Indice
Parte 1. Introduzione 3
Introduzione 4
Parte 2. Ottica Non Lineare 8
1. Proprietà elettriche molecolari 9
1.1. Caso classico di un gas ideale di elettroni 9
1.2. Proprietà elettriche molecolari statiche e dinamiche 12
1.2.1. Proprietà statiche 13
1.2.2. Teoria QM delle proprietà elettriche molecolari statiche 14
1.2.3. Proprietà dinamiche 16
1.3. Da proprietà elettriche molecolari a proprietà elettriche macroscopiche 18
2. Effetti NLO in molecole organiche 19
2.1. Ingegneria molecolare 19
2.2. Effetto del solvente 21
3. Tecniche sperimentali 23
3.1. Forma contratta dei tensori di suscettibilità 24
3.2. Metodo delle polveri di Kurtz 24
3.3. EOAM (ElectroOptical Absorption Measurements) 26
Parte 3. Approssimazioni Utilizzate 31
4. Descrizione QM dei sistemi molecolari in soluzione 32
5. SCF Hartree-Fock (Metodo HF) 35
6. Teoria del funzionale densità (DFT) 39
7. Metodo Møller-Plesset di ordine II (MP2) 41
INDICE 2
8. Metodo PCM (Polarizable Continuum Model) 44
8.1. Soluzione del problema elettrostatico 44
8.2. Soluzione del problema QM 46
9. Metodi “Time Dependent” 50
9.1. Teoria della Risposta lineare 50
9.2. Metodo Time Dependent Hartree-Fock (TDHF) e DFT (TDDFT) 57
10. Modello a due stati (Two Levels Model) 58
Parte 4. Risultati Numerici 63
11. Strategia adottata 64
12. Analisi a due stati 66
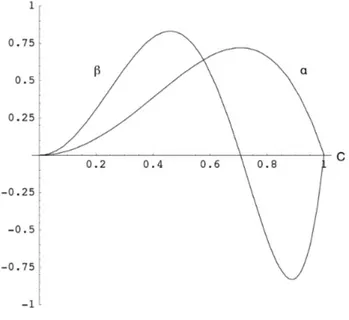
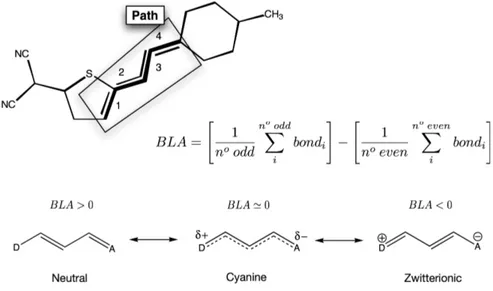
12.1. Analisi della Bond Length Alternation (BLA) 66
12.2. Assorbimenti UV-Vis 73
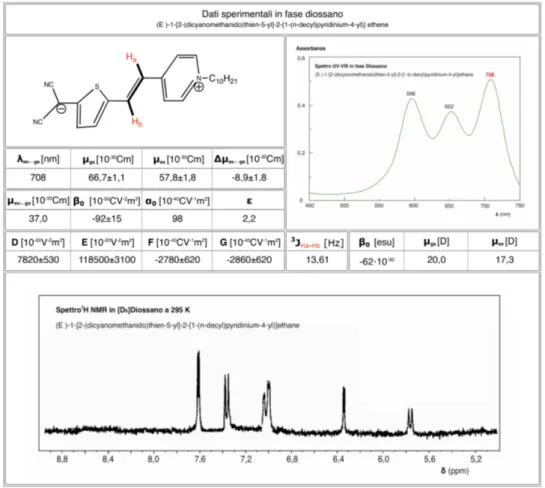
12.3. Spettrometria NMR 76
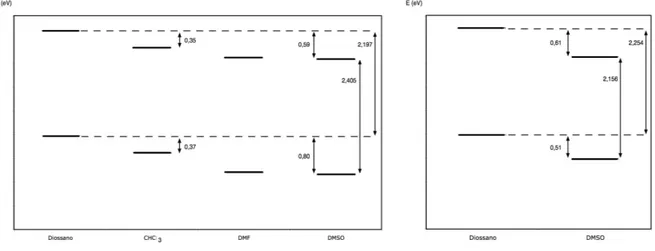
12.4. Dipendenza della prima iperpolarizabilità statica βo dal solvente 81
12.5. Analisi di Popolazione 90
12.5.1. Analisi NBO 91
12.5.2. Analisi di popolazione basata sul potenziale elettrostatico 92
12.5.3. Risultati delle analisi di popolazione 93
13. Analisi a campi finiti (FFT) 99
Parte 5. Conclusione 107
Conclusione 108
Appendice A: Termodinamica di solvatazione 110
Bibliografia 115
Bibliografia 115
Elenco delle figure 121
Parte 1
Introduzione
Si parla comunemente di materiali dotati di proprietà ottiche non lineari qualora la loro interazione con un campo elettromagnetico renda possibile la generazione di nuovi campi elettromagnetici alterati in frequenza, fase ed altre proprietà fisiche. Sostanze in grado di elaborare efficientemente l’informazione sono di notevole importanza per lo sviluppo di vari tipi di tecnologie, che includono telecomunicazioni, optoelettronica, olografia e fotonica.
