4. PARTE SPERIMENTALE
Tutte le operazioni furono effettuate in atmosfera di argon. La vetreria e i tubi NMR furono seccati in stufa, evacuati (10-2 mmHg) e riempiti con argon al momento dell’uso.
4.1. Solventi
• Diclorometano, cloroformio (J. T. Baker): furono rifluiti e distillati su P4O10.
• Eptano (Panreac), pentano (Carlo Erba): furono rifluiti e distillati su LiAlH4.
• Cloroformio deuterato (Armar Chemicals), diclorometano deuterato (CIL): furono filtrati su γ-Al2O3 e conservati in atmosfera di argon.
4.2. Reagenti
• Molibdeno pentacloruro MoCl5, tungsteno esacloruro WCl6 (Aldrich): i prodotti
commerciali furono trasferiti e conservati in fiale chiuse alla fiamma in atmosfera di argon.
• Dimetilformammide HCONMe2 (DMF, Aldrich): fu mantenuta 12 ore in agitazione su BaO e successivamente distillata a pressione ridotta.
• Metilformiato HCO2Me (Fluka) : il prodotto commerciale fu usato come tale.
• Acetone Me2CO (J.T.Baker): fu rifluito e distillato su carbonato di potassio.
• Benzofenone Ph2CO (Aldrich): il prodotto commerciale fu usato come tale.
• o-tolualdeide (Aldrich): il prodotto commerciale fu usato come tale.
• Dimetilmalonato CH2(CO2Me)2, dimetilmaleato cis-(MeO2C)CH=CH(CO2Me),
dietilfumarato trans-(EtO2C)CH=CH(CO2Et), Metilmetossiacetato
MeOCH2CO2Me (Aldrich): i prodotti commerciali furono usati come tali.
• Tetraidropirano C5H10O (THP), 1,4-diossano C4H8O2 (Aldrich): furono rifluiti e
distillati su sodio metallico.
• 1,2-dimetossietano C4H10O2 (DME), 1,2-dietossietano C6H14O2,
1,2-dimetossipropano C5H12O2 (Aldrich): furono rifluiti e distillati su P4O10.
• Tetrametilurea CO(NMe2)2 (TMU, Aldrich): il prodotto commerciale fu conservato su setacci molecolari.
• Etere etilico Et2O (Carlo Erba): fu mantenuto 12 ore in agitazione su CaCl2 e
successivamente rifluito e distillato su LiAlH4.
• 1,3-diossolano C3H6O, 1,1-dimetossimetano CH2(OMe)2, 1,1-dietossimetano
CH2(OEt)2, 1,1-dietossietano CHMe(OEt)2, 2,2-dimetossipropano CMe2(OMe)2,
1,1,1-trimetossimetano CH(OMe)3 (Fluka): i prodotti commerciali furono
4.3. Misure analitiche e chimico-fisiche
Gli spettri infrarossi furono registrati allo stato solido a 298 K mediante spettrofotometro FT-IR Spectrum One Perkin Elmer munito di unità UATR. Le posizioni delle bande furono espresse in cm−1 e le relative intensità indicate con d (debole), m-d (media-debole), m (media), m-f (media-forte), f (forte) e mf (molto forte).
Gli spettri di risonanza magnetica nucleare 1H e 13C furono registrati con uno spettrometro Varian Gemini 200 BB su soluzioni in cloroformio deuterato a 298 K, a meno di diversa specificazione. I valori di chemical shift, espressi in ppm, sono riferiti al residuo non deuterato del solvente.
Le analisi GC/MS furono effettuate con un gas-cromatografo HP6890 munito di colonna Phenonex Zebron e interfacciato con uno spettrometro di massa HP 5973.
Le conduttività molari (ΛM) furono calcolate basandosi su misure di resistenza effettuate con un Metrohm AG Konduktometer E382 (costante di cella = 0.815 cm−1) su soluzioni ca. 0.010 M dei distinti prodotti in diclorometano.66
L’analisi elementare non fu effettuata a causa dell’estrema sensibilità all’aria mostrata dai composti.
Le strutture molecolari, determinate mediante diffrazione di raggi X su cristallo singolo, furono risolte dal Dott. Stefano Zacchini del Dipartimento di Chimica Fisica e Inorganica dell’Università di Bologna.
4.4. Reattività di MoCl
5con composti carbonilici, eteri e dieteri
4.4.1. Reazione di MoCl5 con DMF, HCONMe2: isolamento e caratterizzazione
di [MoOCl4{O=CH(NMe2)}]− − − − [ClCHNMe2]+, 2.
In un provettone codato, furono introdotti MoCl5 (0.293 mmol), CH2Cl2 (ca. 7
mL) e DMF (0.586 mmol). La miscela fu lasciata in agitazione per ca. 20 ore. La soluzione verde risultante fu stratificata con eptano e mantenuta a -30 °C per 19 giorni. Si ottenne così un precipitato cristallino di colore verde, corrispondente a 2, che fu isolato, seccato in corrente di argon e conservato in atmosfera inerte.
Resa: 0.096 g (78 %). IR (cm−1): 3059d, 3005d, 2938d, 1715d, 1658f (νC=O), 1634f (νC=N), 1493d, 1434m, 1373m-f, 1307d, 1201d, 1156d, 1118m, 1055d, 1044d, 971mf (νMo=O), 921m, 905m, 861d, 829d, 810d, 703d, 681f.
ΛM = 3.17S·cm2·mol−1.
I cristalli di 2 furono utilizzati per la determinazione della struttura molecolare mediante diffrazione di raggi X.
In un secondo esperimento, furono introdotti in un tubo NMR MoCl5 (0.447
mmol), CD2Cl2 (0.70 mL) e DMF (0.894 mmol). Il tubo fu sigillato alla fiamma e la
miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 12 giorni. Si ottennero una soluzione verde brillante e cristalli di uguale colore. I cristalli furono isolati trasferendo la soluzione in un secondo tubo NMR, quindi entrambi i campioni furono sigillati. Alla soluzione fu aggiunto un largo eccesso di acqua (ca. 10 mmol). Dopo agitazione del campione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. L’unica specie chiaramente riconoscibile fu DMF.33
4.4.2. Reazione di MoCl5 con HCOOMe: isolamento e caratterizzazione di
[MoOCl3(HCO2Me)]2, 4.
In un provettone codato, furono introdotti MoCl5 (0.516 mmol), CH2Cl2 (ca. 10
mL) e HCOOMe (1.032 mmol). La miscela fu lasciata in agitazione per ca. 17 ore. La soluzione verde-marrone risultante fu stratificata con eptano e mantenuta a -30 °C per 21 giorni. Il solido verde scuro ottenuto, corrispondente a 4, venne isolato, seccato in corrente di argon e conservato in atmosfera inerte. Resa: 0.104 g (72 %). IR (cm−1): 3020d, 2961d, 1654m, 1602f (νC=O), 1550m, 1446d, 1429m-f, 1367m-f, 1312f, 1295m-f, 1176m, 994mf (νMo=O), 878m-f, 809m, 731m.
I cristalli di 4 furono utilizzati per la determinazione della struttura molecolare mediante diffrazione di raggi X.
In un secondo esperimento, furono introdotti in un tubo NMR MoCl5 (0.483
mmol), CDCl3 (0.70 mL) e HCOOMe (0.966 mmol). Il tubo fu sigillato alla fiamma
e la miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 12 giorni. Quindi, il tubo fu aperto e alla sospensione marrone fu aggiunto un largo eccesso di acqua (ca. 10 mmol). Al momento della apertura del tubo si osservò lo sviluppo di gas. Dopo agitazione del campione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. Fu così possibile identificare HCOOMe e MeCl.33
4.4.3. Reazione di MoCl5 con R2CO (R = Me, Ph): isolamento e caratterizzazione
di [MoOCl3(L)]2 (L = OCMe2, 6a; L = OCPh2, 6b).
