1
FACOLTA’ DI FARMACIA
CORSO DI LAUREA SPECIALISTICA IN FARMACIA
TESI DI LAUREA
Studio del ruolo di PGC1α nel diabete
RELATORE:
Prof. Antonio Lucacchini
CORRELATORE:
Prof. Gino Giannaccini
CANDIDATA Ferretti Ilaria
3
AI miei genitori che mi hanno accompagnato lungo questo percorso , senza i quali oggi non avrei potuto esser qui: a mio padre che con il suo motto “Studia bimba che la testa ce l’hai” mi ha incoraggiato semore e comunque a credere nelle mie capacità e a pensare positivo; a mia madre che mi insegna ogni giorno a credere che c’è sempre una soluzione a tutto e che ha tanto atteso questo giorno per veder avverare il sogno che lei non è riuscita a realizzare. A Franco, amico e maestro di vita che avrei voluto tanto veder qui oggi, che con la sua saggezza mi ha trasmesso il piacere per lo studio e mi ha insegnato
4
E’ una follia odiare tutte le rose perché una spina ti ha punto, abbandonare tutti i sogni perché uno di loro non si è realizzato, rinunciare a tutti i tentativi perché uno è fallito. E’ una follia condannare tutte le amicizie perché una ti ha tradito, non credere in nessun amore solo perché uno di loro è stato infedele, buttare via tutte le possibilità di essere felici solo perché qualcosa non è andato per il verso giusto. Ci sarà sempre un’altra opportunità, un’altra amicizia, un altro amore, una nuova forza. Per ogni fine c’è un nuovo inizio.
6
STUDIO DEL RUOLO DEL COATTIVATORE 1 DEL
PROLIFERATORE γ MITOCONDRIALE PEROSSOSIMIALE (PGC1α)
Introduzione
Il diabete di tipo 2 e le sue complicanze, come le malattie cardiovascolari, rappresentano un serio problema di salute e hanno raggiunto proporzioni epidemiche nei paesi industrializzati. Il diabete di tipo 2 (DT2) è una delle principali cause di morbilità e mortalità negli Stati Uniti e in molti altri paesi del mondo. La prevalenza di diabete tipo 2 è raddoppiata, dal 4% al 8%, negli ultimi 40 anni negli Stati Uniti(1). L'aumento di diabete tipo 2 nei paesi in via di sviluppo in Asia, compresa la Cina, è altrettanto allarmante(2,3). Con forniture alimentari prontamente disponibili e notevoli miglioramenti della medicina e delle moderne tecnologie si è verificato nel tempo un cambiamento nella diffusione di diverse patologie; dalle malattie infettive e acute nel passato a quelle legate all’età come diabete, malattie neurodegenerative e alcuni tipi di cancro che crivellano la nostra società contemporanea(4). Fin dagli inizi della ricerca molecolare sull’invecchiamento, il ruolo dei mitocondri è stato subito sospettato come fattore chiave dell’insorgenza delle malattie legate ad esso. Infatti se inizialmente l’attenzione venne destata dai ROS prodotti durante la respirazione mitocondriale,
7
successivamente la ricerca ha identificato molti più aspetti legati ai mitocondri che influenzano positivamente e negativamente le funzioni cellulari. Per esempio è stato dimostrato che la restrizione calorica (CR), regolata dalle sirtuine(5,6) e che include in particolare un aumento della funzione mitocondriale, aumenta la durata della vita in tutte le specie esaminate, inclusi i primati non umani. Questo aspetto è solo un esempio che ci permette di intuire la complessità del processo di invecchiamento e delle malattie legate all’età, specialmente in relazione alla funzione mitocondriale. In particolare, si è iniziato a investigare sul ruolo dell’ NO come mediatore in alcuni aspetti del metabolismo ,quali risposta all’esercizio, obesità, differenziazione delle cellule adipose. NO sembra stimolare la biogenesi mitocondriale in alcune situazioni tramite l’attivazione di proteine quali PPARγ e PGC1α e la frammentazione dei mitocondri, la quale può essere causata da una eccessiva fissione o da una ridotta fusione. NO funziona come un messaggero molecolare in vari sistemi fisiologici e si converte in specie radicaliche tossiche (RNS) che possono danneggiare le cellule tramite un processo conosciuto come stress nitrosativo. NO venne inizialmente identificato come un fattore di rilascio endoteliale che media la vasodilatazione(7). A livello del SNC media importanti funzioni quali il rilassamento nervo mediato durante la digestione(8), innervazione delle arterie cerebrali e del pene(9-11), prevenzione dell’eccitotossicità da parte della S nitrosilazione sui recettori NMDA del glutammato(12,13). Inoltre, le cellule del sistema immunitario devono la loro attività di combattere i patogeni all’abilità tossica dell’ NO(14,15)
. In questa trattazione daremo risalto al ruolo dell’ NO nella biogenesi mitocondriale e quindi alla sua importanza nel mantenimento della funzionalità cellulare, della salute e dell’insorgenza delle patologie legate all’età.
8
Invecchiamento
E’ una complessa ed eterogenea condizione che comprende cambiamenti che si presentano nel tempo: l’incapacità dell’organismo a soddisfare le richieste energetiche, le disfunzioni a livello di PGC1α e PGC1β dovute alla loro riduzione durante la disfunzione dei telomeri sono condizioni
associate all’invecchiamento(16). Una importante teoria
sull’invecchiamento è la “teoria dei radicali liberi” secondo la quale l’incremento della produzione di ROS da parte dei mitocondri e il danno
ossidativo che ne risulta sono fattori determinanti
dell’invecchiamento(17)
. Si pensa che mutazioni a livello del mtDNA abbiano un ruolo centrale nel declino delle funzioni mitocondriali associato all’età. La POLG (polimerasi gamma mitocondriale) dei topi modello è stata di valido aiuto per chiarire l’importanza dei mitocondri nell’invecchiamento(18,19)
. Questa è localizzata nei mitocondri dove replica mtDNA e regola la sua riparazione. I topi con POLG mutata hanno mostrato alopecia, osteoporosi e cardiomiopatie, tutte condizioni associate all’invecchiamento(18,19). Per verificare se l’incremento del
metabolismo mitocondriale, attraverso l’espressione della PGC1α, possa migliorare il fenotipo della POLG dei topi, questi sono stati incrociati con MCK-PGC1α tg mice(20). I topi che esprimono sia POLG che PGC1α mutate hanno aumentato l’attività mitocondriale nel tessuto muscolare e nel cuore, il che porta ad un miglioramento delle funzioni dei detti tessuti, a confronto di quanto avviene nei topi che esprimono solo POLG mutata(21). Questi dati chiariscono che elevate funzioni mitocondriali possono avere effetti benefici sull’invecchiamento, indipendentemente dalle mutazioni del mtDNA. E’ stato dimostrato che l’elevata espressione della PGC1α nel tessuto muscolare nel corso della
9
all’invecchiamento, come la sarcopenia(22)
, determinando così un miglioramento delle funzioni mitocondriali dovuto ad un loro minor decremento e alla riduzione dell’accumulo del danno ossidativo. Nel loro insieme, quindi, gli studi svolti dimostrano che PGC1α può ritardare l’inizio delle condizioni associate all’invecchiamento e contribuire ad attenuare l’impatto del danno ossidativo, quando presente.
Mitocondri
I mitocondri occupano una porzione sostanziale del volume citoplasmatico delle cellule eucariotiche e sono elementi essenziali per l’evoluzione degli animali complessi. Essi sono organelli mobili e plastici con una forma in continuo cambiamento in grado di fondersi l’uno con l’altro e dividersi. Dal momento che i mitocondri si muovono nel citoplasma sembrano essere associati ai microtubuli, i quali ne determinano l’orientamento e la distribuzione nei diversi tipi cellulari. In alcune cellule, come quelle cardiache, restano fermi insaccati in miofibrille adiacenti, e forniscono energia direttamente ad un sito ad alto consumo di ATP. Il mitocondrio effettua la maggior parte delle ossidazioni cellulari e produce la maggior parte dell’ATP. La matrice dei mitocondri contiene una grande varietà di enzimi, inclusi quelli che convertono il piruvato e gli acidi grassi ad acetil-CoA e quelli che ossidano la CO2 mediante il ciclo dell’acido citrico. Da queste reazioni
di ossidazione vengono prodotte grandi quantità di NADH e di FADH2
che forniscono elettroni reattivi con cui si combina l’ossigeno molecolare. Tale combinazione permette la produzione di energia attraverso la catena respiratoria nella matrice della membrana mitocondriale. In particolare, la catena respiratoria pompa ioni H+ fuori dalla matrice per creare un gradiente elettrochimico transmembrana , determinato sia dal potenziale di membrana che dalla differenza di pH.