Per ottenere progressi nelle aree di ricerca suddette si è reso necessario avere disponibilità di nuovi materiali caratterizzati da risposte ottiche non lineari sufficienemente grandi. Una estesa ricerca è stata infatti improntata alla sintesi di sistemi molecolari in grado di interagire efficientemente con la radiazio-ne elettromagradiazio-netica. Negli ultimi decenni tale tipo di ricerca ha sfruttato efficacemente i nuovi mezzi messi a disposizione dalle procedure di calcolo di derivazione teorico-quantistico. Affrontare la progetta-zione di nuovi sistemi molecolari, avvalendosi di tecniche computazionali in grado di razionalizzare una correlazione tra struttura molecolare e proprietà di interesse, ha radicalmente accelerato la scoperta di intere famiglie di cromofori. Al giorno d’oggi, un chimico sperimentale ha a disposizione due mezzi per elaborare una strategia di sintesi: l’intuizione, ed i risultati qualitativi forniti da una pre-analisi di tipo computazionale.
La chimica dei materiali è di per se un campo vasto e di difficile comprensione, la parte che in essa possono giocare le tecniche computazionali non è appunto quella di riuscire a fornire dei risultati accurati, ma piuttosto di essere strumento al servizio dell’interpretazione, correlazione e previsione di osservazioni sperimentali. La sfida non consiste nel riuscire a determinare con precisione alla seconda o terza cifra decimale le componenti dei tensori di iperpolarizzabilità, bensì di riuscire a razionalizzare quali caratte-ristiche della struttura elettronica di una molecola siano causa della risposta ottica non lineare. Il campo della ricerca di nuovi materiali NLO1è paradigmatico per l’attuale chimica, in essa abbiamo sforzi, da
una parte volti al miglioramento di disegno, sintesi ed analisi, e dall’altra volti all’elaborazione di possibili
1NLO è acronimo di Non Linear Optic.
INTRODUZIONE 5
applicazioni pratiche per i nuovi materiali prodotti. Per poter raggiungere questo obiettivo è necessario avere una conoscenza a livello teorico degli aspetti che si celano sotto la risposta NLO dei singoli cromo-fori, riuscire ad elaborare un modello che spieghi il differente comportamento delle molecole qualora esse siano soggette ad interazioni con l’ambiente circostante ed infine conoscere la risposta del materiale a livello macroscopico. Tutti quanti questi aspetti sono sfide per la chimica teorica. Ottenere delle osserva-bili ottiche non lineari per sistemi complessi è tuttora un’impresa di per sè difficile, anche se il progresso tecnologico dei sistemi di elaborazione ha reso la questione meno problematica di un tempo. Anche per il problema della relazione tra proprietà del cromoforo ed ambiente circostante è necessario tenere in considerazione interazioni cromoforo-¯solvente, cromoforo-matrice polimerica e cromoforo-cromoforo; al giorno d’oggi per affrontare questa problematica, con parziale successo ed in tempi di calcolo ragionevoli, abbiamo la possibilità di scegliere tra dei modelli di solvatazione denominati “continui”. Infine, l’analisi dei risultati computazionali alla luce di dati sperimentali misurati su campioni macroscopici complica ulteriormente la questione. Le tecniche sperimentali utilizzate più comunemente per la determinazioni di proprietà NLO (ad esempio la tecnica EFISH) presentano sia dei problemi di riproducibilità (il che complica non poco le procedure di acquisizione del dato), che una non completamente rigorosa definizione di una convenzione univoca per procedure, definizioni e unità di misura.
La data di nascita storica per il campo dell’ottica non lineare è il 1875, anno in cui J. Kerr pubblicò un lavoro sulle sue osservazioni riguardo la variazione dell’indice di rifrazione del CS2indotta da una campo
elettrico. A breve distanza di tempo, nel 1893, un ulteriore effetto ottico non lineare venne scoperto da F. Pockels mentre studiava l’interazione tra quarzo e radiazione luminosa. Ancora oggi chiamiamo queste tipologie di effetti ottici non lineari rispettivamente “effetto-Kerr” ed “effetto-Pockels”. In realtà l’interesse per l’utilizzo di fenomeni di ottica non lineare si può dire essersi scatenato a partire dal 1960, anno in cui il primo dispositivo Laser venne inventato e seguito a distanza di un anno dalla pubblicazione pionieristica di P. Franken [1] riguardo al fenomeno della generazione armonica di secondo ordine in un cristallo di quarzo. Dopo questi due eventi il fenomeno dell’ottica non lineare esplose letteralmente, venne rifinito e parzialmente razionalizzato teoricamente negli anni ’70. Verso la fine di questa decade si assistette infatti all’avvento delle fibre ottiche come nuovo veicolo dell’informazione nell’ambito delle telecomunicazioni. Da quel momento in poi un rinnovato interesse venne riversato nella costruzione e progettazione di materiali polimerici in grado di essere utilizzati per generazione armonica di ordine
INTRODUZIONE 6
Figura 1. Sistemi merocianinici oggetto di questo lavoro di tesi con relativa nomen-clatura. Nell’ultima illustrazione è enfatizzata la caratteristica principale di questo tipo di sistemi: le anulazioni successive al fine di modularne la risposta NLO.
secondo e per dispositivi elettro-ottici. Il resto è storia dei giorni nostri, notevoli sforzi sono stati impiegati recentemente nella miniaturizzazione su scala molecolare e nei criteri che determinano la resistenza al consueto esercizio di lavoro di dispositivi di nuova generazione come diodi e transistori molecolari[2].
All’interno di questo lavoro di tesi è stata focalizzata l’attenzione sulla categoria di composti mero-cianinici riportati in figura (1).