In un provettone codato, furono introdotti MoCl5 (0.436 mmol), CH2Cl2 (ca. 10
mL) e R2CO (R = Me, Ph) (0.872 mmol). La miscela fu lasciata in agitazione per ca.
24 ore. La soluzione marrone risultante fu stratificata con eptano e mantenuta a -30 °C per 23 giorni. Il solido ottenuto venne isolato, seccato in corrente di argon e conservato in atmosfera inerte.
6a. Resa: 0.083 g (69 %). IR (cm−1): 3412d, 2958m, 2923m, 2873m-d, 2857m-d, 1997d, 1616f (νC=O), 1467m-d, 416m-d, 1382m, 1361m, 1347m, 1255m, 1081m, 1000mf (νMo=O), 882d, 735m-d.
6b. Resa: 0.129 g (74 %). IR (cm−1): 3061d, 1593m, 1581m, 1538m-f, 1522f (νC=O), 1491m, 1446m, 1329f, 1316m, 1293f, 1181m-d, 1159m-d, 1104d, 1086d, 1074d, 1028d, 990mf (νMo=O), 941m-d, 923m, 838d, 808d, 770m-f, 734d, 696mf, 678m-f.
4.4.4. Reazione di MoCl5 con MeOCH2COOMe.
In un tubo NMR furono introdotti MoCl5 (0.472 mmol), CDCl3 (0.70 mL), CH2Cl2
(0.468 mmol) e MeOCH2COOMe (0.475 mmol). Il tubo fu sigillato alla fiamma e la
miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 10 giorni. Quindi, il tubo fu aperto e alla sospensione fu aggiunto un largo eccesso di acqua (ca. 10 mmol). Al momento della apertura del tubo si osservò la fuoriuscita di una abbondante quantità di gas. Dopo agitazione del campione, la soluzione incolore risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. Fu così possibile identificare MeOCH2COOMe
e MeCl.33 Rapporti molari CH2Cl2 : MeOCH2COOMe : MeCl = 10 : 6 : 3.
4.4.5. Reazione di MoCl5 con THF, C4H8O: isolamento e caratterizzazione di
[MoOCl3(THF)]2, 9.
In un provettone codato, furono introdotti MoCl5 (0.520 mmol), CH2Cl2 (ca. 10
mL) e THF (1.040 mmol). La miscela fu lasciata in agitazione per ca. 5 ore. La soluzione rossa risultante fu stratificata con eptano e mantenuta a -30 °C per 4 giorni. Il solido verde ottenuto, corrispondente a 9, venne isolato, seccato in corrente di argon e conservato in atmosfera inerte. Resa: 0.129 g (85%). IR (cm−1): 3411m-d, 3154d, 2972d, 2950d, 2903d, 2875d, 1587m, 1481d, 1469d, 1449d,1359d, 1346m-d, 1317d, 1298d, 1246d, 1231d, 1171d, 1042d, 992mf (νMo=O), 956m-d, 924d, 916d, 835f, 735m-d, 680m-d
I cristalli di 9 furono utilizzati per la determinazione della struttura molecolare mediante diffrazione di raggi X.
In un secondo esperimento, furono introdotti in un tubo NMR MoCl5 (0.428
temperatura ambiente per 12 giorni e successivamente analizzato mediante 1H-NMR. Lo spettro rivelò la presenza di 1,4-diclorobutano.33 Quindi, il tubo fu aperto e alla sospensione rossa fu aggiunto un largo eccesso di acqua (ca. 10 mmol). Dopo agitazione del campione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. Fu così possibile identificare1,4-diclorobutano.33
4.4.6. Reazione di MoCl5 con 1,2-dialcossialcani.
4.4.6.1. Reazione di MoCl5 con DME, 1,2-dimetossietano.
In un tubo NMR furono introdotti MoCl5 (0.465 mmol), CDCl3 (0.70 mL), CH2Cl2
(0.468 mmol) e 1,2-DME (0.471 mmol). Il tubo fu sigillato alla fiamma e la miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 10 giorni. Quindi, il tubo fu aperto e alla sospensione fu aggiunto un largo eccesso di acqua (ca. 10 mmol). Dopo agitazione del campione, la soluzione incolore risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR. Fu così possibile identificare 1,2-DME, MeCl e ClCH2CH2Cl.33
Rapporti molari CH2Cl2 : 1,2-DME : MeCl : ClCH2CH2Cl = 10 : 9 : 4 : 1.
4.4.6.2. Reazione di MoCl5 con 1,2-dietossietano.
In un provettone codato, furono introdotti MoCl5 (0.498 mmol), CH2Cl2 (ca. 10
mL) e 1,2-dietossietano (0.499 mmol). La miscela fu lasciata in agitazione per ca. 19 ore. La soluzione giallo-verde risultante fu stratificata con eptano e mantenuta a –30 °C per 6 giorni. Il solido marrone ottenuto venne isolato, seccato in corrente di argon e conservato in atmosfera inerte. IR (cm−1): 2983d, 2936d, 1602d, 1467d, 1444m-d, 1386m, 1304d, 1264d, 1236d, 1167d, 1089m-d, 1056m, 1018m-f, 988mf (νMo=O), 921m-f, 911m-f, 832f, 809m, 792mf, 774m-f.
4.4.6.3. Reazione di MoCl5 con 1,2-dimetossipropano.
In un provettone codato, furono introdotti MoCl5 (0.655 mmol), CH2Cl2 (ca. 10
mL) e 1,2-dimetossipropano (0.657 mmol). La miscela fu lasciata in agitazione per ca. 20 ore. La soluzione giallo-verde risultante fu stratificata con eptano e mantenuta
a -30 °C per 5 giorni. Il solido marrone ottenuto venne isolato, seccato in corrente di argon e conservato in atmosfera inerte. IR (cm−1): 2949d, 1598d, 1445m-d, 140d, 1385d, 1348d, 1266d, 1194d, 1161d,1149d,1114d,1066m-d,1043d, 1012m, 987mf (νMo=O), 928m, 900m-f, 766m.
4.4.7. Reazione di MoCl5 con THP, C5H10O: isolamento e caratterizzazione di
MoCl4(THP)2, 19.
In un provettone codato, furono introdotti MoCl5 (0.622 mmol), CH2Cl2 (ca. 7
mL) e THP (1.248 mmol). La miscela fu lasciata in agitazione per ca. 4 ore. La soluzione arancione-scuro risultante fu stratificata con eptano e mantenuta a -30 °C per 19 giorni. Si ottenne così un precipitato cristallino di colore arancione corrispondente a 19, che fu isolato, seccato in corrente di argon e conservato in atmosfera inerte. Resa: 0.204 g (80 %). IR (cm−1): 2944m, 2925m-d, 2872d, 1590d, 1557d, 1492d, 1470d, 1452d, 1436m, 1429m, 1377d, 1364d, 1355d, 1325d, 1304m, 1272d, 1186m, 1150m, 1069d, 996mf, 988mf, 954m, 939f, 884d, 849f, 788f, 733f.
ΛM = 1.72S·cm2·mol−1.
I cristalli di 19 furono utilizzati per la determinazione della struttura molecolare mediante diffrazione di raggi X.
