
pazienti trattati con rFVIII rispetto a quelli trattati con prodotti plasma derivati ad alto contenuto di vWF. Tale differenza potrebbe essere spiegata con l’impiego nello studio di Goudemand di prodotti plasma derivati meno immunogeni rispetto a quelli utilizzati nello studio CANAL, dove peraltro sono stati impiegati 23 tipi differenti di prodotti [8,25].
Un aspetto che merita ulteriori chiarimenti è quello relativo alla minore incidenza di inibitori nel caso dell’utilizzo di prodotti plasma derivati a basso o nullo contenuto di vWF risultato dallo studio CANAL.
Tale risultato infatti contrasta con i dati ottenuti da altri studi sulla base dei quali è stato invece ipotizzato un ruolo protettivo del vWF. E’ stato supposto che il vWF possa modulare l’immunogenicità del FVIII mascherando gli epitopi riconosciuti dalle cellule T e B o alterando la struttura terziaria del FVIII [20,25,27].
Sia da studi in vitro che da osservazioni cliniche è stato suggerito che il vWF protegga gli epitopi sul dominio C2 del FVIII dal legame con gli anticorpi inibitori. Quindi avrebbe un ruolo protettivo nei riguardi solo degli inibitori diretti contro la catena leggera del FVIII. Inoltre topi trattati con prodotti senza vWF rispetto a quelli trattati con prodotti contenenti il vWF sviluppano un alto titolo di inibitori [20,25]. Si è recentemente ipotizzato che l’effetto immunoprotettivo del vWF sia dovuto alla sua capacità di legare il FVIII impedendo che questo venga processato dalle cellule dendritiche e di conseguenza presentato ai linfociti T CD4+ FVIII-specifici [8,19,20]. Dallo studio di Salvagno et al. si ricavano altre evidenze a supporto di una funzione ‘positiva’ del vWF. In questo studio si mette in relazione il titolo di inibitori, il tipo di prodotto plasma derivato impiegato e la capacità di generare trombina. Ciò che si osserva è che l’impiego di prodotti a più alto contenuto di vWF si traduce in una minore capacità degli inibitori di inibire la produzione di trombina [8,28].
A differenza dello studio CANAL, che non mostra diversa incidenza di inibitori nel caso di impiego di rFVIII o prodotti plasma derivati, una recente review di Wigth e Paisley riporta i seguenti dati: PUP trattati con differenti prodotti plasma derivati sviluppano inibitori nel 20.3-33% dei casi, pazienti trattati con un solo tipo di prodotto plasma derivato contenente vWF sviluppano inibitori nello
0-12.4% dei casi. Pazienti ai quali è stato somministrato un solo tipo di rFVIII hanno sviluppato inibitori nel 32-38.7% dei casi, anche se dati più recenti indicano che l’incidenza è del 16.7% impiegando rFVIII di seconda generazione (privo di albumina umana). Tutti i risultati di questi studi hanno il limite di essere ricavati da dati ottenuti da studi di coorte retrospettivi e quindi non possono essere considerati conclusivi [5,27,29].
Sulla base di tale background è stato pianificato lo studio SIPPET, attualmente in corso. Il SIPPET è uno studio internazionale, multicentrico, prospettico, controllato, randomizzato sulla frequenza di inibitori in pazienti precedentemente non trattati (PUP) ed esposti per la prima volta a prodotti plasma derivati contenenti FVIII o a rFVIII. L’obiettivo principale è quello di stabilire, appunto, se c’è una maggiore sicurezza di un prodotto rispetto all’altro [29].
In riferimento alla possibile relazione tra rischio di sviluppare inibitori e marca di prodotto utilizzato nel 2003 e nel 2004 la FDA (Food and Drug Administration) ha promosso due studi con l’obiettivo appunto di stabilire tale relazione. Sono stati utilizzati due diversi modelli statistici e ciò ha portato ad avere dati non conclusivi. Infatti nel primo studio solo due dei cinque prodotti esaminati risultavano aver un rischio accettabile, essi sono il REFACTO e l’ADVATE, mentre nel secondo studio tutti e cinque i prodotti avevano rischio accettabile; gli altri tre prodotti sono Original KOGENATE, RICOMBINATE e KOGENATE Bayer [7].
Alla luce degli aspetti fin qui valutati nuovi quesiti si pongono in merito ai benefici della terapia genica e della terapia sostituiva. La terapia genica potrebbe infatti fornire la quantità minima di FVIII necessario da un punto di vista coagulativo, ma potrebbe non essere in grado di garantire una quantità tale di FVIII che potrebbe essere vantaggiosa in termini di induzione di immunotolleranza.