INTRODUZIONE 7
Questi sistemi molecolari appartengono alla categoria di composti π-coniugati in cui le due zone alle estremità della molecola si comportano rispettivamente come donatore e accettore di elettroni, da qui la denominazione “push-pull”. La transizione tra i due orbitali HOMO e LUMO di queste molecole si dice essere a “trasferimento di carica”, e con ciò si vuole intendere che nello stato iniziale la distribuzione di carica elettrica si trova localizzata prevalentemente su una estremità della molecola (gruppo dona-tore), mentre nello stato finale è l’estremità opposta ad avere una più alta densità elettronica (gruppo accettore). Cromofori che possiedono una struttura di questo tipo presentano anche una risposta ottica non lineare di ordine I di entità elevata. Per operare un controllo su questo tipo di risposta una delle strade possibili consiste nella variazione della forza relativa dei due gruppi donotore e accettore, cercando cioè di aumentare la asimmetria elettronica delle molecole. Il percorso intrapreso con questa categoria di composti è invece totalmente differente, si vuole cioè osservare l’andamento della risposta ottica non lineare attraverso anulazioni successive dell’estremità azotata.
Una panoramica sui fondamenti dei fenomeni di risposta ottica non lineare in molecole organiche viene fornita nella parte II di questo elaborato di tesi. Per una migliore comprensione del fenomeno si parte da un approccio di tipo classico al problema (pag.9) per poi successivamente passare ad un approccio più rigoroso e di tipo quanto-meccanico. Fondamentalmente l’approccio quanto-meccanico al giorno d’oggi consiste in due alternative: la consueta somma sugli stati2 (a pag.12) ed il metodo a
campi finiti3che consiste nella derivazione numerica degli elementi della serie perturbativa utilizzata per
esprimere il momento di dipolo indotto da una perturbazione elettrica esterna.
Successivamente si parla di sistemi organici, di come in essi il trasferimento di carica, CT, sia la principale causa degli effetti non lineari di ordine I, e di quali siano le leve tramite cui sia possibile modulare questo CT (pag.19). Per ultimo, ma non per importanza, abbiamo poi l’effetto del solvente (pag.21).
La parte III elenca tutte le approssimazioni utilizzate nell’ambito di questo lavoro, con ciascun mo-dello illustrato attraverso i fondamenti fisici che lo giustificano. Nella parte IV sono invece riportati e commentati i dati sperimentali ottenuti con i modelli precedentementu introdotti.
2Metodo “Sum Over State” o SOS 3Metodo “Finite Fields”
Parte 2
1. Proprietà elettriche molecolari 1.1. Caso classico di un gas ideale di elettroni4.
Per introdurre i concetti di polarizzabilità ed iperpolarizzabilità si parte da una situazione ideale. Si immagini di immergere un gas di N particelle all’interno di una camera contenente un campo elettrico −→F oscillante a frequenza ω. In una situazione simile le particelle si comporteranno, in prima approssimazione, come fossero N oscillatori armonici smorzati, ovvero come se ciascun elettrone fosse tenuto al suo posto da una molla smorzata dalla sola forza d’attrito, soggetti cioè ad una forza proporzionale allo spostamento più un contributo di smorzamento proporzionale alla loro velocità (una sorta di forza di resistenza al moto). La forza totale agente su di un singolo elettrone è riassumibile in un’equazione differenziale di risposta lineare: (1) −→F = qe−→F = qe· −→ F0 2 ! eiωt+ e−iωt"= m−→¨x + mγ−→˙x + mω20−→x dove:
• qeè la carica della particella ed m è la sua massa
La soluzione dell’equazione (1), se la particella è un elettrone, sarà data dall’espressione:
(2) −→x =−qe· −→ F0 2m · eiωt iγω + ω2 0− ω2 + complesso coniugato
Per una singola particella il dipolo indotto dal campo elettrico, −→p(1), sarà di conseguenza ricavabile
dal prodotto tra carica elettrica qe e la (2):
(3) −→p(1)= −qe−→x = # −→F 0 2 $q2 e m· 1 iγω + ω2 0− ω2 % eiωt & + complesso coniugato
1. PROPRIETÀ ELETTRICHE MOLECOLARI 10 (4) −→p(1) = −qe−→x = # −→F 0 2 ' α(1)(eiωt & + complesso coniugato dove: • α(1) indica la polarizzabilità
Ad un livello di risposta lineare, il dipolo è dato dalla somma di due contributi, uno di tipo permanente ed un altro indotto dal campo elettrico:
(5) −→p = −→p0+ −→p(1)= −→p0+
) α(1)
−→ F0
2 eiωt+ complesso coniugato *
Per descrivere classicamente il coefficiente di iperpolarizzabilità del I ordine è necessario introdurre un termine aggiuntivo anarmonico all’equazione (1):
(6) −→F = qe−→F = qe· −→ F0 2 ! eiωt+ e−iωt"= m−→¨x + mγ−→˙x + mω02−→x + + mB−→x2,
Ciascun elettrone è dotato di una frequenza di oscillazione intrinseca ω0, di un suo fattore di
smorza-mento γ, e di una costante di anarmonicità B. La posizione dell’elettrone si ottiene adesso dalla somma di due termini, uno identico a quello ricavato precedentemente (2), che chiamo −→x(1), ed uno aggiuntivo,
che chiamo −→x(2), dovuto all’introduzione del termine anarmonico.