In un secondo esperimento, furono introdotti in un tubo NMR MoCl5 (0.538
mmol), CDCl3 (0.70 mL) e THP (0.542 mmol). Il tubo fu sigillato alla fiamma e la
miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 12 giorni. Si ottennero una soluzione marrone e cristalli di uguale colore. Dagli spettri NMR della miscela fu identificato THP coordinato al centro metallico [1H-NMR(CDCl3): δ = 3.68, 1.70 (CH2) ppm]. Alla miscela fu
aggiunto un largo eccesso di acqua (ca. 10 mmol). Dopo agitazione del campione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. Fu così possibile identificare THP e 1,5-dicloropentano.33 Rapporti molari THP : 1,5-dicloropentano = 10: 1.
4.4.8. Reazione di MoCl5 con trans-(EtO2C)CH=CH(CO2Et): isolamento e
caratterizzazione di [MoCl4{{{{κκκκ2-trans-(EtO2C)CH=CH(CO2Et)}}}}]∞, 20.
In un provettone codato, furono introdotti MoCl5 (0.619 mmol), CH2Cl2 (ca. 10
mL) e trans-(EtO2C)CH=CH(CO2Et) (0.617 mmol). La miscela fu lasciata in
agitazione per ca. 18 ore e successivamente filtrata per eliminare del solido rimasto in sospensione.. La soluzione rosso-scuro risultante fu stratificata con eptano e mantenuta a -30 °C per 4 giorni. Il solido arancione ottenuto, corrispondente a 20, venne isolato, seccato in corrente di argon e conservato in atmosfera inerte.
Resa: 0.211 g (83%). IR (cm−1): 3093d, 2960d, 2926d, 2874d, 2859d, 1628mf (νC=O), 1598f, 1462m, 1400m, 13273m, 1308mf, 1283m, 1243m-f, 1208d, 1094m-d, 1020m, 992f, 972f, 961f, 923f, 858m, 801d, 772m, 735m, 703d, 667d.
ΛM = 0.13 S·cm2·mol−1.
I cristalli di 20 furono utilizzati per la determinazione della struttura molecolare mediante diffrazione di raggi X.
In un secondo esperimento, furono introdotti in un tubo NMR MoCl5 (0.450
mmol), CDCl3 (0.70 mL) e trans-(EtO2C)CH=CH(CO2Et) (0.452 mmol). Il tubo fu
sigillato alla fiamma e la miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 14 giorni. Si ottennero una soluzione marrone e cristalli di uguale colore. Il campione fu quindi riscaldato a ca. 100 °C per 1 ora ottenendo un solido arancione. A questa miscela fu aggiunto un largo eccesso di acqua (ca. 10 mmol) e dopo agitazione del campione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR. Fu così possibile identificare trans-(EtO2C)CH=CH(CO2Et) e EtCl.33 Rapporti molari
4.4.9. Reazione di MoCl5 con diesteri.
In un provettone codato, furono introdotti MoCl5, CH2Cl2 (ca. 10 mL) e diestere.
La miscela fu lasciata in agitazione per ca.18 ore e successivamente filtrata per eliminare del solido rimasto in sospensione. La soluzione marrone risultante fu stratificata con eptano e mantenuta a -30 °C per 4 giorni. Il solido marrone ottenuto venne isolato, seccato in corrente di argon e conservato in atmosfera inerte.
Reazione di MoCl5 con CH2(CO2Me)2
MoCl5 (mmol) CH2(CO2Me)2 (mmol) 0.637 0.761 IR (cm−1): 2998d, 2871d, 1687f ,1653mf (νC=O), 1618f, 1603f, 1458f, 1447f, 1389m, 1371m, 1356f, 1241f, 1197m, 1173m, 995f, 954m- d, 927m, 890f, 780d, 736d, 697d. ΛM = 0.21 S·cm2·mol−1
Reazione di MoCl5 con cis-(CO2Me)CH=CH(CO2Me)
MoCl5
(mmol)
cis-(CO2Me)CH=CH(CO2Me)
(mmol)
0.695 0.838
IR (cm−1): 3053d, 2957d, 1687m-f, 1651f (νC=O), 1622f, 1453f, 1439f, 1400m-f, 1327m, 1261mf, 1232f, 1182m, 1150d, 992f, 945f, 906m-d, 834f, 818m, 656m-d.
In un secondo esperimento, furono introdotti in un tubo NMR MoCl5, CDCl3 (0.70
mL) e diestere. Il tubo fu sigillato alla fiamma e la miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 14 giorni. Quindi, fu riscaldato a ca. 100 °C per 1 ora e alla miscela risultante fu aggiunto un largo eccesso di acqua (ca. 10 mmol). Dopo agitazione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e
GC/MS. L’identificazione dei composti organici fu condotta confrontando gli spettri NMR ottenuti con i dati riportati in letteratura.33 Di seguito sono riportati i risultati ottenuti.
Reazione di MoCl5 con CH2(CO2Me)2
MoCl5
(mmol)
CH2(CO2Me)2
(mmol)
0.399 0.394
Dopo riscaldamento e idrolisi CH2(CO2Me)2, MeCl a
a Uniche specie chiaramente riconoscibili.
Reazione di MoCl5 con cis-(CO2Me)CH=CH(CO2Me)
MoCl5
(mmol)
cis-(CO2Me)CH=CH(CO2Me)
(mmol)
0.533 0.555
Dopo riscaldamento e idrolisi
cis-(CO2Me)CH=CH(CO2Me), trans-(CO2Me)CH=CH(CO2Me), MeCl,
2,5-furandione b
b Rapporti molari CH
3Cl : cis-(CO2Me)CH=CH(CO2Me) : trans-(CO2Me)CH=CH(CO2Me) : 2,5-furandione = 10 : 3 : 0.5 : 5
4.5. Reattività di WCl
6con composti carbonilici, eteri e dieteri
4.5.1. Reazione di WCl6 con DMF, HCONMe2: isolamento e caratterizzazione di
WOCl4(O=CHNMe2), 3.
In un provettone codato, furono introdotti WCl6 (0.646 mmol), CH2Cl2 (ca. 7 mL)
e DMF (1.292 mmol). La miscela fu lasciata in agitazione per ca. 6 ore. La soluzione rossa-arancione risultante fu stratificata con eptano e mantenuta a -30 °C per 19 giorni. Il solido giallo ottenuto, corrispondente a 3, venne isolato, seccato in corrente di argon e conservato in atmosfera inerte. Resa: 0.204 g (76 %). IR (cm−1): 3060d, 2981d, 2938d, 1720d, 1652mf (νC=O), 1486d, 1428m-f, 1361f, 1302d, 1244d, 1153d, 1121m, 1057d, 978f (νW=O), 921m, 862 m, 687f.
I cristalli di 3 furono utilizzati per la determinazione della struttura molecolare mediante diffrazione di raggi X.
In un secondo esperimento, furono introdotti in un tubo NMR WCl6 (0.625
mmol), CD2Cl2 (0.70 mL) e DMF (1.253 mmol). Il tubo fu sigillato alla fiamma e la
miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 12 giorni. Si ottenne una soluzione arancione che fu
analizzata mediante 1H-NMR. Fu possibile identificare DMF coordinata [1H-NMR(CD2Cl2): δ = 8.45 (s, 1 H, CH), 3.35 (s, 3 H, CH3), 3.18 (s, 3 H, CH3)
ppm]. Quindi, alla soluzione fu aggiunto un largo eccesso di acqua (ca. 10 mmol) e dopo agitazione del campione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. L’unica specie chiaramente riconoscibile fu DMF.33
4.5.2. Reazione di WCl6 con chetoni e aldeidi.
4.5.2.1. Reazione di WCl6 con Me2CO: caratterizzazione di WOCl4(Me2CO) 5a.
In un tubo NMR furono introdotti WCl6 (0.492 mmol), CDCl3 (0.70 mL) e
Me2CO (0.981 mmol). Il tubo fu sigillato alla fiamma e la miscela omogeneizzata
14 giorni. Si ottenne una soluzione arancione che fu analizzata mediante NMR. Lo
spettro rivelò la presenza di Me2CO coordinato al centro metallico
[1H-NMR(CDCl3): δ = 2.68 (s, 6 H, CH3) ppm; 13C-NMR(CDCl3): δ = 223.1(C=O),
32.6 (CH3) ppm] e di 2,2-dicloropropano33 in rapporto molare ca. 1 : 1.