10
La grande quantità di energia rilasciata durante il passaggio dei protoni, attraverso la membrana interna nella matrice fornisce la base per la produzione di ATP mediante un complesso proteico chiamato ATP sintasi. Inoltre, il gradiente elettrochimico transmembrana viene sfruttato per guidare il trasporto attivo dei metaboliti, attraverso la membrana mitocondriale interna, incluso lo scambio ATP-ADP tra mitocondrio e citoplasma che mantiene il pool di ATP della cellula ad alti livelli. La produzione di ATP è un’attività del mitocondrio comune a tutte le cellule, ma come suggerito da un’indagine proteomica quasi la metà delle proteine mitocondriali sono tessuto specifiche(22). Queste differenze proteiche si traducono nelle diversità che i diversi tipi cellulari mostrano nel numero, nelle dimensioni e nella forma dei mitocondri(23). Il numero dei mitocondri varia da una singola unità nella retina a centinaia negli epatociti.
Biogenesi mitocondriale
I mitocondri si dividono durante la mitosi, fornendo alle cellule figlie un normale numero di essi, oppure la divisione può avvenire al di fuori del ciclo cellulare come nel caso dei mitocondri del muscolo. Questi proliferano durante la biogenesi e in seguito all’esercizio fisico(24)
.
La divisione mitocondriale può essere indotta da numerose sostanze quali le benzodiazepine, inibitori della fosforilazione ossidativa, esteri del forbolo, e flussi di calcio(25). Inoltre l’esposizione dei mammiferi a basse temperature e per prolungati periodi di tempo induce un marcato aumento nella massa mitocondriale negli adipociti bruni; questo fornisce un importante meccanismo per il mantenimento del bilancio energetico dell’organismo e della temperatura interna(26)
. I mitocondri sono tubulari nelle cellule della maggior parte dei tessuti ma la fusione, la fissione e la traslocazione producono cambiamenti dinamici nella morfolgia(23), per
11
cui i mitocondri vengono risaldatati a formare filamenti allungati o una rete. In ogni istante possono essere considerati in uno stato di transizione e alcune prove suggerirebbero che esiste una intricata relazione tra funzione e struttura, tra la morfologia dei mitocondri e la loro attività di produzione di energia.
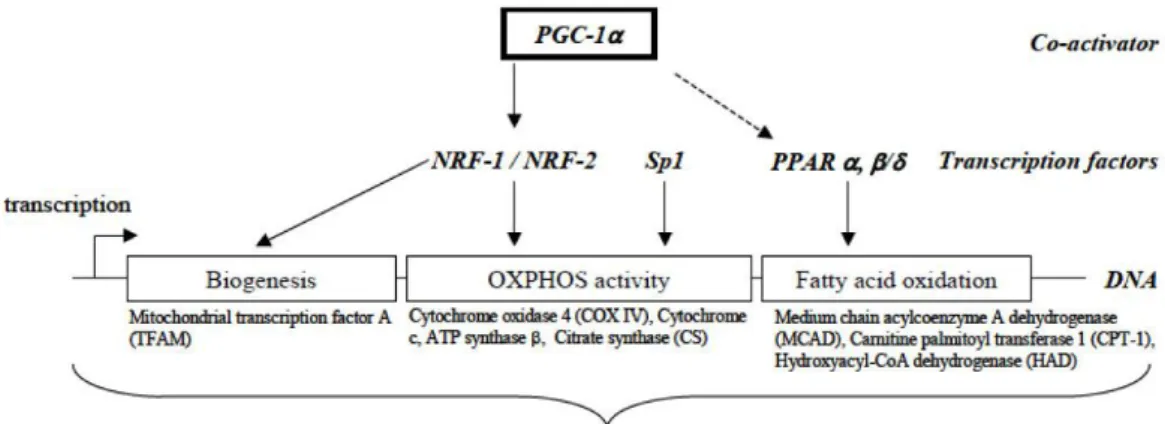
Il termine biogenesi mitocondriale si riferisce alla formazione di nuovi mitocondri nelle cellule, e include la coordinata regolazione tra la trascrizione dei complessi delle proteine respiratorie, che consiste sia nella codificazione nucleare dei componenti sia nella produzione enzimatica, e la replicazione del mtDNA(27). Dalla trascrizione del mtDNA il mitocondrio ottiene 13 molecole di mRNA che codificano per i componenti proteici della catena respiratoria. Alcune agiscono singolarmente, altre si combinano con proteine codificate dal nucleo per formare complessi proteici come COX o NADH deidrogenasi. La trascrizione e la replicazione richiedono l’importazione di prodotti del genoma nucleare, che agiscono come polimerasi o fattori di trascrizione. Un mitocondrio possiede copie multiple di una piccola molecola di DNA circolare (mtDNA) che può replicare in modo indipendente dal DNA nucleare, ed è inoltre presente in un alto numero di copie. La sua replicazione richiede la presenza della DNA polimerasi γ, delle proteine che legano il DNA a singola elica, Tfam, che inizia la trascrizione e genera primers per permettere la replicazione del DNA e una endonucleasi per la processazione dell’RNA mitocondriale (RNase MRP)(28). Poichè i mitocondri sono fondamentali per la salute delle cellule, elevati livelli della biogenesi mitocondriale sono indicativi di una intatta funzionalità metabolica e bioenergetica della cellula e del suo benessere.
12
La biogenesi mitocondriale è un meccanismo che richiede l’intervento di numerosi fattori, il primo dei quali è l’attivazione del peroxisome proliferator-actived receptor γ coactivator 1α (PGC-1α), fattore di trascrizione che a sua volta induce l’attivazione del nuclear respiratory factor (NRF-1) e poi il mitochondrial transcription factor A(Tfam) che promuove la replicazione dell’mtDNA e quindi l’aumento della massa mitocondriale.
Disordini metabolici, resistenza insulinica e funzionalità mitocondriale Il metabolismo e il consumo energetico sono importanti processi coinvolti nel globale mantenimento della salute, come evidenziato dalla relazione tra obesità (che deriva da un ridotto consumo energetico in rapporto all’energia assorbita) e un ampio spettro di disturbi legati all’età come: diabete, cancro, danni cardiaci e vascolari e neurodegenerazione. Al contrario, le diete di restrizione calorica, l’ esercizio fisico(entrambi aumentano la funzione mitocondriale), il metabolismo dei lipidi e del glucosio e la spesa energetica sono fattori associati all’aumento della longevità e ad una riduzione dei rischi dei disturbi legati all’età. Tutti i disordini metabolici sono indotti da un’alterazione a carico di tessuti specifici: sangue, tessuto adiposo, fegato e muscoli. Un eccesso di intake calorico, l’iperinsulinemia, il colesterolo, gli acidi grassi liberi(FFA) e i trigliceridi(TG), le citochine proinfiammatorie, l’accumulo di grasso e l’infiammazione cronica sono infatti i principali contribuenti alla sindrome metabolica che induce lo sviluppo di malattie cardiovascolari. Nel 1988 Gerald M. Reaven(29) ha definito sindrome X la manifestazione simultanea di insulino-resistenza, iperinsulinemia, stati pre-diabetici o diabete mellito di tipo 2 conclamato, dislipidemia, obesità centrale, iperuricemia (una concentrazione alta di acido urico nel sangue), e ipertensione arteriosa, considerandola una condizione clinica che
13
precede lo sviluppo di complicanze vascolari. Ad essa si associa un'aumentata incidenza di cardiopatia ischemica, disfunzioni del ventricolo sinistro e scompenso cardiaco. Tutto ciò comporta un forte incremento del rischio di mortalità per cause cardiovascolari. Tale sindrome era conosciuta anche con il nome di Sindrome di Reaven, in suo onore.
Le stesse patologie cardiovascolari inducono, a loro volta, insulino-resistenza e aumentano la probabilità che si sviluppi nel tempo un diabete mellito di tipo 2. La sindrome X è stata definita anche sindrome da insulino-resistenza e successivamente sindrome metabolica cardiovascolare. Attualmente la sindrome è stata rinominata plurimetabolica e comprende l'associazione di insulino-resistenza, iperinsulinemia, obesità centrale, intolleranza glucidica o diabete mellito di tipo 2, iperuricemia, dislipidemia e ipertensione arteriosa. Su quasi tutti i testi è ancora comune trovare la dicitura di sindrome metabolica, mentre in Australia tale sindrome è conosciuta con il nome di CHAOS.
14
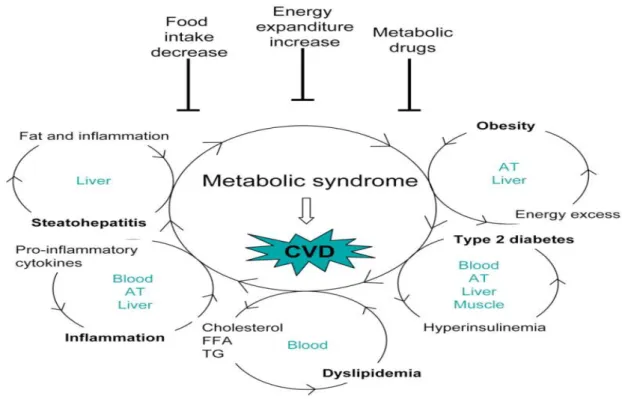
Figura 1 Sindrome metabolica. Schematizzazione dei fattori contribuenti alla sindrome metabolica che induce patologie cardiovascolari (CVD): obesità, diabete di tipo 2, infiammazione e steatosi epatica. Ogni disordine metabolico deriva da un’alterazione, come un eccesso di intake calorico, l’iperinsulinemia, acidi grassi liberi e trigliceridi e citochine proinfiammatorie. Gli organi o tessuti coinvolti nella sindrome metabolica sono sangue, tessuto adiposo, il fegato e i muscoli. Restrizione calorica, dispendio energetico e/o farmaci che agiscono a livello metabolico costituiscono una strategia terapeutica per contrastare la sindrome metabolica.