(7) −→x = −→x(1)+ −→x(2) (8) −→x(1)= ) qe· −→ F0 2m · 1 iγω + ω2 0− ω2 eiωt+ cc * (9) −→x(2) = ) q2 e· −→ F0·−F→0 2m · B [iγω + ω2 0− ω2] 2[2iγω + ω2 0− 4ω2] 2e i2ωt+ cc *
1. PROPRIETÀ ELETTRICHE MOLECOLARI 11
Con un potenziale così modificato il dipolo differisce da (5) per un termine aggiuntivo ricavabile dal prodotto tra la carica elettrica qee la (9):
(10) −→p(2)= # − −→ F0·−F→0 2 ) q3 e m· B [iγω + ω2 0− ω2] 2 [2iγω + ω2 0− 4ω2] 2 * ei2ωt & + cc
Pertanto il vettore di polarizzazione totale per una singola particella sarà:
(11) −→p = −→p0+ −→p(1)+ −→p(2) = −→p0+ −→ F0 2 -α(1)(−ω; ω) eiωt+ cc.+ −→ F0·−F→0 2 -α(2)(−2ω; ω, ω) ei2ωt+ cc. dove polarizzabilità ed iperpolarizzabilità compaiono con il loro consueto formalismo:
α(1)(−ω; ω) e α(2)(−2ω; ω, ω) .
Con un’approccio di tipo classico si ottiene una descrizione intuitiva ma limitata del problema della polarizzazione.
Essendo il gas composto da N particelle identiche, per ottenere la risposta macroscopica al campo elettrico esterno basterà moltiplicare polarizzabilità ed iperpolarizzabilità della singola particella per N ed ottenere i rispettivi termini macroscopici di suscettibilità di primo e secondo ordine, χ(1)(−ω; ω) e
χ(2)(−2ω; ω, ω): (12) χ(1)(−ω; ω) = N · α(1)(−ω; ω) = N $q2 e m· 1 iγω + ω2 0− ω2 % (13) χ(2)(−2ω; ω, ω) = N · α(2)(−2ω; ω, ω)
1. PROPRIETÀ ELETTRICHE MOLECOLARI 12
Nel caso semplice fin qui considerato, in cui un singolo campo elettrico solleciti il sistema, il vettore di polarizzazione macroscopica per il gas di particelle, −→P, sarà:
(14) −→P = χ0+ # χ(1)(−ω; ω) · −→ F0 2 eiωt+ cc & + # χ(2)(−2ω; ω, ω) · −→ F0·−F→0 2 ei2ωt+ cc &
Qualora la trattazione per il moto oscillatorio degli elettroni sia di tipo quantistico si ottiene un risultato apparentemente simile a quello classico, ma con delle differenze di sostanza: gli elettroni in MQ hanno parecchie frequenze naturali di oscillazione (modi di oscillazione), con diversa intensità l’una dall’altra, e ciascuna dotata della propria costante dissipativa. Per tenere conto delle diverse intensità si introducono fattori di peso fk specifici per ogni k-¯esimo modo di oscillazione e, analogamente, una serie di fattori γk di smorzamento. L’espressione quanto-meccanica per (12) si complica ad esempio in:
(15) χ(1)(−ω; ω) = N q 2 e m )modi/ k fk iγkω + ω2 0− ω2 *
1.2. Proprietà elettriche molecolari statiche e dinamiche.
Analizzare il problema dell’ottica non lineare con l’approccio seguito nel sottoparagrafo 1.1 è un utile strumento per avere una visione qualitativa dell’interazione tra materiali NLO e campi elettromagnetici. Questo modello non è tuttavia utilizzabile per descrivere dei sistemi di interesse chimico. Nella realtà i materiali sono ben più complessi di un gas di particelle cariche, sono costituiti da atomi che interagendo l’uno con l’altro formano strutture molecolari o cristalline. Per interesse ci riferiamo ai soli composti molecolari per i quali, dato il numero piccolo di atomi che li compongono, in linea di principio è anche possibile calcolare osservabili microscopiche NLO senza ricorrere ad eccessive approssimazioni. Per una accurata descrizione della risposta NLO di sistemi molecolari di tipo organico occorre prestare particolare attenzione agli effetti dovuti alla correlazione elettronica. Nel corso della successiva trattazione viene sottaciuta l’approssimazione di fondo tramite cui si considerano trascurabili le interazioni con la parte magnetica della radiazione luminosa.
1. PROPRIETÀ ELETTRICHE MOLECOLARI 13
1.2.1. Proprietà statiche.
Come descritto nella sezione precedente, le proprietà di una molecola variano in caso di interazione con fattori esterni. Sia λ un generico fattore esterno che induce una risposta della proprietà molecolare E. Espandendo in serie E in funzione di λ si ottiene E(λ), la risposta del sistema alla perturbazione:
(16) E(λ) = E(λ = 0) + $ ∂E ∂λ % λ=0 + 1 2! $ ∂2E ∂λ2 % λ=0 λ2+ 1 3! $ ∂3E ∂λ3 % λ=0 λ3+ . . .
Per il caso particolare in cui il fattore esterno sia rappresentato da un campo elettrico statico −→F e la proprietà di cui ci interessa la risposta sia l’energia5, E:
(17) E(−→F ) = E(−→F = 0) + $∂E ∂−→F % − → F =0 F + 1 2! $∂2E ∂−→F2 % − → F =0 − →F2+ 1 3! $∂3E ∂−→F3 % − → F =0 − →F3+ . . .