4.5.2.2. Reazione di WCl6 con C6H4(CHO)(2-Me): caratterizzazione di
WOCl4(C6H4(CHO)(2-Me)), 5b.
In un tubo NMR furono introdotti WCl6 (0.310 mmol), CDCl3 (0.70 mL) e
o-tolualdeide (0.620 mmol). Il tubo fu sigillato alla fiamma e la miscela omogeneizzata mediante breve mescolamento. Il tubo NMR fu mantenuto a temperatura ambiente per 3 giorni, quindi fu aperto e vi fu introdotto un largo eccesso di acqua (ca. 10 mmol). Dopo agitazione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante 1H-NMR e GC/MS. Fu così possibile identificare o-tolualdeide e 1-diclorometil-2-metilbenzene [1H-NMR(CDCl3): δ = 7.22-7.81 (m,
4 H, anello benzenico), 6.96 (s, 1 H, CH), 2.49 (s, 3 H, CH3) ppm].67
4.5.3. Reazione di WCl6 con MeOCH2COOMe: isolamento e caratterizzazione
di WCl4(OCH2CO2Me), 7 e caratterizzazione di WCl5[O(Me)CH2CO2Me], 8.
In un tubo NMR furono introdotti WCl6 (0.396 mmol), CDCl3 (0.70 mL), CH2Cl2
(0.390 mmol) e MeOCH2COOMe (0.394 mmol). Il tubo fu sigillato alla fiamma e la
miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 10 giorni. Si ottennero una soluzione rossa e cristalli di uguale colore. I cristalli furono isolati trasferendo la soluzione in un secondo tubo NMR, quindi entrambi i campioni furono sigillati. Lo spettro 1H-NMR della soluzione rivelò la presenza di MeCl, del composto 7 [1H-NMR(CDCl3): δ = 6.21 (s,
2 H, CH2), 4.14 (s, 3 H, CO2Me) ppm] e di MeOCH2COOMe coordinato al centro
metallico presumibilmente nel complesso WCl5[O(Me)CH2CO2Me], 8
[1H-NMR(CDCl3): δ = 4.31 (s, 2 H, CH2), 4.14 (s, 3 H, CO2Me), 3.62 (s, 3 H, OMe)
ppm]. Rapporti molari CH2Cl2 : 7 : 8 : MeCl = 10 : 2 : 4 : 5. Alla soluzione fu
soluzione incolore risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. Fu così possibile identificare MeOCH2COOMe e MeCl.33
La struttura del composto 7 fu determinata mediante uno studio di diffrazione di raggi X sui cristalli isolati nel suddetto esperimento.
4.5.4. Reazione di WCl6 con DME, 1,2-dimetossietano: caratterizzazione di
WCl5(OCH2CH2OMe), 10.
In un tubo NMR furono introdotti WCl6 (0.449 mmol), CDCl3 (0.70 mL), CH2Cl2
(0.452 mmol) e 1,2-DME (0.452 mmol). Il tubo fu sigillato alla fiamma e la miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 10 giorni e successivamente analizzato mediante NMR. Fu possibile identificare WCl5(OCH2CH2OMe), 10 [1H-NMR(CDCl3): δ = 5.72 (t, 2H, 3
JHH = 5 Hz, WOCH2); 4.27 (t, 2H, 3JHH = 5 Hz, CH2); 4.03 (s, 3H,CH3) ppm ; 13
C-NMR(CDCl3): δ = 79.0 (WOCH2), 75.6 (CH2), 63.5 (Me) ppm], e MeCl.33
Rapporti molari CH2Cl2 : 10 : CH3Cl = 10 : 5 : 11. Alla soluzione fu aggiunto un
largo eccesso di acqua (ca. 10 mmol). Dopo agitazione del campione, la soluzione gialla risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. Fu così possibile identificare MeOCH2CH2Cl [1H-NMR(CDCl3): δ = 4.01,
3.99 (CH2), 3.44 (CH3) ppm ; 13C-NMR(CDCl3): δ = 77.8, 74.4 (CH2), 62.3 (CH3)
ppm], ClCH2CH2Cl e MeCl.33 Rapporti molari CH2Cl2 : MeOCH2CH2Cl :
ClCH2CH2Cl : MeCl = 10 : 4 : 6 : 8.
4.5.5. Reazione di WCl6 con TMU, (NMe2)2CO: isolamento e caratterizzazione
di [WCl6]− − − − [(NMe2)2COH]+, 18.
In un provettone codato, furono introdotti WCl6 (0.426 mmol), CH2Cl2 (ca. 10
mL) e TMU (0.426 mmol). La miscela fu lasciata in agitazione per ca. 6 ore e successivamente filtrata per eliminare una piccola quantità di solido giallo presente in sospensione. La soluzione arancione risultante fu stratificata con eptano e
mantenuta a -30 °C per 19 giorni. Il solido rosso ottenuto, corrispondente a 18 venne isolato, seccato in corrente di argon e conservato in atmosfera inerte.
Resa: 0.103 g (47 %). IR (cm−1): 3018d, 2937d, 2810d, 1766m-f, 1656f, 1602m, 1548m-f, 1463m-f, 1443m-f, 1404f, 1284m, 1238m, 1225m, 1159mf, 1119f, 1062f, 1001m, 979m, 957m, 909m, 892m-f, 868f, 784f, 763f, 734f.
I cristalli di 18 furono utilizzati per la determinazione della struttura molecolare mediante diffrazione di raggi X.
In un secondo esperimento, furono introdotti in un tubo NMR WCl6 (0.411
mmol), CDCl3 (0.70 mL) e TMU (0.411 mmol). Il tubo fu sigillato alla fiamma e la
miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 12 giorni. Si ottenne una soluzione rossa che fu analizzata mediante 1H-NMR. Lo spettro rivelò la presenza di una risonanza molto intensa a 3.25 ppm. Quindi, alla soluzione fu aggiunto un largo eccesso di acqua (ca. 10 mmol) e dopo agitazione del campione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. L’unica specie chiaramente riconoscibile fu TMU.33
4.5.6. Reazione di WCl6 con diesteri.
In un provettone codato, furono introdotti WCl6, CH2Cl2 (ca. 10 mL) e diestere.
La miscela fu lasciata in agitazione per ca.36 ore e successivamente filtrata per eliminare del solido rimasto in sospensione. La soluzione arancione risultante fu stratificata con eptano e mantenuta a -30 °C per 4 giorni. Il solido nero ottenuto venne isolato, seccato in corrente di argon e conservato in atmosfera inerte.