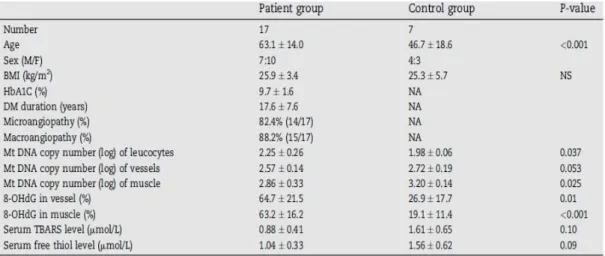
Tra i meccanismi implicati nella patogenesi dell’obesità, della resistenza insulinica e della loro progressione verso il diabete di tipo 2 è stata ipotizzata l’esistenza di un danno della funzione mitocondriale(30)
. Le principali alterazioni che insorgono nei soggetti diabetici(31) sono la riduzione del trasporto del glucosio, della sua fosforilazione e della capacità della sintesi del glucosio. A queste si aggiungono anche un aumento del metabolismo degli acidi grassi, dell’accumulo dei trigliceridi e una ridotta ossidazione dei lipidi(32,33). Inoltre l’insulino resistenza è associata a un ridotto contenuto mitocondriale e a una ridotta attività della e-NOS, che si presume essere il primo fattore implicato
15
nella catena di signalling. Come vedremo in seguito, studi funzionali e quantitativi hanno dimostrato che il contenuto di mtDNA nel tessuto muscolare di soggetti obesi insulino resistenti(34), e in pazienti con diabete di tipo 2(35), è minore e questo può contribuire all’alterazione della trasduzione del segnale, del trasporto e dell’ossidazione dei substrati(33).
Soggetti insulino resistenti hanno meno mitocondri muscolari a causa, forse, della diminuzione dell’espressione di geni nucleari che regolano la biogenesi mitocondriale, come PGC-1α(36) e PGC-1β(37). Studi che sfruttano microarray supportano l’idea che sia negli obesi che nei diabetici PGC-1α e PGC-1β sono down-regolati(38). Inoltre studi recenti hanno dimostrato che anche il ridotto contenuto mitocondriale nel tessuto adiposo può contribuire all’insorgenza del diabete di tipo 2(39)
. La presenza di danno mitocondriale nella progenie insulino resistente di pazienti affetti da diabete di tipo 2 ne ha suggerito un ruolo primario nella patogenesi: i cambiamenti sono infatti associati alla riduzione dell’attività ossidativa mitocondriale e della sintesi dell’ATP. Questo fa nascere l’ipotesi che l’insulino resistenza sia originata da difetti nell’ossidazione degli acidi grassi a livello mitocondriale, che a loro volta conducono a incrementi dei propri metaboliti come acetil-CoA e diacilglicerolo, che influenzano negativamente le vie del segnale insulinico.
16
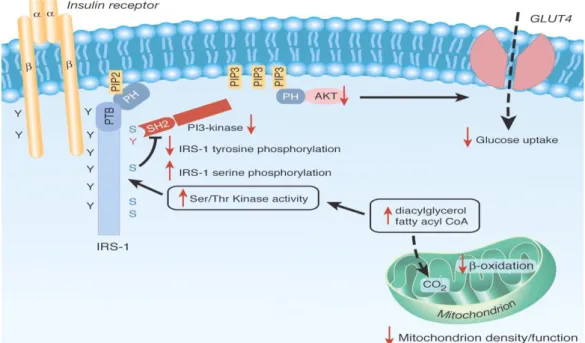
Figura 2. Potenziale meccanismo con cui la disfunzione mitocondriale induce insulino resistenza nei muscoli scheletrici. Una diminuzione dell’ossidazione mitocondriale degli acidi grassi, causata da una disfunzione dei mitocondri o da una diminuzione del loro contenuto nella cellula produce l’aumento dei livelli di acetilCoA e diacilglicerolo che attivano la protein chinasi C che a sua volta innesca una cascata di fosforilazioni su residui di compresi quelli del substrato del recettore dell’insulina IRS-1. Questo evento inibisce la fosforilazione dell’IRS1 sui residui di tirosina da parte del recettore insulinico (che in seguito al legame con l’insulina acquisisce attività tirosin chinasica) che a sua volta blocca l’attività della fosfatidil-inositolo 3-chinasi sulla membrana. Questo determina una soppressione del trasporto di glucosio in risposta all’insulina.
OSSIDO NITRICO
NO è un gas prodotto in diverse cellule che ha un importante ruolo, sia fisiologico, a livello della muscolatura liscia dove media la vasodilatazione, sia nel SNC, dove agisce come neurotrasmettitore, infine nel sistema immunitario e nella cicatrizzazione delle ferite. Ha
anche un ruolo fisiopatologico nello stress nitrosativo,
nell’eccitotossicità e nella modificazione delle proteine. A causa della sua breve emivita l’NO è in grado di agire solo in prossimità delle cellule
17
che lo producono per cui la sua attività è considerata autocrina e paracrina. Essendo una molecola neutra di piccole dimensioni NO diffonde liberamente nelle membrane.
Sintesi dell’ossido nitrico e regolazione dell’attività enzimatica
NO viene sintetizzato a partire dalla L arginina da uno specifico enzima denominato sintasi dell’ossido nitrico (NOS).
Sono state identificate 3 isoforme sintetizzate da altrettanti geni: e-NOS (endoteliale), n-NOS (neuronale), i-NOS (inducibile). Le diverse isoforme mostrano specifiche distribuzioni tissutali e specifiche attività che riflettono i loro ruoli fisiologici. L’e-NOS è una isoforma costitutiva, attiva nel tessuto endoteliale dei vasi, dove produce NO a ritmo costante per lunghi periodi, il quale determina la vasodilatazione e il rilasciamento del tessuto muscolare liscio. L’i-NOS è attiva principalmente nelle cellule immunitarie e gliali e produce NO in seguito al riconoscimento dell’agente patogeno e al rilascio di citochine. Il suo principale ruolo è quello di mediare la morte cellulare in risposta ai patogeni, generando NO ad alte concentrazioni per periodi brevi, fino ai livelli tossici. L’n-NOS è una isoforma costitutiva attiva nei neuroni centrali e periferici ,che produce basse concentrazioni di NO per brevi periodi, il quale a questo livello agisce come un importante neurotrasmettitore nella comunicazione cellula-cellula e nella plasticità neuronale. Il membro della famiglia delle NOS riconosciuto più recentemente è la mtNOS, che si trova nella membrana interna dei mitocondri dove ha un ruolo nella regolazione bioenergetica e nel buffering del calcio. Le isoforme della NOS catalizzano la seguente reazione:
18
I geni che codificano per le rispettive isoforme della NOS presentano una struttura simile costituita da due domini, con un dominio ossigenasico e uno reduttasico. Il primo è il dominio N terminale e contiene siti di legame per l’eme, la tetrabioperidina (BH4) E L-arginina.
Il dominio ossigenasico è legato al dominio reduttasico C terminale attraverso un sito di riconoscimento della calmodulina (CaM) e porta siti di legame per flavinadenindinucleotide (FAD), flavinmononucleotide (FMN) e nicotinamin-deadenindinucleotide fosfato (NADPH). Inoltre sembra che tutte le isoforme presentino un centro di legame dello zinco che stabilizzerebbe il dimero della NOS, in quanto il monomero è inattivo.
La dimerizzazione avviene tra i domini ossigenasici. I cofattori flavinici accettano gli elettroni dal NADPH cedendoli all’eme, mentre CaM regola l’attività enzimatica delle 3 isoforme le quali differiscono per la dipendenza dal calcio. N-NOS e e-NOS necessitano di una maggiore quantità di calcio rispetto a quella della i-NOS. ll legame del calcio con la calmodulina determina la velocità di trasferimento degli elettroni dal NADPH alle flavine del dominio reduttasico e da questo all’eme. Un altro tipo di regolazione della NOS è la fosforilazione. L’ attività della n-NOS diminuisce con la fosforilazione; al contrario l’attività della e-n-NOS aumenta con la fosforilazione.