L’espansione (17) può essere riscritta in modo da far comparire esplicitamente la componente µi del momento di dipolo6nel caso in cui il campo elettrico abbia una sola componente, F
i, diversa da zero: (18) −[E(Fi) − E(F[F i= 0)] i− 0] = µi= − $ ∂E ∂Fi % Fi=0 −2!1 $ ∂2E ∂F2 i % Fi=0 Fi− 1 3! $ ∂3E ∂F3 i % Fi=0 Fi2− . . .
per poi venire condensata in una forma più compatta7:
(19) µi= µ0,i+ αiiFi+
1
2!βiiiFi2+ . . .
5Effect of a uniform electric Field di [4].
6Vedi teorema in equazione (26) che estende questo risultato anche al caso quantistico
7Ricavando la funzione di risposta del momento di dipolo, µ(F ), si otterrebbe direttamente la serie (19). Tra gli elementi
della serie (17) a pag.13, della serie (19) e gli elementi dei tensori di polarizzabilità ed iperpolarizzabilità valgono le seguenti relazioni: • 1 2 „ ∂2E ∂Fi2 « =“∂µi ∂Fi ” = αii, • 1 3! „ ∂3E ∂Fi3 « =2!1 „ ∂2µi ∂Fi2 « =2!1βiii, • 1 4! „ ∂4E ∂Fi4 « = 1 3! „ ∂3µi ∂Fi3 « = 1 3!γiiii.
1. PROPRIETÀ ELETTRICHE MOLECOLARI 14
Campo elettrico e momento di dipolo sono dotati di tre componenti, mentre polarizzabilità ed iper-polarizzabilità di ordine I sono dei tensori, rispettivamente di rango II e III. Qualora il campo elettrico abbia tutte e tre le componenti non nulle l’espressione generale per una singola componente del momento di dipolo indotto sarà:
(20) µi = µi,0+ / j αijFj+ 1 2! / j / k βijkFjFk+ 1 3! / j / k / l γijklFjFkFl+ . . .
Con la notazione di Einstein (regola della somma su indici ripetuti) le somme vengono invece sottintese: (21) µi= µ0,i+ αijFj+ 1 2!βijkFjFk+ 1 3!γijklFjFkFl+ . . . 1.2.2. Teoria QM delle proprietà elettriche molecolari statiche.
In meccanica quantistica lo stato di un sistema viene descritto come un vettore di norma unitaria, indicato con |ψ(t)#, in uno spazio di Hilbert, Tale vettore si definisce come combinazione lineare delle funzioni linearmente indipendenti in tale spazio. L’evoluzione temporale di questo vettore si ottiene dopo trasformazione con operatori unitari. L’Hamiltoniana è un operatore unitario e operando con essa sullo stato di un sistema se ne ottiene l’evoluzione temporale: l’equazione di Schrödinger.
(22) H |ψ(t)# = iˆ ∂t∂ |ψ(t)#
Qualora l’Hamiltoniana sia indipendente dal tempo le soluzioni dell’equazione precedente si chiamano soluzioni stazionarie:
1. PROPRIETÀ ELETTRICHE MOLECOLARI 15
Il valore di aspettazione dell’Hamiltoniana dello stato ψ(t) è anche il valore medio dell’energia, $E#, per tale stato:
(24) $E# = $ψ(t)| ˆH |ψ(t)#
$ψ(t)| ψ(t)#
Seguendo un approccio di tipo perturbativo [5], per introdurre l’effetto di un campo elettrico −→F si aggiunge una perturbazione ˆH(1)all’Hamiltoniana modello ˆ
H(0): (25) H = ˆˆ H(0)+ ˆ H(1)= ˆ H(0)+ ) / i ˆ µiFi *
Utilizzando il teorema di Hellmann-Feynman si possono ricavare le componenti µi 8del momento di dipolo: (26) $ ∂E ∂Fi % = 0∂'ˆ H(0)+ ˆ H(1)( ∂Fi 1 = 0∂'ˆ H(0) − ˆµi· Fi ( ∂Fi 1 = − $µi#
Al fine di ottenere espressioni per derivata prima e seconda dell’energia rispetto a variazioni del campo elettrico, tramite cui ricavare il momento di dipolo, si utilizzano le equazioni di ordine I dell’espansione perturbativa dell’energia9: (27) E0= E0(0)− / i $ψ0| ˆµi|ψ0# Fi+ / i / j / n#=0 $ψ0| ˆµi|ψn# $ψn| ˆµj|ψ0# E0− En FiFj Adesso le espressioni per le derivate che compaiono in (18) sono note, ad esempio:
(28) $ ∂E0 ∂Fi % Fi=0 = − $ψ0| ˆµi|ψ0# + 2 / j / n#=0 $ψ0| ˆµi|ψn# $ψn| ˆµj|ψ0# E0− En Fj Fi=0
8Si utilizza i per indicare genericamente una delle tre coordinate spaziali x, y e z 9Rayleigh-Schrödinger Perturbation Theory [6]
1. PROPRIETÀ ELETTRICHE MOLECOLARI 16
Dalla derivata prima e seconda si possono ricavare momento di dipolo permanente e polarizzabilità:
(29) µi,0= $ψ0| ˆµi|ψ0# (30) αij= − $ ∂2E ∂Fi∂Fj % = −2/ n#=0 $ψ0| ˆµi|ψn# $ψn| ˆµi|ψ0# E0− En
Troncando al primo ordine l’espansione perturbativa che porta alla (27), è possibile ricavare soltanto (29) e (117). Espressioni per le iperpolarizzabilità statiche si potranno quindi ottenere con una espansione perturbativa che includa anche termini di secondo ordine e successivi.