Reazione di WCl6 con CH2(CO2Me)2 WCl6 (mmol) CH2(CO2Me)2 (mmol) 0.709 0.849 IR (cm−1): 2985d, 1737m, 1680f (νC=O), 1617m, 1475m, 1463m, 1444m, 1402m, 1371m, 1354m, 1248m, 1213m, 1204m, 1162m, 1013m, 989m, 968m, 947m, 903mf, 892mf, 790m. ΛM = 0.28 S·cm2·mol−1
Reazione di WCl6 con cis-(CO2Me)CH=CH(CO2Me)
WCl6
(mmol)
cis-(CO2Me)CH=CH(CO2Me)
(mmol) 0.651 0.777 IR (cm−1): 3061d, 2960d, 1727d, 1646f (νC=O), 1614m, 1471f, 1456f, 1448f, 1406m, 1382m, 1349m, 1298f, 1276f, 1217m, 1191m, 1159m, 1146m, 1050m, 1000m, 941m, 897mf, 848mf. ΛM = 2.21 S·cm2·mol−1
Reazione di WCl6 con trans-(EtO2C)CH=CH(CO2Et)
WCl6
(mmol)
trans-(EtO2C)CH=CH(CO2Et)
(mmol) 0.731 0.880 IR (cm−1): 2986d, 1728d, 1634mf (νC=O), 1462m, 1443m, 1397d, 1377m, 1311f, 1285m, 1262d, 1241m, 1172d, 1094m, 1019m, 1003mf, 977m-f, 959d, 893m, 855m, 773m. ΛM = 0.35 S·cm2·mol−1
In un secondo esperimento, furono introdotti in un tubo NMR WCl6, CD2Cl2 (0.70
mL) e diestere. Il tubo fu sigillato alla fiamma e la miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 3 giorni. Si ottennero una soluzione arancione e un solido nero. La miscela di reazione fu analizzata mediante NMR a temperatura ambiente e a bassa temperatura (-80 °C). Successivamente, i campioni furono riscaldato a ca. 100 °C per 2 ore e alla miscela risultante fu aggiunto un largo eccesso di acqua (ca. 10 mmol). Dopo agitazione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. L’identificazione dei composti organici fu condotta confrontando gli spettri NMR ottenuti con i dati riportati in letteratura.33 Di seguito sono riportati i risultati ottenuti.
Reazione di WCl6 con CH2(CO2Me)2
WCl6 (mmol) CH2(CO2Me)2 (mmol) 0.401 0.405 Prima dell’idrolisi (NMR a -80 °C)
CH2(CO2Me)2 coordinato al centro metallico [1H-NMR(CD2Cl2): δ = 4.16, 3.95,
3.73 ppm ; 13C-NMR(CD2Cl2): δ = 176.2, 166.1, 58.4, 54.38, 41.3 ppm], MeCl a
Dopo riscaldamento e idrolisi CH2(CO2Me)2, MeCl b
a
MeCl presente in tracce b Rapporti molari CH
Reazione di WCl6 con cis-(CO2Me)CH=CH(CO2Me)
WCl6
(mmol)
cis-(CO2Me)CH=CH(CO2Me)
(mmol) 0.577 0.578 Prima dell’idrolisi (NMR a -80 °C) 1 H-NMR(CD2Cl2): δ = 7.20, 6.84, 6.73, 6.47, 4.18, 3.84, 3.76, 3.02 13 C-NMR(CD2Cl2): δ = 176.0, 172.3, 166.0, 165.3, 139.2, 133.8, 130.9, 130.0, 58.0, 53.5
Dopo riscaldamento e idrolisi
MeCl, 2,5-furandione, ClOCCH=CHCOCl, cis-(CO2Me)CH=CH(CO2Me),
trans-CH(COOMe)=CH(OOMe) a a
Rapporti molari MeCl: 2,5-furandione: ClOCCH=CHCOCl: trans-CH(COOMe)=CH(OOMe) : cis-(CO2Me)CH=CH(CO2Me) = 10: 2: 3 : 5 :tracce
Reazione di WCl6 con trans-(EtO2C)CH=CH(CO2Et)
WCl6
(mmol)
trans-(EtO2C)CH=CH(CO2Et)
(mmol) 0.376 0.378 Prima dell’idrolisi (NMR a -80 °C) 1 H-NMR(CD2Cl2): δ = 7.19, 6.83, 4.62, 4.16, 1.43, 1.26 13 C-NMR(CD2Cl2): δ = 171.8, 165.6, 164.4, 139.4, 134.0, 129.9, 68.1, 62.9, 62.4, 14.4
Dopo riscaldamento e idrolisi trans-(EtO2C)CH=CH(CO2Et)
4.5.7. Reazione di WCl6 con eteri ciclici.
4.5.7.1. Reazione di WCl6 con THP, C5H10O: caratterizzazione di WCl5(THP), 21a.
In un tubo NMR furono introdotti WCl6 (0.308 mmol), CDCl3 (0.70 mL), THP
(0.307 mmol) e CH2Cl2 (0.312 mmol). Il tubo fu sigillato alla fiamma e la miscela
omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 4 giorni. Si ottennero una soluzione rossa e un solido marrone. Dagli spettri NMR della miscela fu identificato THP coordinato al centro metallico [1H-NMR(CDCl3): δ = 4.03 (s, 4H, CH2), 1.73 (s,6H, CH2) ppm ; 13
C-NMR(CDCl3): δ = 107.7, 26.6, 22.9 (CH2) ppm]. Rapporti molari CH2Cl2 :
THPcoord. = 1 : 1. Il tubo fu mantenuto a 60 °C per 1 ora e 30 minuti e poi a 95 °C per
25 minuti, lo spettro 1H-NMR risultò identico a quello registrato a temperatura ambiente.
4.5.7.2. Reazione di WCl6 con THF, C4H8O: caratterizzazione di WCl5(THF), 21b.
In un tubo NMR furono introdotti WCl6 (0.328 mmol), CDCl3 (0.70 mL), THF
(0.321 mmol) e CH2Cl2 (0.328 mmol). Il tubo fu sigillato alla fiamma e la miscela
omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 12 giorni e successivamente analizzato mediante NMR. Lo spettro rivelò la presenza di 21b [1H-NMR(CDCl3): δ = 2.16 (OCH2), 4.57
(CH2CH2) ppm ; 13C-NMR(CDCl3): δ = 74.1 (OCH2), 26.2 (CH2CH2) ppm].
Rapporti molari CH2Cl2 : 21b = 1 : 1. Il tubo fu mantenuto a ca. 80 °C per 1 ora e
quindi analizzato mediante 1H-NMR. Lo spettro risultò analogo a quello acquisito prima del riscaldamento.
4.5.7.3. Reazione di WCl6 con 1,4-diossano: caratterizzazione di
WCl5[O(CH2)2O(CH2)2], 21c.
In un tubo NMR furono introdotti WCl6 (0.421 mmol), CDCl3 (0.70 mL) e
1,4-diossano (0.422 mmol). Il tubo fu sigillato alla fiamma e la miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 12 giorni. Si ottennero una soluzione rossa e un solido marrone. La miscela di reazione fu analizzata mediante NMR a temperatura
ambiente e a bassa temperatura (-60 °C). Lo spettro 1H-NMR a temperatura ambienta mostrò un singolo picco largo a 3.94 ppm. Gli spettri NMR registrati a -60 °C
permisero di identificare 1,4-diossano coordinato al centro metallico [1H-NMR(CDCl3): δ = 4.89 (2H), 4.25 (2H), 3.99 (4H) ppm ; 13C-NMR(CDCl3):
δ = 72.9 (OCH2), 67.5 (CH2) ppm]. Quindi, il campione fu riscaldato a 60 °C per
1 ora e 30 minuti e successivamente a 95 °C per 25 minuti. Lo spettro 1H-NMR risultò analogo a quello precedentemente registrato a temperatura ambiente.
4.5.8. Reazione di WCl6 con Et2O: isolamento e caratterizzazione di
WCl6(Et2O), 22.