NO e biogenesi mitocondriale
Studi recenti hanno stabilito che aumenti cronici piccoli o moderati di NO stimolano la biogenesi mitocondriale(27). Per esempio il trattamento di colture di precursori di adipociti bianchi e bruni del topo con donatori di NO incrementa il contenuto di mtDNA, la fluorescenza di mitotracker (indicatore del potenziale della membrana mitocondriale), l’espressione della subunità IV della citrocomo C ossidasi della catena respiratoria
19
mitocondriale, il citocromo C e la massa dei mitocondri(39). I cambiamenti osservati sono dovuti all’aumento dell’espressione della PGC-1α, il principale regolatore della biogenesi mitocondriale, NRF-1, NRF-2 e Tfam(40) e all’effetto del secondo messaggero su cui NO agisce, cioè Cgmp. Inoltre le cellule HeLa transfettate con e-NOS mostrano lo stesso incremento nella biogenesi mitocondriale, mentre l’inibizione della e-NOS abolisce l’effetto. Per di più, la carenza di e-NOS esibita dai topi riduceva i livelli di mtDNA, COX IV e citocromo C nel cervello, nel cuore e nel fegato e quindi della biogenesi mitocondriale nonostante il fatto che i topi esprimessero le altre isoforme come n-NOS e i-NOS(39). Infine, la biogenesi mitocondriale NO dipendente incrementava la produzione di ATP e il consumo energetico(41).
Poichè l’attività mitocondriale gioca un ruolo centrale in importanti processi, quali transizioni delle fibre muscolari scheletriche con metabolismo da glicolitico a quello ossidativo, rigenerazione dei muscoli cardiaco e scheletrico, NO è indirettamente coinvolto nell’aumento del trasporto di glucosio nel muscolo scheletrico(42)
. I meccanismi responsabili di questo evento sono ancora in parte sconosciuti ma si pensa possano coinvolgere l’interazione con altri percorsi di segnalazione o avere effetti sul trasporto vescicolare di GLUT4, processi mediati da AMPK. L’NO è un mediatore che si trova a valle della via di segnalazione di AMPK.
NO ed esercizio
L’esercizio cronico include una serie di adattamenti che coinvolgono in particolare il muscolo scheletrico ma anche cuore, sistema vascolare e metabolico: durante l’attività fisica si verifica l’aumento dell’espressione della e-NOS e quindi di NO che va a influenzare l’attività dei mitocondri, sia positivamente che negativamente. Di conseguenza, è
20
facile ipotizzare che un difetto nel metabolismo dell’ NO causi un’alterazione della funzionalità mitocondriale(43)
; la composizione proteica mitocondriale può cambiare in risposta all’esercizio cronico(44,45). Il contenuto mitocondriale è stato valutato, in vitro, mediante il cambiamento dell’attività di un enzima marcatore, come la citrato sintetasi, o di una proteina simile al citocromo C(46). Partendo dal fatto che il comportamento proteico riflette quello del mitocondrio, la misura delle proteine è stata utilizzata per determinare il contenuto mitocondriale. Sono stati misurati marcatori proteici in risposta ad uno stimolo fisico di allenamento e si è visto che occorrono diverse settimane per ottenere un nuovo e maggiore contenuto mitocondriale; inoltre, si è verificato che gli adattamenti avvengono solo a livello delle fibre muscolari reclutate nella contrazione. I nuovi trascritti mitocondriali si traducono in variazioni del mtDNA(47). Il principale fattore di trascrizione per mtDNA è Tfam e i suoi livelli sono correlati dalla presenza, più o meno sostanziosa, dello stesso mtDNA. Inoltre, nel muscolo scheletrico l’espressione di Tfam è regolata dall’attività contrattile quindi se ne deduce che l’induzione mitocondriale è indotta da una stimolazione muscolare cronica. Inoltre, visto il legame tra NO e PGC1α e considerando il fatto che i livelli di quest’ultimo aumentano nel muscolo in seguito all’esercizio, è ragionevole pensare che NO possa mediare questa risposta fisiologica. Per esaminare questa possibilità, da un recente studio che ha misurato l’effetto dell’inibizione farmacologica di NOS da parte della L-NAME (NG-nitro-L-arginine methyl ester), è emerso che questa non ha attenuato l’up regulation di PGC1α indotta dall’esercizio. Comunque, sarebbero necessari ulteriori studi che utilizzassero modelli NOS knockout per confermare il fatto che NO non abbia un ruolo sostanziale nell’aumento dei livelli di PGC1α come risposta fisiologica indotta dall’esercizio.
21
NO e tessuto adiposo
Il profilo della composizione del grasso corporeo e la differenziazione delle cellule adipose sono aspetti importanti del metabolismo e del consumo energetico dei mammiferi. La maggior parte del grasso corporeo negli adulti è costituita dal grasso bianco, che è localizzato prevalentemente nell’addome e nelle aree sottocutanee, ed ha funzione di deposito delle riserve di energia. L’altro, meno comune è il tessuto adiposo bruno (BAT), che è confinato in piccole cavità ed è coinvolto nella produzione di calore soprattutto in risposta alle fredde temperature. Al contrario del grasso bianco, BAT è un tessuto altamente vascolarizzato contenente un’abbondante quantità di mitocondri che operano nella produzione di calore per il mantenimento della temperatura corporea, e generano calore tramite il disaccoppiamento della respirazione mitocondriale mediato dalla UCP1(48). Mentre la respirazione accoppiata si riferisce alla respirazione alla produzione di ATP, il disaccoppiamento della respirazione si riferisce alla respirazione non accoppiata alla produzione di ATP e il calore viene generato dalla dissipazione del gradiente protonico mitocondriale. Nei mammiferi, in condizioni standard l’80% della respirazione mitocondriale è accoppiata e il 20% è disaccoppiata. Il tessuto adiposo svolge un ruolo attivo nei meccanismi di adattamento all’esercizio fisico che determina un aumento della produzione di NO. Questo incremento ha un’azione positiva nella stimolazione della lipolisi a livello del tessuto adiposo e nella riduzione del grasso sottocutaneo addominale(tessuto adiposo bianco)(49),e contemporaneamente aumenta la quantità di BAT e la termogenesi. Ciò suggerisce un potenziale ruolo indiretto dell' NO nella differenziazione delle cellule adipose brune e risulta molto importante per meglio comprendere il ruolo dell’ NO nella mitocondriogenesi.
22
L’azione positiva dell’esercizio fisico sui tessuti bersaglio lo suggerisce come una strategia terapeutica da seguire nella prevenzione di diabete di tipo 2 e obesità, benché i cambiamenti con cui il tessuto adiposo è influenzato dagli adattamenti fisiologici non siano ancora chiari.
Sulla base della relazione tra NO e BAT sono state fatte recenti osservazioni per approfondire il fatto che la e-nos dei topi knock-out comprometta la biogenesi mitocondriale e che questi presentino le caratteristiche tipiche della sindrome metabolica come obesità, dislipidemie, insulino-resistenza e ipertensione. Inoltre, se sottoposti a dieta ipocalorica mostrano un conseguente calo ponderale ma non un aumento della biogenesi mitocondriale; al contrario, nei topi wild type con lo stesso trattamento migliora anche la sensibilità insulinica(50).
La ridotta espressione della e-NOS nel tessuto adiposo sia bianco che bruno di topi diabetici e obesi, dimostra che può esistere un legame tra la disfunzione mitocondriale e l’insorgenza del diabete di tipo 2. Inoltre, l’inattivazione di e-NOS riduce l’attività e la massa dei mitocondri anche nei tessuti che mostrano un livello basale di n-NOS, come nel cervello, nel fegato, nei muscoli scheletrici e nel cuore determinando così un ridotto consumo basale, e a riposo, di ATP sia nei tessuti con metabolismo ossidativo che glicolitico; quindi l’effetto è generalizzato. Per concludere, i topi e-NOS rappresentano un modello valido per studiare fenomeni metabolici e adattativi in presenza delle tipiche alterazioni della sindrome metabolica e delle disfunzioni mitocondriali.
NO e restrizione calorica
La riduzione dell’introito calorico è legata all’allungamento della durata della vita e alla prevenzione delle malattie come diabete e patologie neurodegenerative in tutte le specie, inclusi primati non umani.
23
Le sirtuine, una classe 3 delle istoni deacetilasi aiutano a mediare gli effetti positivi di CR. Le sirtuine localizzate nei mitocondri e partecipano alla coordinazione della regolazione metabolica. In particolare SIRT 3 aumenta l’ossidazione degli acidi grassi in risposta al digiuno, mentre SIRT 1 (nucleare) deacetila e attiva la PGC1α. L’attivazione di SIRT1 e PGC1α da parte di CR è stata messa in relazione all’ aumento della biogenesi mitocondriale. Il ruolo dell’ NO nella regolazione della risposta cellulare è da chiarire, ma è stato dimostrato che CR induce l’espressione di e-NOS, la formazione dei complessi, l’espressione della SIRT 1 e stimola la biogenesi mitocondriale. Di conseguenza si verifica anche l’aumento delle altre proteine mitocondriali come NRF1, Tfam, mitofusina 1 e 2, COX IV e citocromo C.