Inoltre la stessa risposta immunitaria verso il FVIII in caso di terapia genica, come per la terapia sostitutiva, risente di numerose variabili come il tipo di vettore e
promotore usato, la quantità e la sede dell’espressione genica, il back-ground genetico del ricevente, l’età al momento della terapia [9].
3. EMOFILIA CONGENITA ED EMOFILIA ACQUISITA: ASPETTI CLINICO DIAGNOSTICI
3.1 Emofilia Congenita
L’emofilia congenita, distinta nelle due forme A e B, è una malattia emorragica ereditaria X-linked, nella quale risultano alterati quantitativamente o qualitativamente il FVIII e il FIX rispettivamente.
La patogenesi delle sindromi emofiliche rimase avvolta nel mistero fino al 1893, quando Wright sviluppò una tecnica per misurare il tempo di coagulazione e dimostrò che il plasma dei pazienti emofilici coagulava molto più lentamente. Tuttavia la miscela di plasma emofilico con plasma normale coagulava normalmente. Questa osservazione portò Brinkhouse e Quick a scoprire nel 1947 che la causa dell’emofilia A consisteva nella mancanza di una globulina, il FVIII, necessaria per la generazione di tromboplastina. Nello stesso anno Pavlovsky osservò che il plasma di alcuni emofilici poteva correggere la prolungata coagulazione in vitro di altri emofilici clinicamente indistinguibili dai primi. Questa osservazione permise di identificare l’emofilia B, dovuta a carenza del FIX, e fu così distinta dall’emofilia A nel 1952 da Aggeler e Biggs. Le due entità risultavano indistinguibili clinicamente entrambe con ereditarietà recessiva legata al sesso [30].
L’incidenza dell’emofilia A e B è rispettivamente di 15-20 e 2-5 casi su 100.000 maschi all’anno senza che siano state rilevate differenze etniche o geografiche. L’emofilia A è da quattro a sei volte più frequente dell’emofilia B.
L’emofilia A viene distinta in tre forme sulla base del livello del FVIII: - Grave: livello di FVIII inferiore all’1% del normale;
- Moderata: livello di FVIII compreso fra 1% e 5% del normale; - Lieve: livello di FVIII compreso fra 6% e 30-40% del normale.
Trattandosi di una malattia trasmessa come carattere recessivo legato al cromosoma X, tutti i figli maschi di donne portatrici hanno il 50% di possibilità di essere malati; la gravità del quadro dipende dal tipo di mutazione, interessante il
gene codificante per il FVIII, e dalla conseguente variabile attività funzionale residua.
Si possono distinguere due tipi principali di difetti genetici:
- difetti genici dovuti a grossolani rimaneggiamenti del DNA che danno invariabilmente luogo ad emofilia A grave. Essi comprendono, oltre ad un difetto comune legato ad una inversione completa della sequenza genica fra l’esone 1 e l’esone 22 con completa impossibilità di sintetizzare FVIII (questa anomalia è alla base del 40% circa dei casi di emofilia A grave), delezioni intrageniche di varia entità coinvolgenti 100 o più nucleotidi o addirittura l’intero gene; inserzione di retrotrasposomi; duplicazioni; - difetti meno grossolani che possono dar luogo sia a forme gravi, come nei
casi dovuti a mutazioni nonsenso o a inserzioni o delazioni di sequenze con meno di 100 basi, sia a forme intermedie o lievi, dovute a mutazioni missenso. Alcune mutazioni missenso permettono una sintesi e secrezione di FVIII con attività residua che risulta tuttavia assente o variabilmente ridotta. In questi casi nel plasma del paziente si rinviene un FVIII anomalo, ancora in grado di reagire con anticorpi specifici, denominato “cross reacting material” (CRM) [30].
Le manifestazioni emorragiche dipendono dal livello residuo di attività del FVIII circolante.
L’emofilia A grave si manifesta, generalmente, entro il primo anno di vita se non già alla nascita con gravi sanguinamenti cerebrali. Il quadro clinico è caratterizzato da emorragie spontanee intramuscolari, nei tessuti molli, negli organi parenchimali e, con l’inizio della deambulazione, a livello articolare. Le estrazioni dentarie e gli interventi chirurgici anche lievi possono essere seguiti da emorragie irrefrenabili. Nella forma moderata le emorragie spontanee sono rare, gli emartri sono sempre possibili anche se meno frequenti. La forma lieve solitamente si presenta nell’adolescenza o anche nell’età adulta, essendo le emorragie spontanee assenti e la principale manifestazione è un imponente sanguinamento post-operatorio, qualora non vengano correttamente identificati.