In un provettone codato furono introdotti WCl6 (0.436 mmol), CH2Cl2 (ca. 7 mL)
e Et2O (0.438 mmol). La miscela fu lasciata in agitazione per ca. 4 ore e
successivamente filtrata per eliminare una piccola quantità di solido rimasta in sospensione. La soluzione rossa risultante fu stratificata con eptano e mantenuta a -30 °C per 26 giorni. Il solido ottenuto venne isolato, seccato sottovuoto e conservato in atmosfera inerte. Resa: 0.167 g (81%). IR (cm−1): 2987d, 2939d, 1520d, 1441d, 1385d, 1294d, 1185d, 1106d, 1055d, 1013d, 997m, 938m, 855mf (νC−O−C), 748f. ΛM = 0.89S·cm2·mol−1.
In un secondo esperimento, furono introdotti in un tubo NMR WCl6 (0.519
mmol), CDCl3 (0.70 mL) e Et2O (0.524 mmol). Il tubo fu sigillato alla fiamma e la
miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 12 giorni. Si ottenne una soluzione rossa e un solido nero. Tale miscela fu analizzata mediante NMR che rivelò la presenza di Et2O coordinato
al centro metallico [1H-NMR (CDCl3): δ = 4.00 (CH2), 1.35 (CH3) ppm] e EtCl.
Dopo ca. 24 ore fu aggiunto Me2CO (0.518 mmol) alla miscela di reazione. Il
campione fu agitato e nell’arco di 2 minuti la soluzione assunse una colorazione gialla e iniziò a formarsi un solido giallo. L’analisi 1H-NMR mostrò la presenza di Et2O, EtCl, 5a e 2,2-dicloropropano.33
4.6. Reattività di MoCl
5con 1,1-dialcossialcani e CH(OMe)
34.6.1. Reazioni condotte in tubo NMR
In un tubo NMR furono introdotti MoCl5, CDCl3 (0.70 mL), 1,1-dialcossialcano e
CH2Cl2. Il tubo fu sigillato alla fiamma e la miscela omogeneizzata mediante breve
mescolamento. Il tubo NMR fu mantenuto a temperatura ambiente, quindi fu aperto e alla sospensione marrone fu aggiunto un largo eccesso di acqua (ca. 10 mmol). Dopo agitazione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. L’identificazione dei composti organici fu condotta confrontando gli spettri NMR ottenuti con i dati riportati in letteratura;33 i risultati sono stati raccolti in Tabella 12 specificando i valori di chemical shift dei composti non presenti nella banca dati.33
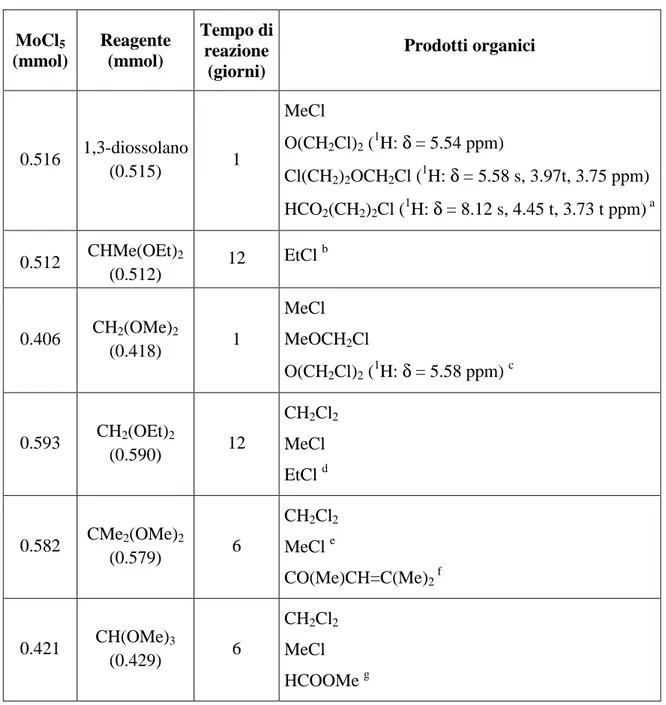
Tabella 12: Reazione di MoCl5 con 1,1-dialcossialcani e con CH(OMe)3, a 298 K. MoCl5 (mmol) Reagente (mmol) Tempo di reazione (giorni) Prodotti organici 0.516 1,3-diossolano (0.515) 1 MeCl O(CH2Cl)2 (1H: δ = 5.54 ppm) Cl(CH2)2OCH2Cl (1H: δ = 5.58 s, 3.97t, 3.75 ppm) HCO2(CH2)2Cl (1H: δ = 8.12 s, 4.45 t, 3.73 t ppm) a 0.512 CHMe(OEt)2 (0.512) 12 EtCl b 0.406 CH2(OMe)2 (0.418) 1 MeCl MeOCH2Cl O(CH2Cl)2 (1H: δ = 5.58 ppm) c 0.593 CH2(OEt)2 (0.590) 12 CH2Cl2 MeCl EtCl d 0.582 CMe2(OMe)2 (0.579) 6 CH2Cl2 MeCl e CO(Me)CH=C(Me)2 f 0.421 CH(OMe)3 (0.429) 6 CH2Cl2 MeCl HCOOMe g
a Rapporti molari MeCl: O(CH
2Cl)2 : Cl(CH2)2OCH2Cl : HCO2(CH2)2Cl = 2 : 7 : 10 : 1 b Unica specie chiaramente riconoscibile nello spettro NMR
c
Rapporti molari MeCl : MeOCH2Cl : O(CH2Cl)2 = 10 : 1 : tracce d Rapporti molari CH 2Cl2 : MeCl : EtCl = 10 : 1 : 8 e Rapporti molari CH 2Cl2 :MeCl= 10 : 10 f Identificato via GC/MS g
4.6.2. Reazioni a scopo preparativo
4.6.2.1. Reazioni di MoCl5 con CH2(OMe)2, CH2(OEt)2, CH(OMe)3.
Tali reazioni condussero a miscele di prodotti difficilmente separabili e identificabili. Di seguito si riporta la procedura relativa alla reazione fra MoCl5 e
CH2(OMe)2 : in un provettone codato, furono introdotti MoCl5 (0.545 mmol),
CH2Cl2 (ca. 7 mL) e CH2(OMe)2 (0.543 mmol). La miscela fu lasciata in agitazione
per ca. 6 ore. Quindi, la soluzione marrone risultante fu stratificata con eptano e mantenuta a -30 °C per 2 giorni. Fu ottenuto un precipitato marrone oleoso, molto sensibile all’aria, che non fu possibile cristallizzare e identificare.
4.6.2.2. Reazione di MoCl5 con 1,3-diossolano: isolamento e caratterizzazione di
[MoOCl3(HCO2CH2CH2Cl)]2, 11.
In un provettone codato, furono introdotti MoCl5 (0.842 mmol), CH2Cl2 (ca. 7
mL) e 1,3-diossolano (0.844 mmol). La miscela fu lasciata in agitazione per ca. 24 ore e successivamente filtrata per eliminare una piccola quantità di solido rimasta in sospensione. La soluzione verde-marrone risultante fu stratificata con eptano, quindi mantenuta a -30 °C per 18 giorni. Si ottenne così un precipitato cristallino di colore verde, corrispondente a 11, che fu seccato in corrente di argon e conservato in atmosfera inerte. Resa: 0.135 g (49 %). IR (cm−1): 2968d, 1644mf (νC=O), 1614f, 1440m, 1429m-d, 1391m-d, 1374m, 1302f, 1288f, 1271f, 1238mf, 1196m, 1065m-d, 1000mf (νMo=O), 959m, 942f, 825m-d, 805m, 782m-d, 736m-d, 661f.