Il fatto che CR induca l’espressione di e-NOS è curioso perché l’e-NOS null mice non induce lo stesso effetto ma al contrario riduce la longevità, compatibilmente all’incapacità di generare nuove proteine mitocondriali e di attivare i fattori di risposta allo stress come le sirtuine.
NO, obesità, diabete e infiammazione
L’obesità costituisce una minaccia per la salute dell’uomo(51)
poiché è
correlata a numerose patologie gravi come patologie CV,
neurodegenerazione, diabete e cancro. E’ diventato sempre più chiaro che l’obesità e il diabete sono due patologie strettamente correlate in cui l’infiammazione rappresenta una figura chiave(52)
. Uno dei primi studi a riguardo, ha riferito come principale promotore dell’infiammazione il TNFα che veniva sovraespressa nei topi obesi, i quali mostravano una ridotta sensibilità insulinica. Oltre al ruolo della citochina pro infiammatoria, è stato studiato il potenziale ruolo dello stress del reticolo
endoplasmatico (ER), indotto dall’NO(53), nel meccanismo
24
della qualità delle proteine nelle cellule, e l’accumulo di proteine unfolded può innescare una cascata di percorsi di trasduzione del segnale così come l’unfolded protein response (UPR) (51)
. Nello specifico, l’ NO S nitrosylated protein-disulphide isomerase nel cervello può causare lo stress dell’ ER e innescare l’UPR(54)
. L’ i-NOS invece, che media l’infiammazione è implicata nell’insulino resistenza, complicazione comune dell’obesità che accompagna il diabete. La sua aumentata espressione è associata all’obesità nella sensibilità insulinica nei tessuti e i livelli della NO S nitrosylated protein-disulphide isomerase è elevata negli obesi e nei diabetici(55). Infine, la disgregazione dell’i-NOS può proteggere dall’insulino resistenza indotta dall’obesità(56)
.
NO e malattie neurodegenerative
Nelle cellule sane, i cicli di fissione e fusione si ripetono e sono regolati da una serie di GTPasi. DPR1 innesca la fissione e MFN1, MFN2 e OPA1 regolano la fusione. Mutazioni a carico delle GTPasi determinano un danno della fusione mitocondriale provocando così l’accumulo dei mitocondri divisi e conseguentemente patologie neurodegenerative. Oltre a questo, studi recenti hanno iniziato a indagare per comprendere se anche la rottura dell’equilibrio fusione-fissione ha un ruolo in merito. Nel sistema nervoso l’ NO è un importante neurotrasmettitore ed è prodotto dai neuroni e dalle cellule gliali. Nelle più comuni condizioni neurodegenerative come stroke, alzheimer, parkinson e sclerosi laterale amiotrofica NO si accumula formando RNS altamente neurotossici come ONOO- che può modificare covalentemente residui di Cys e Tyr nelle proteine e alterare la loro struttura e la loro funzione. Numerosi studi sostengono che questa forma di stress nitrosativo abbia un ruolo causale in queste patologie. I meccanismi che spiegherebbero l’aumento di RNS includono l’infiammazione, l’attivazione delle cellule del microglia e
25
l’attivazione dell’i-NOS mediata dalle citochine. Un altro probabile meccanismo è l’accumulo di amminoacidi eccitatori nelle sinapsi come il glutammato, a causa di un deficit del suo reuptake da parte dei trasportatori degli astrociti. La sovrastimolazione dei recettori neuronali del glutammato provoca l’attivazione della NOS mediata dal calcio e induce l’aumento di NO/ ONOO
-. Il calcio in eccesso che affluisce penetra nel mitocondrio, determinando perdita del suo potenziale di membrana e diminuzione della produzione di ATP per disaccoppiamento della fosforilazione ossidativa con la sintesi di ATP. Questo fa si che le pompe di membrana ATP dipendenti responsabili del mantenimento della depolarizzazione, smettano di funzionare e in una sorta di circolo vizioso l’ingresso di calcio aumenta e la produzione di NO, che inibisce la catena di trasporto mitocondriale viene stimolata. E’ stato dimostrato che lo stress nitrosativo innesca una frammentazione persistente nei mitocondri (come risultato dell’attivazione della fissione o dell’inattivazione della fusione) prima nelle cellule neuronali morte, nei neuroni isolati in vitro, e poi in un modello sperimentale di danno ischemico in vivo(57). Lo stress nitrosativo altera l’organizzazione dei microtubuli “deragliando” proteine motorie e provocando, come risultato, un cambiamento morfologico dei mitocondri, l’arresto del trasporto assonale e un danno alle sinapsi causato della deplezione dei mitocondri dai terminali nervosi.
Un danno delle sinapsi, degli organelli e del trasporto vescicolare è stato attestato nell’Alzheimer e in altre patologie neurodegenerative. E’ stato visto che il peptide beta amiloide, principale componente delle placche senili del cervello affetto da AD, induce la frammentazione mitocondriale dei neuroni corticali(58). In generale nelle patologie neurodegenerative la frammentazione mitocondriale persistente è
26
accompagnata da un compromesso bioenergetico, in quanto induce un ulteriore danno mitocondriale dovuto all’aumento dei ROS e contemporaneamente la riduzione dei livelli di ATP(57). Gli spazzini dei radicali liberi come il glutatione ridotto prevengono la frammentazione NO mediata e la morte cellulare. Mediante immagini timelaps è stato visto che la frammentazione mitocondriale NO mediata può essere reversibile nei neuroni che esprimono geni survival come BC1-XL(59), e che la frammentazione mitocondriale a breve termine rappresenta solo una risposta allo stress a rimuovere gli organelli danneggiati da parte degli autofagosomi, incrementando perciò la sopravvivenza della cellula. Al contrario quando BAX, membro della famiglia delle pro-apoptotic BCL2, si sposta nei siti della fissione i mitocondri non vengono rifusi e si verifica la morte della cellula(60) a causa dell’attivazione di percorsi di trasduzione del segnale di tipo downstream come la ricollocazione nei mitocondri. Uno dei meccanismi che induce la frammentazione mitocondriale NO mediata nelle malattie neurodegenerative e in particolare nell’ Alzheimer sembra essere dovuto alla nitrosilazione della tirosina del recettore nucleare PPARγ di cui viene inibita la traslocazione nel nucleo e di conseguenza l’espressione dell’ NRF1 e delle proteine mitocondriali. Aspetto interessante è che PPARγ è un attivatore di PGC1α, che a sua volta è un fattore di regolazione della MFN2. L’ inibizione del PPARγ NO mediata determina quindi la ridotta espressione della MFN2(61,62). In particolare, recentemente è stato svolto uno studio sul ruolo dell’ NO nell’ AD. Sulla base della scoperta che il peptide beta amiloide induce la frammentazione mitocondriale, lo studio sostiene che la S nitrosilazione della DPR1 sulla Cys 644 porta alla sua dimerizzazione e all’attivazione enzimatica, meccanismo principale della fissione mitocondriale NO indotta, del compromesso bioenergetico e del danno neuronale in AD, ma non in PD. Tale studio rimane però
27
controverso e in disaccordo sull’attuale letteratura riguardo alla DPR1. Per prima cosa gli autori non hanno chiaramente determinato l’attività GTPasica della DPR1 ma hanno semplicemente equiparato la densità ottica in un singolo punto del tempo con l’attività dell’enzima tramite il saggio Malachite Green. Inoltre hanno riferito un valore di assorbanza inaspettato per la gtp defective mutant DPR1K38A. Sulla base del saggio gli autori hanno proposto un duplice incremento dell’attività GTPasica della DPR1 rispetto alla S nitrosilazione della Cys 644. Secondo, gli autori sostengono che la formazione del dimero di DPR1 sia un riflesso dell’aumentata attività enzimatica. Non ci sono altri studi che hanno suggerito che l’attività enzimatica sia dipendente dalla dimerizzazione. Invece esperimenti cross-linking e filtrazione su gel hanno dimostrato che DPR1 in condizioni fisiologiche è un tetramero che si assembla in un ordine oligmerico superiore di anelli e spirali, che stimola l’idrolisi del GTP. Per di più l’ossidazione non sembra mediare l’oligomerizzazione.
Al contrario, gli agenti riducenti sembrano richiesti per
l’oligomerizzazione di proteine come dinamina e OPA1. Terzo, gli autori sostengono che tutti i campioni AD ma nessuno dei PD mostrano un incremento dei livelli di SNO-DPR1. Questo ha destato curiosità in quanto sappiamo che lo stress nitrosativo è coinvolto anche nel Parkinson. Per risolvere la controversia e per conciliare le relazioni contrastanti è stata studiata la relazione tra NO e DPR1 nell’ Alzheimer. Contrariamente al resoconto di Cho è stato osservato che la S nitrosilazione della DPR1 non aumenta né l’attività GTPasica della DPR1 né la dimerizzazione. Inoltre è stato dimostrato che DPR1 nel cervello (in vivo) è un tetramero capace di formare in vitro oligomeri come spirali o anelli. I risultati negativi non sono causati dalla overossidazione della proteina, quindi i residui di Cys non sono ostruiti dall’ossidazione e sono disponibili per la S nitrosilazione da parte dell’
28
NO. L’aspetto importante è che non sono state trovate differenze significative nei livelli di SNO-DPR nel cervello di individui sani, affetti da PD e AD post mortem. Come ultimo aspetto OPA1 è stata S nitrosilata nello stesso gruppo di campioni, a dimostrazione del fatto che la S nitrosilazione non è specifica per DPR1. Questi risultati, nell’insieme, forniscono prove interessanti a dimostrazione del fatto che il meccanismo alla base della frammentazione mitocondriale indotta dallo stress nitrosativo nell’ AD non sia la S nitrosilazione della DPR1.