Il sanguinamento intrarticolare, per i suoi esiti, è probabilmente la manifestazione più grave. Clinicamente si presenta con tutti i segni della flogosi: dolore via via più inteso, tumefazione articolare, calore e impotenza funzionale con flessione antalgica. Gli episodi tendono a recidivare e col tempo si instaura un’anchilosi altamente invalidante. Le sedi articolari più colpite, in ordine decrescente di frequenza, sono: ginocchio, tibio-tarsica, gomito, polso, spalla e anca [30].
Gli ematomi muscolari si realizzano generalmente dopo traumi anche banali. A seconda del distretto possono verificarsi fenomeni di compressione di tronchi nervosi o vascolari con sintomi clinici correlati (parestesie e ischemie). In caso di ematomi alla lingua o alla muscolatura della laringe o del collo si possono avere fenomeni di soffocamento o difficoltà respiratorie.
La diagnosi di emofilia A si basa in prima istanza sull’anamnesi familiare, che risulta positiva nel 70% circa dei casi, e sull’anamnesi personale, da cui emergono pregresse emorragie sia spontanee sia post-traumatiche, soprattutto nella forma grave della malattia. L’anamnesi può essere negativa nelle forme lievi.
Si riscontrerà allungamento dell’aPTT con un PT normale, il dosaggio del FVIII permette di stabilirne anche la gravità.
L’emofilia B presenta la stessa modalità di trasmissione dell’emofilia A anche se c’è una maggiore eterogeneità genetica. Sono stati descritti almeno 80 tipi di difetti molecolari diversi. Nella maggior parte dei casi il difetto è quantitativo, nel 30%, invece il deficit è prevalentemente funzionale, con presenza di CRM. Nell’emofilia BM, forma riscontrabile in circa il 15% dei casi, il PT, normale con le tromboplastine di uso più comune, si presenta allungato con l’impiego di tromboplastina bovina, probabilmente per una particolare inibizione sul FXa. Una forma particolare è l’emofilia B Leiden, in cui con il progredire dell’età dei pazienti il deficit di FIX diminuisce fino alla normalizzazione; questa variante è generalmente causata da mutazioni a livello del promoter del gene che lasciano intatta la zona responsiva agli androgeni, per cui il difetto tende a correggersi dopo la pubertà [30].
Il quadro clinico e la suddivisione in tre forme con diversa gravità è sovrapponibile a quello dell’emofilia A.
La diagnosi si basa sul riscontro dell’allungamento dell’aPTT con normale PT e poi sul dosaggio specifico del FIX.
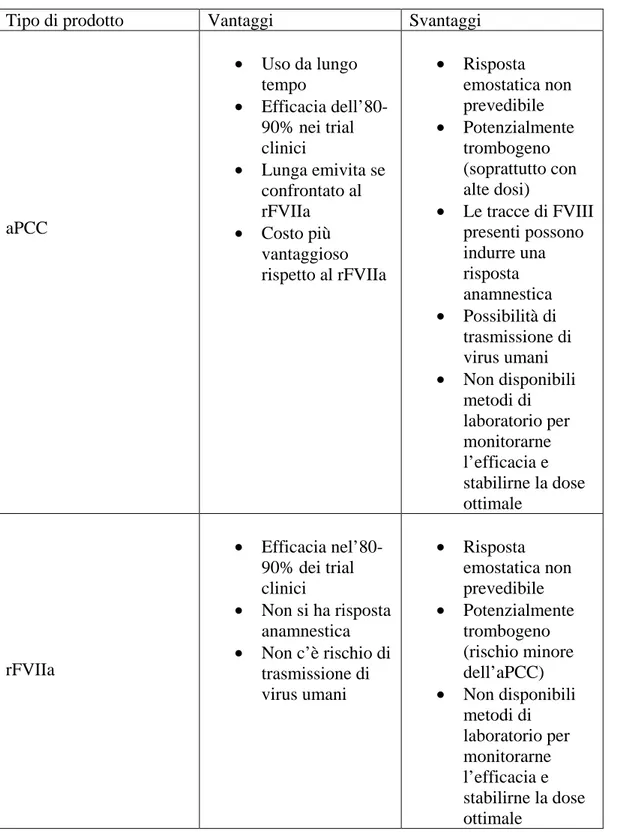
Il trattamento del paziente emofilico si fonda essenzialmente sull’infusione del fattore della coagulazione carente, praticata dal paziente stesso, dai suoi familiari o dal medico di medicina generale. Tale modalità, infatti, consente la massima tempestività d’intervento e si associa alla maggiore efficacia della terapia e ad una migliore qualità di vita. I prodotti disponibili, per l’emofilia A, sono rappresentati da concentrati di FVIII plasma derivati e da rFVIII, ottenuto con tecniche di ricombinazione genica. I concentrati plasma derivati possono presentare un diverso grado di purezza, si possono così avere prodotti ad intermedia purezza ( ottenuti mediante tecniche convenzionali di precipitazione-adsorbimento), concentrati purificati mediante cromatografia a scambio ionico e concentrati purificati impiegando anticorpi monoclonali; in quest’ultimo tipo il preparato è privato totalmente del vWF. Per quanto riguarda i prodotti ricombinanti si può avere l’intero FVIII oppure il FVIII privato del dominio B; inoltre, la continua evoluzione tecnologica ha recentemente condotto alla registrazione di prodotti privi di albumina umana nella formulazione finale e sono già stati sviluppati nuovi farmaci ricombinanti il cui processo produttivo non prevede l’impiego di albumina umana ed è esente da contaminanti proteici di origine animale [31]. Per il trattamento dell’emofilia B si dispone di concentrati purificati mediante cromatografia anionica e per affinità, concentrati purificati con anticorpi monoclonali e FIX ricombinante, inoltre possono essere utilizzati concentrati del complesso protrombinico (PCC) contenenti, oltre al FIX, il FII e FX. L’uso di PCC si associa a rischio trombotico, in particolare trombosi venosa profonda, embolia polmonare, CID ed infarto del miocardio. Si ritiene che queste complicanze siano dovute alla presenza di fattori della coagulazione in forma attivata e l’entità del rischio trombotico è correlata al dosaggio di PCC impiegato e alla concomitanza di altri fattori di rischio. Pertanto, la chirurgia, la prolungata immobilizzazione, l’epatopatia e le malattie cardiovascolari aumentano la suscettibilità dei pazienti trattati con PCC a sviluppare tali complicanze [31].
3.2 Emofilia Acquisita
L’emofilia acquisita è una malattia rara con un’incidenza annua stimata di 0.2-1/1.000.000 di abitanti. E’ dovuta alla formazione di autoanticorpi diretti contro gli epitopi funzionali dei fattori VIII e/o IX della coagulazione, che ne determinano la neutralizzazione e/o ne accelerano la scomparsa dal plasma [32]. E’ così denominata in quanto sono gli stessi fattori carenti nell’emofilia congenita. Come quest’ultima è caratterizzata clinicamente da sanguinamenti potenzialmente mortali. Ha una distribuzione bimodale con un primo picco d’incidenza in giovani donne, 20-30 anni, nel periodo del post-partum tipicamente, e un secondo nella popolazione anziana, 64-80 anni, nella quale si trova frequentemente associata a malattie autoimmuni, neoplasie maligne e reazioni allergiche a farmaci [33]. Si possono, difatti, distinguere una forma idiopatica di emofilia acquisita, nella quale non si ritrovano patologie associate, e una forma secondaria, rappresentante il 40-50% dei casi, nella quale si ritrovano numerose condizioni associate tra le quali: gravidanza e post-partum, malattie autoimmuni, malattie infiammatorie croniche intestinali, diabete, malattie dermatologiche, malattie dell’apparato respiratorio, reazioni allergiche a farmaci, epatite acuta da HBV o HCV, neoplasie maligne (Tab.3).
C’è una eguale distribuzione fra i 2 sessi, benché le femmine predominino nell’età più giovane in rapporto alle gravidanze, mentre i maschi costituiscono la maggioranza degli affetti di età superiore ai 60 anni [32].