I cristalli di 11 furono utilizzati per la determinazione della struttura molecolare mediante diffrazione di raggi X. Inoltre, per idrolisi di una soluzione di 11 in CDCl3
fu possibile identificare HCO2(CH2)2Cl [1H-NMR(CDCl3): δ = 8.12 s, 4.45 t, 3.73 t
ppm].
4.6.2.3. Reazione di MoCl5 con CHMe(OEt)2: isolamento e caratterizzazione di
Mo2Cl5(OMe)5, 14, e [MoOCl3(HCO2Me)]2, 4.
In un provettone codato, furono introdotti MoCl5 (0.615 mmol), CH2Cl2 (ca. 7
mL) e CHMe(OEt)2 (0.615 mmol). La miscela fu lasciata in agitazione per ca. 20 ore
sospensione. La soluzione marrone risultante fu stratificata con eptano e mantenuta a -30 °C per 7 giorni. Si ottenne una miscela solida di colore marrone, contenente in larga prevalenza il complesso [MoOCl3(HCO2Me)]2, 4. Resa: 0.068 g (40 %). IR
(cm−1): 2967d, 1643mf (νC=O), 1445d, 1429m, 1369m, 1285f, 1174d, 1044m, 987mf (νMo=O), 963f, 884f, 794m, 730d.
La struttura del composto cristallino 14 fu determinata mediante uno studio di diffrazione ai raggi X. Inoltre, per idrolisi di una soluzione della miscela di 14 e 4 in CDCl3, fu possibile identificare mediante spettroscopia 1H-NMR i composti
HCOOMe e MeOH.33 Rapporto molare HCOOMe : MeOH = 10:1.
4.6.2.4. Reazione di MoCl5 con CMe2(OMe)2: isolamento e caratterizzazione di
[MoOCl3(MeC(O)CH=CMe2)]2, 16a.
In un provettone codato, furono introdotti MoCl5 (0.597 mmol), CH2Cl2 (ca. 7
mL) e CMe2(OMe)2 (0.596 mmol). La miscela fu lasciata in agitazione per ca. 24
ore. La soluzione risultante, di colore arancione-scuro, fu stratificata con eptano e mantenuta a -30 °C per 18 giorni. Fu ottenuto un olio marrone scuro che mostrava il seguente spettro IR (cm−1): 2954m-d, 2844d, 1584f (νC=O), 1519m (νC=C), 1484m-d, 1448m, 1428m, 1361m, 1337m, 1302m, 199d, 1101m-d, 978mf (νMo=O), 893d, 735m-d.
4.7. Reattività di WCl
6con 1,1-dialcossialcani e CH(OMe)
34.7.1. Reazioni condotte in tubo NMR
4.7.1.1. Reazione di WCl6 con 1,1-dialcossialcani.
In un tubo NMR furono introdotti WCl6, CDCl3 (0.70 mL), 1,1-dialcossialcano e
CH2Cl2. Il tubo fu sigillato alla fiamma e la miscela omogeneizzata mediante breve
mescolamento. Il campione fu mantenuto a temperatura ambiente e successivamente analizzato con NMR. Il tubo fu quindi aperto e alla soluzione fu aggiunto un largo eccesso di acqua (ca. 10 mmol). Dopo agitazione, la soluzione risultante fu separata dal solido tramite filtrazione e analizzata mediante NMR e GC/MS. L’identificazione dei prodotti organici fu condotta confrontando gli spettri NMR ottenuti con i dati riportati in letteratura;33 i risultati sono stati raccolti nelle Tabelle 13, 14, 15, 16, 17, 18, specificando i valori di chemical shift dei composti non presenti nella banca dati.33
Tabella 13: Reazione di WCl6 con 1,3-diossolano.
WCl6
(mmol) 1,3-diossolano (mmol) Tempo di reazione
0.492 0.487 12 giorni
Prodotti prima dell’idrolisi a
CH2Cl2, O(CH2Cl)2 (1H: δ = 5.56; 13C: δ = 82.7 ppm), O(CH2CH2Cl)2,
Cl(CH2)2OCH2Cl (1H: δ = 5.58 s, 3.94 t, 3.71 t ppm), WCl5[O(CH2)2OCH2Cl] (12 ; 1
H: δ = 5.84 s, 5.74 t, 4.48 t ; 13C: δ = 80.6, 78.1, 70.5 ppm) Prodotti dopo idrolisi b
CH2Cl2, O(CH2Cl)2 (1H: δ = 5.56 ; 13C: δ = 82.7 ppm), O(CH2CH2Cl)2,
Cl(CH2)2OCH2Cl (1H: δ = 5.58 s, 3.94 t, 3.71 t ; 13C: δ = 83.0, 68.7, 43.6 ppm) a Rapporti molari CH
2Cl2: O(CH2Cl)2: O(CH2CH2Cl)2 : Cl(CH2)2OCH2Cl : 12 = 10 : 3 : 2 : 1 : 3 b Rapporti molari CH
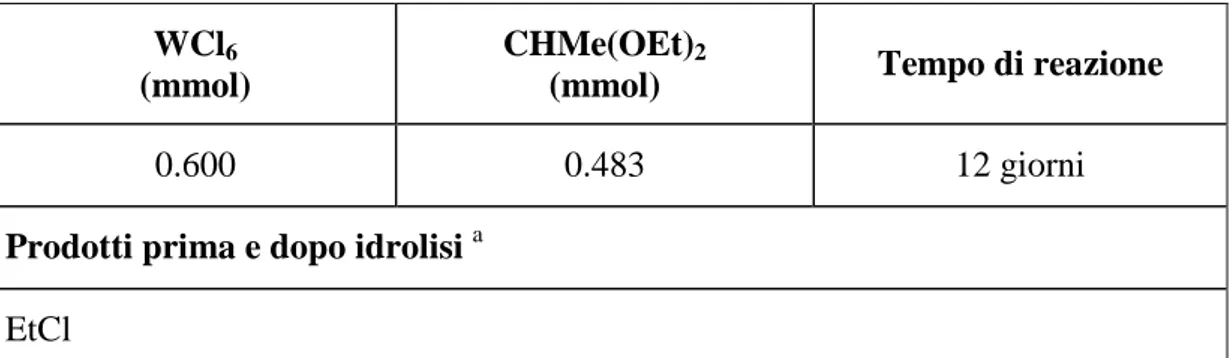
Tabella 14: Reazione di WCl6 con CHMe(OEt)2.
WCl6
(mmol)
CHMe(OEt)2
(mmol) Tempo di reazione
0.600 0.483 12 giorni
Prodotti prima e dopo idrolisi a EtCl
a
Unica specie chiaramente riconoscibile negli spettri NMR.
Tabella 15: Reazione di WCl6 con CH2(OMe)2.
WCl6
(mmol)
CH2(OMe)2
(mmol) Tempo di reazione
0.295 0.339 2 giorni
Prodotti prima dell’idrolisi a
MeCl, WOCl4[O(Me)CH2Cl] (15a ; 1H: δ = 5.71, 3.82 ; 13C: δ = 84.8, 57.7 ppm)
Prodotti dopo idrolisi b MeCl, MeOCH2Cl a Rapporti molari MeCl
: 15a = 1 : 10 b
Tabella 16: Reazione di WCl6 con CH2(OEt)2. WCl6
(mmol)
CH2(OEt)2
(mmol) Tempo di reazione
0.482 0.479 12 giorni
Prodotti prima dell’idrolisi
CH2Cl2, EtCl, ClCH2OCH2CH3, WOCl4[O(Et)CH2Cl] (15b ; 1H: δ = 5.69 s , 4.48 q ,
1.41 t ; 13C: δ = 84.0 , 69.8 , 14.4 ppm) Prodotti dopo idrolisi a
CH2Cl2, EtCl, ClCH2OCH2CH3 a
Rapporti molari CH2Cl2 : EtCl : ClCH2OCH2CH3 = 10 : 6 : 13
Tabella 17: Reazione di WCl6 con CMe2(OMe)2
WCl6
(mmol)
CMe2(OMe)2
(mmol) Tempo di reazione
0.436 0.433 6 giorni
Prodotti dopo idrolisi a
CH2Cl2, MeCl, MeC(O)CH=CMe2 a Rapporti molari CH
Tabella 18: Reazione di WCl6 con CH(OMe)3.