PGC1α: CARATTERISTICHE FUNZIONALI E STRUTTURALI
PGC1α è un coattivatore trascrizionale e un induttore centrale della biogenesi mitocondriale nelle cellule. Questa proteina promuove la trascrizione e l’espressione di numerosi geni, tra i quali quelli responsabili della regolazione della biogenesi mitocondriale e dell’ossidazione dei grassi. La famiglia di tali coattivatori è composta da tre membri: PGC1α, PGC1β e PRC che interagiscono con i fattori di trascrizione e con i recettori nucleari per esercitare le loro funzioni biologiche. PGC1α è il membro più conosciuto e più studiato ed è codificato dal gene PPARGC1A. E’ espresso nei tessuti ad elevato consumo energetico cioè muscolo striato cardiaco e scheletrico, tessuto adiposo bruno, fegato e cervello. Le sue funzioni sono la regolazione positiva della biogenesi e della respirazione mitocondriale, della termogenesi adattativa e della gluconeogenesi. La sua espressione è indotta da segnali fisiologici come esercizio, freddo e digiuno. Nel tessuto adiposo PGC1α regola la termogenesi stimolando l’ espressione della UCP e mediante l’ interazione con PPARγ induce la differenziazione del tessuto adiposo da bianco a bruno. Gli altri due membri della famiglia sono stati scoperti sulla base di analogie nella identità di sequenza. Mentre PRC ha meno omologie con PGC1α e
29
regola una serie di componenti della catena respiratoria, PGC1β possiede un ampio raggio di analogie ed è coinvolta nell’espressione del programma lipogenico nel fegato e a livello delle fibre muscolari di tipo IIX oltre ad avere un ruolo nella difesa dell’ospite indotta dall’interferone gamma e nell’attivazione degli osteoclasti.
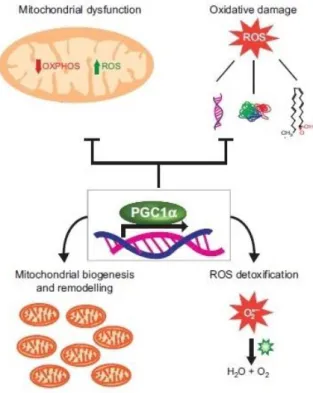
Da recenti studi, che hanno chiarito la capacità di PGC1α di modulare la composizione e la funzione dei mitocondri, è emerso che tale proteina è implicata nel controllo del globale metabolismo ossidativo, tramite due tipi di rimodellamento: 1) rimodellamento cellulare attraverso la biogenesi mitocondriale 2) rimodellamento degli organelli tramite l’alterazione delle proprietà intrinseche dei mitocondri. Inoltre l’elevato metabolismo ossidativo associato all’incremento dell’attività della PGC1α può essere accompagnato dall’aumento di specie reattive dell’ ossigeno (ROS) prodotte principalmente dai mitocondri e PGC1α è un importante regolatore dell’eliminazione dei ROS mediante l’espressione di enzimi ROS detossificanti. Quindi oltre che con il rimodellamento dei mitocondri e con la biogenesi mitocondriale, PGC1α contribuisce al mantenimento del metabolismo ossidativo anche regolando l’omeostasi dei ROS. In questo modo PGC1α aumenta il metabolismo ossidativo e minimizza l’impatto dei ROS sulle cellule. In particolare, il rimodellamento mitocondriale ha importanti conseguenze fisiologiche in quanto costituisce una via effettiva della regolazione del metabolismo. Inoltre PGC1α sembra avere un ruolo chiave nelle condizioni patologiche, quali malattie neurodegenerative e legate all’età, dal momento che esse sono associate alla compromissione delle funzioni mitocondriali e dai livelli di ROS.
30
Sono fattori trascrizionali implicati nella regolazione dell’attività ossidativa del mitocondrio e regolano la trascrizione dei geni coinvolti nel metabolismo degli acidi grassi dopo aver formato un eterodimero con il retinoid X receptor (RXR). PGC1α induce un aumento dell’attività delle isoforme dei PPARs nel muscolo scheletrico. PPARα è un importante fattore di controllo della ossidazione degli acidi grassi ed è espresso prevalentemente nel fegato ma anche nel cuore e nel muscolo. PPARδ sebbene abbia un’ espressione ubiquitaria, è più abbondante nel muscolo scheletrico dove è fortemente associato a PGC1α e funziona come un potente attivatore dei geni implicati nel catabolismo degli acidi grassi e nella termogenesi adattativa. PPARγ è espresso in vari tessuti tra cui il tessuto adiposo dove modula la trascrizione di numerosi geni coinvolti nel metabolismo lipidico tramite l’azione lipogenica e adipogenica. Nel muscolo scheletrico media la down regulation di GLUT 4 (principale trasportatore di glucosio nei tessuti insulino sensibili come cuore e muscolo scheletrico; è racchiuso all’interno di vescicole intracellulari che in seguito allo stimolo insulinico si spostano verso la membrana plasmatica con cui si fondono trasportando il glucosio dallo spazio interstiziale all’interno della cellula) nelle stesse cellule muscolari scheletriche di topi sottoposti a dieta grassa.
PGC1α come coordinatrice di un programma di energia pulita: regolazione metabolismo ossidativo
PGC1α può aumentare il metabolismo ossidativo tramite due diverse vie: 1) Può attuare il rimodellamento cellulare tramite la biogenesi degli
organelli (mitocondri e perossisomi) 2) Può coordinare il
rimodellamento degli organelli modificando la loro composizione e la loro funzione.
31
La scoperta della PGC1α nelle cellule adipose brune ha rivelato l’importante funzione di tale coattivatore trascrizionale nel metabolismo mitocondriale. Gli adipociti bruni sono cellule contenenti molte piccole goccioline lipidiche e molti mitocondri e la loro funzione principale è la generazione di calore tramite il disaccoppiamento della respirazione mitocondriale mediante la proteina UCP1. Il disaccoppiamento della respirazione si riferisce alla respirazione non accoppiata alla produzione di ATP per cui il calore viene prodotto tramite la dissipazione del gradiente protonico. L’espressione ectopica della PGC1α nel tessuto adiposo bianco determina un notevole aumento della biogenesi mitocondriale e della espressione della proteina UCP1(63) , due caratteristiche che rimandano al tessuto adiposo bruno. In seguito a questa scoperta è stato dimostrato che PGC1alfa incrementa l’espressione dei fattori della respirazione nuceare (NRFs) che vanno, a loro volta, a regolare l’espressione di molti geni mitocondriali(36).
NRF1 e NRF2 sono importanti fattori di trascrizione che regolano l’espressione di geni mitocondriali codificati nel nucleo. In particolare PGC1alfa aumenta l’espressione e agisce come coattivatore dell’NRF1 per regolare l’espressione del fattore di trascrizione mitocondriale TFAM, responsabile della trascrizione e della replicazione dei geni mitocondriali dal genoma(36). Infatti i mitocondri consistono di proteine codificate sia dai geni nucleari che mitocondriali.