1.2.3. Proprietà dinamiche.
Se i campi elettrici esterni sono oscillanti la risposta del materiale sarà differente e l’espansione del dipolo avrà molti contributi. D’ora in avanti ci riferiremo ai tensori di iperpolarizzabilità utilizzando una notazione compatta di questo tipo:
• β'-−<2i=1ωi . ; ω1, ω2 ( , • γ'-−<3i=1ωi . ; ω1, ω2, ω3 ( .
Al fine di ricavare proprietà non statiche, si inserisce nella (21) a pag. 14 un campo elettrico oscillante: (31) −→F =−→F0+ −→Fωcos(ωt) =−→F0+
1
2−→Fω!eiωt+ e−iωt"
Il dipolo indotto che se ne ricava è somma di vari contributi: statici (contrassegnati dal pedice 0) e dinamici [7].
Si riporta a titolo d’esempio l’espressione per il dipolo con termini di iperpolarizzabilità, limitandoci nell’espansione all’ordine I:
(32)
(contributi statici) (contributi non statici) µ = = >? @ µ0+ α0−→F0+ 1 2β0−→F20+ = >? @ +α(−ω; ω)−→Fωcos(ωt) +1 4β(−2ω; ω, ω)−→F2ωcos(2ωt)+ +1 4β(0; ω,−ω) − →F2 ω+ β(−ω; ω, 0)−→F0−→Fωcos(ωt) + . . .
1. PROPRIETÀ ELETTRICHE MOLECOLARI 17
Differenti combinazioni delle frequenze dei campi elettrici applicati producono differenti fenomeni di ottica non lineare di ordine II. Nell’espressione (32) il termine β(−2ω; ω, ω) tiene conto del fenomeno SHG10(la generazione di una radiazione di frequenza 2ω in risposta a due campi elettrici di frequenza
degenere pari a ω), mentre β(−ω; ω, 0) è responsabile dell’effetto elettro-ottico lineare. Effetti NLO di ordine successivo si descrivono attraverso le iperpolarizzabilità di ordine II: γ(−3ω; ω, ω, ω) tiene conto del fenomeno THG11, γ(−2ω; ω, ω, 0) il dc-¯SHG (attraverso questa iperpolarizzabilità di ordine II si ottiene
una risposta di generazione armonica di ordine II “indotta” da un campo elettrico statico). Perturbativamente [8] [9] si ricavano gli elementi di (32).
Le espressioni che si ottengono per le varie iperpolarizzabilità hanno bisogno di spazio12, perciò si
riporta, a titolo d’esempio, soltanto l’espressione generale della componenteβxxx del tensore βSHG:
βxxx(−2ω; ω, ω) = !22<Nl#=0<Nm#=0 $0|ˆµx|l%($l|ˆµx|m%−$0|ˆµx|0%δlm)$m|ˆµx|0% (ωmo−2ω)(ωlo−ω) + +$0|ˆµx|l%($l|ˆµx|m%−$0|ˆµx|0%δlm)$m|ˆµx|0% (ωmo+ω)(ωlo−ω) + +$0|ˆµx|l%($l|ˆµx|m%−$0|ˆµx|0%δlm)$m|ˆµx|0% (ωmo+ω)(ωlo+2ω) dove:
• l’insieme delle {l} ed {m} rappresentano gli stati eccitati |l# ed |m# mentre |0# rappresenta lo stato fondamentale;
• ˆµ e l’operatore dipolo di transizione;
• !ω0→{l} o {m}è l’energia necessaria per avere transizione tra due stati.
10SHG è acronimo di Second Harmonic Generation, fenomeno scoperto da P. Franken [1]. 11THG è acronimo di Third Harmonic Generation.
12Le formule generali per la determinazione di β
ijke γijklsono riportate per esteso a pag. 19 di [10]. Quelle per i processi SHG e per gli effetti elettro-ottici sono invece a pag. 32 sempre di [10].
1. PROPRIETÀ ELETTRICHE MOLECOLARI 18
1.3. Da proprietà elettriche molecolari a proprietà elettriche macroscopiche.
Nel caso classico di un gas ideale di elettroni, trattato nella sezione 1.1 a pag.9, passare da una trattazione microscopica ad una macroscopica richiedeva la moltiplicazione per il numero di particelle costituenti il gas. In un sistema di questo tipo non ci sono infatti interazioni inter-particellari, gli elettroni si trovano ad una distanza infinitamente grande l’uno dall’altro. Le suscettibilità (12) e (13) a pag.11 sono frutto di questa operazione. In un sistema costituito da più molecole ci sono delle complicazioni e occorre tenerne di conto. In un sistema reale:
• il campo risentito da una singola particella sarà dato dalla somma di due contributi rappresen-tati dal campo elettrico esterno e dai campi elettrici aggiuntivi generati dall’interazione tra il campo elettrico esterno e le altre particelle costituenti il materiale. Le particelle nel bulk risen-tono quindi di un campo elettrico “locale” diverso dal campo elettrico esterno. Si introducono quindi i tensori di trasformazione F, che modificano gli elementi dei tensori di polarizzabilità ed iperpolarizzabilità in una maniera tale da potersi riferire non ai campi elettrici locali ma a quegli esterni [5] [3];
• le N molecole del campione saranno orientate, l’una rispetto all’altra, in modo differente rispetto alla terna d’assi solidale con il “frame laboratorio”. Si utilizzano perciò delle matrici di rotazione che si occupano di trasformare oggetti riferiti ad assi solidali con la molecola ad oggetti riferiti alla terna d’assi del laboratorio;
• occorre operare una media su tutte le possibili orientazioni delle molecole presenti all’interno di una porzione macroscopica del campione. E’ raro avere dati di distribuzione orientazionale delle molecole, specialmente quando esse sono inserite all’interno di materiali come ad esempio film polimerici, cristalli liquidi o cristalli molecolari [10].