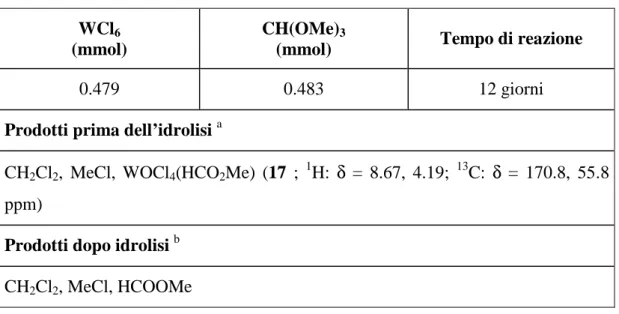
WCl6
(mmol)
CH(OMe)3
(mmol) Tempo di reazione
0.479 0.483 12 giorni
Prodotti prima dell’idrolisi a
CH2Cl2, MeCl, WOCl4(HCO2Me) (17 ; 1H: δ = 8.67, 4.19; 13C: δ = 170.8, 55.8
ppm)
Prodotti dopo idrolisi b CH2Cl2, MeCl, HCOOMe a
Rapporti molari CH2Cl2 : MeCl : 17 = 10 : 16 : 10 b
Rapporti molari CH2Cl2 : MeCl : HCOOMe = 10 : 11 : 8
4.7.1.2. Reazione di WOCl4 con 1,3-diossolano.
In un tubo NMR furono introdotti WOCl4 (0.547 mmol), CDCl3 (0.70 mL) e
1,3-diossolano (0.444 mmol). Il tubo fu sigillato alla fiamma e la miscela omogeneizzata mediante breve mescolamento. Il campione fu mantenuto a temperatura ambiente per 12 giorni e successivamente analizzato con 1H-NMR. Lo spettro rivelò la presenza del complesso WOCl4(C3H6O2), 13 [1H-NMR(CDCl3): δ = 5.86 (s, 2 H, OCH2O),
5.77, 4.50 (t, 4 H, 3JHH = 5.5 Hz, CH2CH2) ; 13C-NMR(CDCl3): δ = 80.6 (OCH2O),
78.2, 70.9 (CH2CH2) ppm]. Il tubo, contenente una soluzione gialla e un solido beige
depositato sul fondo, fu mantenuto a 60 °C per 1 ora e 30 minuti e successivamente a 95 °C per 25 minuti. Il campione fu nuovamente analizzato mediante 1H-NMR e lo spettro risultò identico a quello acquisito prima del riscaldamento. La conseguente idrolisi della soluzione permise di identificare chiaramente 1,3-diossolano.33
4.7.2. Reazioni a scopo preparativo
4.7.2.1. Reazione di WCl6 con 1,3-diossolano.
In un provettone codato, furono introdotti WCl6 (0.514 mmol), CH2Cl2 (ca. 7 mL)
e 1,3-diossolano (0.515 mmol). La miscela fu lasciata in agitazione per ca. 19 ore. La soluzione risultante, di colore arancione-scuro, fu stratificata con eptano e mantenuta a -30 °C per 12 giorni. Tutti i tentativi di isolare prodotti solidi per evaporazione del solvente fallirono a causa della formazione di prodotti oleosi.
4.7.2.2. Reazione di WCl6 con CH2(OR)2 (R = Me, Et): isolamento e
caratterizzazione di WOCl4[O(R)CH2Cl] (R = Me, 15a; R = Et, 15b).
In un provettone codato, furono introdotti WCl6 (0.376 mmol), CH2Cl2 (ca. 7 mL)
e CH2(OR)2 (0.373 mmol). La miscela fu lasciata in agitazione per ca. 6 ore. La
soluzione gialla risultante fu stratificata con eptano e mantenuta a -30 °C per 3 giorni. Il solido verde-chiaro ottenuto venne isolato, seccato sottovuoto e conservato in atmosfera inerte. 15a. Resa: 0.103 g (65 %). IR (cm−1): 2955d, 1614d, 1442d, 1374d, 1152d, 1109d, 1068m, 999d, 970f (νW=O). 1H-NMR(CDCl3): δ = 5.71 (s, 2 H, CH2), 3.82 (s, 3 H, CH3) ppm; 13C-NMR(CDCl3): δ = 84.8 (CH2), 57.7 (CH3) ppm. 15b. Resa: 0.110 g (67 %). IR (cm−1): 2963d, 1441d, 1396d, 1379d, 1351d, 1260m, 1086m-f, 1018f, 980m-f (νW=O), 935m-f, 934m-f, 855f, 793mf. 1H-NMR(CDCl3): δ = 5.69 (s, 2 H, CH2), 4.48 (q, 2 H, CH2), 1.41 (t, 3 H, CH3) ppm; 13C-NMR (CDCl3): δ = 84.0 (CH2), 69.8 (CH2), 14.4 (CH3) ppm.
4.7.2.3. Reazione di WCl6 con CMe2(OMe)2: isolamento e caratterizzazione di
WOCl4[MeC(O)CH=CMe2], 16b.
In un provettone codato, furono introdotti WCl6 (0.504 mmol), CH2Cl2 (ca. 7 mL)
e CMe2(OMe)2 (0.506 mmol). La miscela fu lasciata in agitazione per ca. 24 ore. La
soluzione marrone risultante fu stratificata con eptano e mantenuta a -30 °C per 18 giorni. Il solido nero ottenuto, corrispondente a 16b, venne isolato, seccato sottovuoto e conservato in atmosfera inerte. Resa: 0.160 g (72 %).IR (cm−1): 2941d,
1595f (νC=O), 1506f (νC=C), 1453d, 1437d, 1403m-d, 1378d, 1342m, 1294d, 1029d, 981f (νW=O), 932d, 872f, 847d, 792d, 733d.
Per idrolisi di una soluzione di 16b in CDCl3, fu possibile identificare
4-metil-3-pentenone, MeC(O)CH=C(Me)2, mediante spettroscopia NMR.33
4.7.2.4. Reazione di WCl6 con CH(OMe)3: isolamento e caratterizzazione di
WOCl4(HCO2Me), 17.
In un provettone codato, furono introdotti WCl6 (0.454 mmol), CH2Cl2 (ca. 7 mL) e
CH(OMe)3 (0.456 mmol). La miscela fu lasciata in agitazione per ca. 18 ore. La
soluzione arancione risultante fu stratificata con eptano e mantenuta a -30 °C per 23 giorni. Il solido marrone ottenuto venne isolato, seccato sottovuoto e conservato in atmosfera inerte. Resa: 0.137 g (75 %). IR (cm−1): 2963d, 1652m (νC=O), 1431m-d, 1417m-d, 1371d, 1289m, 1262m-d, 1148d, 1056f (νW=O), 986m, 886mf, 808f, 675m.
1
H-NMR(CDCl3): δ = 8.67 (CH), 4.19 (CH3) ppm; 13C-NMR(CDCl3): δ = 170.8