Le proteine mitocondriali che sono codificate dai geni nucleari vengono trasportate nei mitocondri, quindi il globale metabolismo mitocondriale è regolato da un controllo coordinato attuato da geni nucleari e mitocondriali. A causa dell’incremento dell’espressione genica mitocondriale, la principale conseguenza fisiologica sui mitocondri, dovuta a PGC1α è l’induzione del disaccoppiamento della respirazione e
32
della biogenesi mitocondriale(37). Esperimenti su tessuti di PPARGC1A null mice che mostravano una ridotta espressione dei geni mitocondriali, in particolare di quelli codificanti varie subunità della catena di trasporto degli elettroni, hanno dimostrato una ridotta respirazione mitocondriale. Ciò ostacola i processi fisiologici legati al metabolismo mitocondriale. In seguito all’esposizione a basse temperature, i topi mostravano sensibilità al freddo associata ad una ostacolata capacità di upregulation dell’espressione della UCP1(63,64)
. Invece, topi transgenici che esprimevano ectopicamente PGC1α nel tessuto cardiaco muscolare e scheletrico, mostravano un’aumentata espressione dei geni mitocondriali e di conseguenza un incremento della biogenesi mitocondriale(20,65,66,67). Un fondamentale organello che supporta la funzione mitocondriale durante il metabolismo ossidativo è il perossisoma, la cui principale funzione nelle cellule dei mammiferi è la regolazione del metabolismo degli acidi grassi che non sono metabolizzabili dai mitocondri. I perossisomi però, non possono degradare completamente i complessi degli acidi grassi e per questo motivo trasportano quelli a catena corta, a livello dei mitocondri, dove viene completato il loro esaurimento. Di conseguenza mitocondri e perossisomi collaborano nel metabolismo dei lipidi che a loro volta sono un importante substrato del metabolismo ossidativo(68). L’idea della relazione tra mitocondri e perossisomi è stata recentemente validata dalla scoperta delle vescicole che si spostano tra i due organelli(69). Mitocondri e perossisomi condividono perciò come fattore comune alla loro biogenesi l’ attività di PGC1α. Nell’insieme, questi dati dimostrano che PGC1α orchestra cambiamenti nel metabolismo cellulare attraverso la biogenesi degli organelli (Figura 3). 2) Rimodellamento degli organelli: modulazione dell’attività intrinseca e della loro composizione
33
PGC1α regola positivamente l’espressione di diverse reti geniche come
quella mitocondriale, perossosimiale e detossificante ROS. L’elevata espressione di tali geni nelle cellule che esprimono ectopicamente PGC1α è stata attribuita all’incremento della biogenesi mitocondriale e conseguentemente all’aumento del numero e del contenuto degli organelli. Oltre a questo, PGC1α modula anche la composizione intrinseca dei mitocondri e dei perossisomi. Infatti, i nuovi mitocondri e i nuovi perossisomi prodotti in presenza di PGC1α mostrano proprietà diverse rispetto a quelli di origine e i cambiamenti sembrano avere un impatto centrale sul profilo dell’espressione genica cellulare e sul metabolismo ossidativo.
Meccanismi di rimodellamento cellulare e degli organelli
I mitocondri consistono di proteine codificate sia da geni mitocondriali che nucleari. Il genoma mitocondriale dei mammiferi codifica subunità della catena respiratoria, RNAs ribosomiale e RNAs transfert. Le proteine mitocondriali codificate dai geni mitocondriali sono trasportate nei mitocondri e il controllo coordinato della loro espressione influenza la regolazione del globale metabolismo mitocondriale. Il fattore 1 nucleare respiratorio NRF1 e il fattore 2 nucleare respiratorio conosciuti anche come GABPA (GA leganti le proteine) e gli ERRs (recettori connessi agli estrogeni) sono importanti fattori di trascrizione che regolano l’espressione dei geni mitocondriali codificati nel nucleo. E’ stato dimostrato che PGC1α aumenta l’espressione e/o la coattivazione di questi fattori al fine di regolare l’espressione dei geni coinvolti nel metabolismo mitocondriale. In particolare PGC1α aumenta l’espressione e agisce come coattivatore di NRF1 per regolare l’espressione del fattore
34
Tfam responsabile della trascrizione e della replicazione dei geni mitocondriali(36). Recentemente è stato dimostrato che PGC1α potrebbe essere localizzata nei mitocondri dove interagirebbe con Tfam. Anche se l’esatta funzione di PGC1α nei mitocondri non è ancora del tutto chiara ci sono evidenze che sia un fattore prossimale che coordina l’espressione dei geni mitocondriali e che quindi controlli il metabolismo mitocondriale. Inoltre, poiché i vari programmi e le varie funzioni metaboliche regolati da PGC1α non necessariamente devono essere regolate da una data condizione fisiologica, è interessante valutare il fatto, come sarà riportato in segiuto, che modificazioni post translazionali potrebbero impartire specificità all’attività di PGC1α.
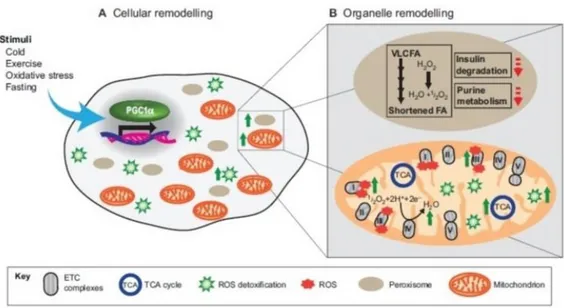
Figura 3. Regolazione del metabolismo ossidativo da parte di PGC1α. Gli stimoli fisiologici inducono l’espressione della PGC1α contribuendo così all’incremento del metabolismo ossidativo tramite il rimodellamento cellulare e degli organelli. (A)Impatto di PGC1α sul rimodellamento cellulare: livelli elevati di PGC1α inducono l’aumento della biogenesi mitocondriale e perossosimiale oltre ai livelli di enzimi ROS
35
detossificanti (illustrato dalle frecce verdi). (B)Impatto della PGC1α sul rimodellamento dei perossisomi e dei mitocondri. Livelli elevati di PGC1α modificano la composizione dei perossisomi in modo che questi possano esibire una riduzione della degradazione dell’insulina e del metabolismo purinico; inoltre alterano la composizione e le funzioni dei singoli mitocondri. Elevati livelli di PGC1α determinano l’ aumento l’attività degli enzimi del ciclo dell’acido citrico (TCA), del contenuto di enzimi ROS detossificanti, della capacità respiratoria, della produzione di ROS, cambiamenti della composizione delle subunità dei complessi ETC (illustrati dalle frecce verdi).
Metabolismo ROS
Dato che i mitocondri insieme al contributo dei perossisomi sono i principali produttori di ROS nelle cellule, le cellule che mostrano l’induzione della PGC1α in risposta a determinati stimoli fisiologici e di conseguenza un maggior numero dei mitocondri e dei perossisomi, devono anche adattarsi all’incremento della produzione di ROS, tra i quali superossido e perossido di idrogeno, altrimenti subirebbero le conseguenze deleterie di un aumentato metabolismo ossidativo. ROS possono infatti aggredire DNA, lipidi e proteine. Nei mitocondri, nel citoplasma e nei perossisomi le cellule contengono enzimi ROS detossificanti di cui PGC1α aumenta l’espressione(37,70,71). E’ stato infatti
dimostrato che l’espressione ectopica di PGC1α nelle cellule muscolari aumenta l’espressione della superossido dismutasi 2 (SOD2) e della
glutatione perossidasi 1(GPX1) rispettivamente implicati nel metabolismo del superossido e del perossido di idrogeno(37). Ulteriori studi hanno ampliato il repertorio degli enzimi ROS detossificanti includendo così altri enzimi perossosimiali, mitocondriali e citoplasmatici(70,71) e hanno dimostrato l’importante ruolo fisiologico di PGCα nel programma metabolico delle cellule; l’espressione ectopica di PGC1α nelle cellule migliora la sopravvivenza in condizioni di stress ossidativo mentre la ridotta espressione sensibilizza le cellule allo stress.
36
A supporto di questi esperimenti, i topi transgenici che esprimono PGC1α nel muscolo mostrano un minor accumulo di danno ossidativo con l’età a confronto dei topi wild type di controllo(67)
. Al contrario, l’ippocampo e la substantia nigra della PPARGC1A null mice mostrano una maggior sensibilità ai fattori di stress ossidativo. In sostanza, nel complesso questi studi hanno dimostrato l’effetto di PGC1α sul metabolismo ossidativo, rivelando che questa incrementa la biogenesi mitocondriale di pari passo all’aumento della capacità cellulare detossificante. In questo modo, la produzione di metaboliti ad alta energia e la rimozione dei metaboliti tossici sono finemente regolate e le cellule possono quindi beneficiare dell’incremento della respirazione e della produzione di ATP senza essere danneggiate.