χ(2)(−ω3; ω1, ω2) = N E Rim!Rjn!Rko!Fωj!1nF ω2 k!oFωi!3mβijk(−ω3; ω1ω2 F dove:
• le matrici di rotazione sono indicate con R; • le matrici di correzione di campo locale sono le F;
• le parentesi (< >) sono il simbolo dell’operazione di media orientazionale; • N il numero di molecole che costituiscono il campione.
2. EFFETTI NLO IN MOLECOLE ORGANICHE 19
2. Effetti NLO in molecole organiche
In molti sistemi molecolari organici gli effetti NLO sono dovuti principalmente ad un trasferimento di carica indotto da una radiazione elettromagnetica. Tali trasferimenti di carica hanno origine dalla partico-lare distribuzione elettronica di talune molecole, e si verificano specialmente quando queste sono dotate di un gruppo elettron-donatore ed un gruppo elettron-accettore connessi attraverso un sistema π-coniugato [11]. È condizione necessaria per avere effetti NLO del II ordine che la molecola non possieda un centro di inversione. Riferendoci all’equazione differenziale (6) a pag.10 si può notare che il potenziale anarmonico aggiuntivo ivi introdotto è dispari13. Se il sistema ha un centro di inversione, il potenziale totale del
sistema dovrebbe risultare invariante rispetto all’inversione spaziale, ovvero UT OT(−→x ) = UT OT(−−→x ), cosa che di per sè implicherebbe l’assenza del termine anarmonico. Anche nel caso di sistemi molecolari senza centri di inversione e dotati di altre proprietà NLO, l’interazione intermolecolare all’interno del bulk può portare alla formazione di dimeri centrosimmetrici e pertanto all’assenza di effetti NLO [12] [13]. Occorre prestare attenzione nel ridurre al minimo questa evenienza, al fine di non vanificare gli sforzi utilizzati nella progettazione di una molecola che, isolata, possieda ottime risposte NLO [14] [15].
2.1. Ingegneria molecolare.
Gli assorbimenti di un cromoforo organico possono modificati variando estensione e tipo di sistema π-coniugato, gruppi elettrondonatori e elettronaccettori (vedi figura 2 a pag.20) e conformazione della mo-lecola. Variando questi parametri si ottiene come risultato uno spostamento delle energie in spettri di as-sorbimento UV-Vis. La variazione delle coppie donatore-accettore non solo comporta shift-solvatocromici, ma anche a una variazione di intensità dell’assorbimento stesso.
Sebbene da prendersi con le dovute precauzioni, esistono delle relazioni empiriche semplici che legano la iperpolarizzabilità all’estensione del ponte π-¯coniugato che lega donatore ed accettore.
13Il potenziale relativo alla forza anarmonica si ottiene per integrazione:
• U(−→x ) =−RB−→x2d−→x =1 3B−→x3
2. EFFETTI NLO IN MOLECOLE ORGANICHE 20
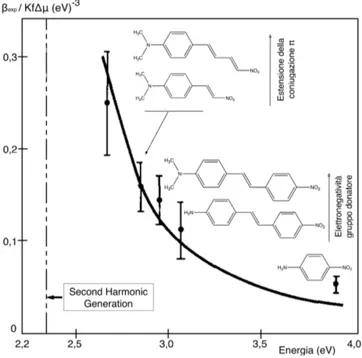
Figura 2. Iperpolarizzabilità βexpcome funzione dell’estensione della coniugazione per derivati di benzene e stilbene. (tratto da J. L. Oudar - Optical Nonlinearities of Conju-gated Molecules. Stilbene Derivatives and HighlyPolar Aromatic Compounds - Journal of Chemical Physic, 67, 446, 1977 [16])
In queste relazioni l’estensione del sistema π-coniugato è quantificata con il numero N di doppi legami π costituenti il ponte. Tra µβ0 ed N la relazione è di tipo cubico, mentre l’andamento di N con µβ si
discosta, sebbene di poco, da questo andamento.
2. EFFETTI NLO IN MOLECOLE ORGANICHE 21
Figura 3. Forme limite di risonanza in sistemi molecolari donatore-accetore o “push-pull”. La lettera D indica il gruppo donatore e la lettera A il gruppo accettore.
Per la scelta di donatore e accettore sono state stilate delle scale di forza relativa. A titolo d’esempio si riportano le relazioni di forza tra i gruppi più comuni:
Scala di forza dei gruppi accettori
⇒ =−CH(CN)2>−NO2>−NO > −COCF>? 3>−CHO > −CN > −SO2CH@3 ⇐
⇒ −N(CH3)2>−NH2>−N2H3>−SCH3>−OCH3>−OC6H5>−Br > −OH > −OCH3
? @= > ⇐
Scala di forza dei gruppi donatori
2.2. Effetto del solvente.
Si ha grande influenza del solvente sulla distribuzione della densità elettronica e sulle proprietà ottinche lineari e non lineari [17]. Si fa riferimento ad un modello semplificato per introdurre l’effetto del solvente sulle proprietà ottiche non lineari. L’effetto solvatocromico, misurabile con la spettroscopia di assorbimento, si può razionalizzare ipotizzando una descrizione del sistema molecolare attraverso le due forme limite di risonanza [18] - vedi figura (3) - o per essere più precisi, in base al peso che esse assumono nella descrizione della funzione d’onda molecolare. Si può modificare l’equilibrio tra la forma chinoide (neutra) e la forma zwitterionica (a separazione di carica) variando l’intorno chimico, il solvente.