Tabella 3. Classificazione Emofilia Acquisita Idiopatica 50-60%
Secondaria 40-50%:
• Gravidanza o post-partum
• Malattie autoimmuni: lupus eritematoso, malattia di Graves, sclerosi multipla, miastenia gravis, artrite reumatoide, sindrome di Sjögren
• Diabete
• Malattie dermatologiche: psoriasi e pemfigo volgare
• Malattie dell’apparato respiratorio: asma, BPCO
• Reazioni allergiche a farmaci: penicillina, sulfonamide, fenitoina, metildopa, etc
• Epatite acuta da HBV o HCV
• Neoplasie maligne (sia tumori solidi che neoplasie linfoproliferative)
Il quadro clinico, pur essendo ad impronta emorragica come nell’emofilia congenita, differisce da quest’ultima per la maggior incidenza di sanguinamenti nei tessuti molli, a livello muscolare, nello spazio retroperitoneale e nel tratto gastrointestinale e genitourinario [7,33,34]. Inoltre, diversamente dall’emofilia congenita, sono rari gli emartri. Un paziente con emofilia acquisita potrà presentarsi nei seguenti modi: emottisi da emorragia dal parenchima polmonare, melena o ematemesi da sanguinamento gastrointestinale, ematuria macroscopica da emorragia dal tratto genitourinario, epistassi intrattabile, eccessivo sanguinamento dopo iniezione intramuscolare, reperimento di un accesso venoso o introduzione di catetere vescicale; questi sono solo alcuni esempi di un quadro emorragico estremamente variabile [35]. I pazienti affetti da emofilia acquisita, in relazione all’emorragia, possono sviluppare gravi complicanze quali una sindrome compartimentale o un ictus emorragico. La mortalità stimata è dell’8-22%, con la maggior parte dei decessi che si verificano nelle prime settimane dall’esordio della malattia [7,32,33,34,36]. In realtà i dati relativi alla mortalità sono precedenti all’introduzione su larga scala di più moderne opzioni terapeutiche, perciò la mortalità potrebbe essere più bassa di quanto riportato. Si deve, tra l’altro, tener presente che circa il 25% dei pazienti con emofilia acquisita ha sanguinamenti relativamente lievi che di solito non richiedono terapia emostatica [37].
Nei pazienti con emofilia acquisita, se confrontati a quelli con emofilia congenita ed inibitori, si osserva, comunque, una mortalità maggiore e ciò sembra dover essere attribuito alla non infrequente associazione con altre condizioni potenzialmente mortali, come la CID, all’insorgenza in soggetti anziani, all’estrinsecarsi con un sanguinamento incontrollato che può essere causa di grave morbilità e mortalità prima che venga diagnosticato e curato [14].
Oltre al meccanismo fisiopatologico, all’età d’esordio e al quadro clinico, un’altra importante differenza con la forma congenita sta nell’assenza nell’emofilia acquisita di una storia familiare o personale di disordini emorragici.
Fondamentale per giungere a diagnosi è il sospetto clinico, non così scontato in quanto, soprattutto nel paziente anziano, potrebbero entrare in gioco fattori confondenti come il riscontro non infrequente di ematomi superficiali in pazienti in terapia con acido acetilsalicilico e una non corretta valutazione dello studio del profilo coagulativo in pazienti in terapia con warfarin.
La diagnosi di certezza si basa quindi sul riscontro di aPTT allungato, non corretto, dopo prolungata incubazione, dall’aggiunta di un uguale volume di plasma normale al plasma del paziente; ridotti livelli di FVIII e, attraverso il test di Bethesda, presenza di inibitori. Mentre risultano normali il PT, il TT e il numero di piastrine. Benché si possa, perciò, considerare facile la diagnosi di emofilia acquisita, sfortunatamente, in pazienti con eccessivi sanguinamenti dopo traumi o interventi chirurgici, non è cosa rara trattarsi di casi mal diagnosticati e riguardanti soggetti sottoposti a trattamenti invasivi o a interventi di chirurgia maggiore [32].
La diagnosi differenziale più tipica è quella con il lupus anticoagulant, classicamente caratterizzato da un aPTT altrettanto allungato e scarsamente corretto da plasma normale. Ma i soggetti con lupus anticoagulant, diversamente da quelli affetti da emofilia acquisita, non presentano gravi sanguinamenti e l’aggiunta di fosfolipidi esogeni corregge il prolungamento dell’aPTT [14,36]. Potrebbe anche essere posta in diagnosi differenziale con la CID, ma in quest’ultimo caso gli esami di laboratorio non lasciano alcun dubbio: aPTT
![Tabella 1. I fattori della coagulazione [2]. Denominazione](https://thumb-eu.123doks.com/thumbv2/123dokorg/7312737.88550/4.892.151.728.633.1054/tabella-fattori-coagulazione-denominazione.webp)