Capacità respiratoria mitocondriale
Il primo studio che ha investigato sull’azione della PGC1α nella capacità respiratoria ha rivelato che i mitocondri isolati dal tessuto muscolare di topi transgenici che esprimono ectopicamente PGC1α (MCK-PPC1α TG mice) mostrano una maggiore capacità ossidativa verso i substrati, rispetto a quella che mostrano i topi wild type(37). Nei mitocondri del tessuto muscolare si verifica cioè, l’aumento dello stato 3 (attività) e dello stato 4 (riposo) della respirazione sui carboidrati malato e piruvato. Per determinare la capacità respiratoria dei mitocondri, questi vengono incubati in presenza di substrati specifici (comunemente carnitina palmitolo e piruvato) e/o farmaci e si fanno misurazioni della respirazione a livello degli stati 3 e 4(72), che vengono poi normalizzate in base ai milligrammi di proteina mitocondriale. Lo stato 3 rappresenta lo stato attivato dei mitocondri che avviene in presenza di substrati e ADP che viene convertita in ATP tramite l’ATP sintetasi. Lo stato 4
37
avviene una volta che l’ADP è stato convertito in ATP e spesso viene aggiunta l’oligomicina per inibire l’ATP sintetasi in modo che la respirazione non sia legata alla produzione di ATP. Questo è lo stato basale o di riposo del mitocondrio. Gli studi presi nell’insieme dimostrano che i mitocondri del tessuto muscolare che esprimono ectopicamente PGC1α presentano un aumento della capacità respiratoria, al contrario dei mitocondri carenti di PGC1α che mostrano invece un ridotto tasso dello stato 3 della respirazione quando respirano sul piruvato o sulla carnitina palmitolo. I cambiamenti della capacità respiratoria mediati da PGC1α possono essere spiegati tramite le alterazioni o dei livelli o dell’attività dei vari enzimi mitocondriali o tramite la combinazione di entrambi gli aspetti. Infatti i mitocondri dei muscoli isolati da MCKPGC1α mice hanno un’ elevata attività della citrato sintetasi, enzima proveniente dal ciclo dell’acido citrico, e della beta idrossiacil CoA deidrogenasi, coinvolta nella ossidazione degli acidi grassi(73). Inoltre, gli stessi mitocondri mostrano elevati livelli dei complessi di alcune subunità della catena di trasporto degli elettroni. A livello dei perossisomi, l’evidenza del loro rimodellamento si basa sui dati dell’espressione genica(74). L’espressione ectopica della PGC1α in
varie linee cellulari aumenta l’espressione di alcuni enzimi come quelli coinvolti nella degradazione e nell’ossidazione degli acidi grassi o la riduzione di altri come la xantina deidrogenasi coinvolta nel metabolismo delle purine(75). Un aspetto da evidenziare è che l’espressione di altri enzimi come quello che degrada l’insulina non viene influenzato. Comunque non sono ancora stati svolti studi specifici sui perossisomi isolati dai tessuti che esprimono ectopicamente PGC1α, per determinare l’impatto di questi cambiamenti sul metabolismo perossisomiale e per chiarire il motivo per cui il rimodellamento di tali organelli si coordini con il rimodellamento e la biogenesi dei mitocondri.
38
Gli studi hanno quindi dimostrato che PGC1α influenza il contenuto e l’attività di numerose proteine all’interno dei mitocondri i quali sono coinvolti in numerosi percorsi, sostenendo così l’idea che PGC1α eserciti un effetto globale sulle funzioni mitocondriali. Per quanto riguarda il controllo che PGC1α esercita sulla respirazione mitocondriale, emerge che può avvenire sia tramite la variazione del numero dei mitocondri nelle cellule, sia tramite l’alterazione della capacità respiratoria dei singoli mitocondri.
Capacità dei mitocondri di produrre ROS e saggi per la loro misurazione
La capacità respiratoria e di produzione di ROS da parte dei mitocondri sono connesse alla ETC, i cui principali siti di produzione di ROS sono i complessi I e III. Il primo produce ROS sul lato della matrice, il secondo produce ROS sia sul lato citoplasmatico che su quello della matrice della membrana. Durante la respirazione mitocondriale gli elettroni possono fuoriuscire dalla catena di trasporto degli elettroni del mitocondrio e reagire con l’ossigeno per formare l’anione superossido. La frazione degli elettroni che sfuggono alla ETC per produrre ROS rimane inalterata dalla presenza di PGC1α ad indicare la stretta associazione tra la loro produzione a livello mitocondriale e la respirazione. I mitocondri isolati dal tessuto muscolare dei MCKPGC1α Tg mice mostrano un aumento della capacità di produrre ROS a livello dei complessi I e III della ETC(70), in accordo all’ aumento della loro capacità respiratoria. Questo può essere spiegato attraverso l’aumento del contenuto delle subunità dei complessi della ETC. Inoltre, un altro fattore chiave che contribuisce al maggior rilascio di ROS da parte dei mitocondri è il loro elevato contenuto di SOD2, enzima che incrementa il tasso di
39
dismutazione del superossido in perossido di idrogeno nella matrice mitocondriale causando il rilascio di livelli elevati di quest’ultimo nel mezzo. Aspetto interessante è che l’elevato rilascio di H2O2 dai
mitocondri sarebbe rimosso da vari enzimi citoplasmatici ROS detossificanti la cui espressione è regolata da PGC1α. Gli studi hanno infatti dimostrato che l’incremento della detossificazione mediata da PGC1α è maggiore rispetto all’incremento della produzione di ROS, questo a sostegno del fatto che PGC1α può essere associata alla riduzione dei livelli basali di ROS nelle cellule. Oltre a questo, i tessuti muscolari dello stesso modello di topi transgenici mostrano un ridotto accumulo di danno ossidativo con l’invecchiamento rispetto al gruppo di controllo, quindi è plausibile che PGC1α abbia un impatto positivo nella protezione dal danno ossidativo. La quantificazione produzione di ROS può essere indiretta, tramite metodi fluorimetrici o diretta, tramite la risonanza di spin elettronico. La prima tecnica si basa sulla reazione tra perossido di idrogeno con la perossidasi di rafano e un substrato che diventa fluorescente. I mitocondri isolati sono incubati in presenza dei substrati che attivano la respirazione. Il superossido che è prodotto a livello della matrice mitocondriale non attraversa la membrana ed è convertito da SOD2 in perossido di idrogeno che diffonde poi nel mezzo
da saggiare. Il superossido prodotto invece sul lato citoplasmatico può essere quantificato aggiungendo SOD2 esogena nel mezzo che lo
converte in perossido di idrogeno. Questo metodo permette la quantificazione di ROS che origina sia dalla matrice che dal citoplasma, e la determinazione della topologia della produzione di superossido nei mitocondri. Se il tasso di perossido di idrogeno aumenta durante il saggio in seguito all’ aggiunta di SOD esogena, significa che il superossido viene prodotto sul lato citoplasmatico. Se invece risulta essere insensibile all’ aggiunta di SOD, il superossido deriva dalla
40
matrice. La seconda tecnica prevede l’ incubazione dei mitocondri in presenza di substrati che attivano la respirazione. Il superossido che origina dal lato citoplasmatico della membrana può essere direttamente individuato e quantificato, al contrario di quello prodotto dalla matrice mitocondriale che non può attraversare la membrana. Per la sua quantificazione si utilizzano trappole di spin che possono essere trasformate e dimostrarsi utili nella valutazione della produzione di ROS in questa sede.
PGC1α e malattie neurodegenerative
Dato che i mitocondri rivestono un ruolo molto importante nell’omeostasi dell’energia e del metabolismo, non è sorprendente il fatto che PGC1α sia coinvolta in molte condizioni associate ad una disfunzione mitocondriale a lungo termine, come malattie legate all’età e malattie neurodegenerative. Per quanto riguarda queste ultime, la prima prova a sostegno del coinvolgimento della PGC1α è stata fornita da tessuti PPARG1Anull mice(64,65) che mostrano neurodegenerazione associata all’iperattività, che ricorda i sintomi della patologia di Hungtington (HD). Questa è caratterizzata dalla produzione della proteina mutante Hungtington (HTT) ed è associata alle ridotte funzioni mitocondriali. I pazienti affetti mostrano una ridotta espressione di alcuni geni mitocondriali di PGC1α(75,76,77). Inoltre è stato dimostrato che HTT si associa con il promotore di PGC1α, limitandone così la sua espressione: questo evento presume un legame meccanicistico tra HD e PGC1α(76). Infine, topi HD riprodotti con PPARGC1A null mice
mostrano un’esacerbazione della neurodegenerazione, mentre
l’espressione ectopica di PGC1α nello striato dei topi transgenici protegge i neuroni dall’atrofia(76)
. Inoltre, PGC1α mostra anche un ruolo protettivo nel Parkinson (PD) patologia caratterizzata dalla perdita dei
41
neuroni dopaminergici nella sostanza nigra e dalla presenza di aggregati come i corpi di Lewy contenenti ubiquitina e α sinucleina(78). Nei pazienti PD si verifica la riduzione dell’espressione di numerosi geni targets di PGC1α, come quelli della catena respiratoria(79). PGC1α, nelle cellule di modelli PD, protegge contro la perdita neuronale e recentemente è stato dimostrato che PARIS, un nuovo substrato della E3 ubiquitina ligasi, spesso mutata nei soggetti PD(78), reprime l’espressione di PGC1α: iniezioni stereotassiche di PARIS nella sosranza nigra dei topi inducono la perdita neuronale, che viene invece attenuata dalla co-iniezione di PGC1α(80). Nel muscolo scheletrico, PGC1α agisce come sensore dei segnali intracellulari del calcio indotti dal motoneurone a livello della placca motrice; pertanto la sua espressione è indotta dalle contrazioni muscolari indotte dall’attività fisica. Durante l’esercizio la combinazione stimolo-contrazione promuove l’espressione di PGC1α. In questo contesto, questa proteina si comporta quindi come un mediatore dell’attività del motoneurone e, attraverso l’interazione con MEF2 e la
calcineurina, aumenta la capacità ossidativa della fibra muscolare promuovendo la biogenesi mitocondriale. Queste osservazioni risultano pertanto utili anche nella potenziale applicazione di PGC1α nella distrofia muscolare di Duchenne(81), in cui sembra stimolare l’espressione dei geni della giunzione neuromuscolare come laminina e utropina, e diminuire il danno tissutale. Nei topi affetti da sclerosi laterale amiotrofica, PGC1α migliora il loro globale fenotipo(82).