Secondo la natura del sistema molecolare in questione, l’effetto solvatocromico può indurre sposta-menti delle linee spettrali a frequenze maggiori o minori [18]. L’entità di tale energia di eccitazione ha diretta ripercussione sulle proprietà NLO. L’iperpolarizzabilità β è infatti proporzionale all’energia di assorbimento, come risulta anche dalle equazioni (??). Al di là del fatto che questo possa rappresentare un vantaggio o uno svantaggio (ciò dipende dall’applicazione tecnologica che si ha in mente), è senza ombra di dubbio necessario ed interessante razionalizzare questa correlazione.
2. EFFETTI NLO IN MOLECOLE ORGANICHE 22
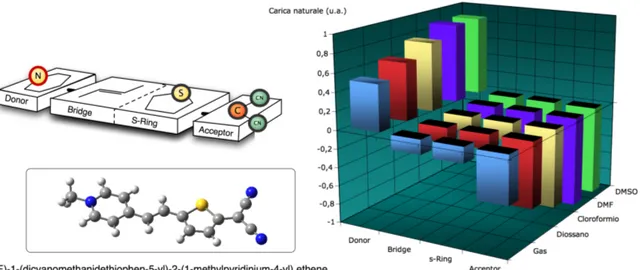
Figura 4. Nell’istogramma si descrive la carica elettrica su diverse zone di una delle molecole oggetto di questo lavoro di tesi. L’analisi di Popolazione adottata è l’NBO ed è condotta sullo stato fondamentale. Il metodo DFT, ivi utilizzato per ricavare la densità elettronica, adotta il funzionale chiamato CAM-B3LYP. Si può notare come all’aumento della polarità del solvente corrisponda un aumento della polarizzazione della molecola e quindi, secondo il nostro modello semplificato, un aumento del peso della forma limite di risonanza di tipo Zwitterionico.
Qualora si volesse utilizzare la risposta NLO di ordine II per la costruzione di un interruttore foto-modulabile, avere una consistente energia di eccitazione, e quindi una β grande, è fondamentale. La somiglianza tra la natura dello stato fondamentale e quella dello stato eccitato implica una minore energia di eccitazione: i due orbitali HOMO e LUMO, se simili, si trovano ad energie che non sono distanti l’una dall’altra. Per illustrare il comportamento di un interruttore fotomodulabile ci rifacciamo al caso ideale di una molecola in cui:
• la transizione HOMO→LUMO sia a trasferimento di carica;
• due delle componenti del momento di dipolo siano nulle ed una soltanto, quella lungo la direzione del CT, sia diversa da zero.
All’accensione di un campo elettrico statico F0,x le molecole risponderanno orientando i loro dipoli lungo
la direzione del campo. Una volta ottenuto quest’ordine orientazionale ciascuna molecola si comporterà come una piccola antenna nei confronti di una radiazione elettromagnetica polarizzata lungo l’asse x e che si sposti lungo l’asse z (direzione perpendicolare a campo elettrico statico). In molecole dotate di proprietà
3. TECNICHE SPERIMENTALI 23
ottiche non lineari, l’accensione del campo elettrico statico non ha la sola funzione dell’orientamento dei dipoli delle molecole. Per avere assorbimento la radiazione elettromagnetica esterna non dovrà più accoppiarsi con il momento di dipolo di transizione della molecola isolata. Il momento di dipolo di transizione avrà infatti due componenti: la prima sarà il dipolo di transizione della molecola isolata, e la seconda è il prodotto tra il tensore di polarizzabilità di transizione ed il campo elettrico statico esterno. Nella nostra trattazione semplificata si assume che in questo caso solo la componente lungo la direzione di F0,x sia diversa da zero, ←→αxx. La frequenza della radiazione eccitante sarà diversa nel caso in cui il
campo elettrico statico esterno sia acceso o spento. Tanto più le frequenze della radiazione differiscono l’una dall’altra e tanto minore sarà la difficoltà di implementazione di un dispositivo di questo tipo.
Lo studio di sistemi donatore-accettore con una adeguata descrizione del solvente può essere una valida linea guida per la sintesi. Il solvente, oltre ad avere la capacità di “sintonizzare” la risposta NLO, ha anche un ruolo fondamentale nel mimare le condizioni in cui si ha il rischio di formazione di dimeri, soprattutto quando le interazioni di tipo dipolo-dipolo apportano il contributo maggiore all’energia libera di formazione del dimero [12].
3. Tecniche sperimentali
La determinazione dell’efficienza NLO di ordine II di molecole organiche e polimeri si valuta in termini di suscettibilità di secondo ordine (χ(2)) e di prima iperpolarizzabilità (β). Il campione può essere sia in
fase solida (polveri o microfilm) che in forma di soluto in una fase liquida organica. Tra le tecniche più note elenchiamo:
(1) il metodo delle polveri o “Powder Method” [19]; (2) il metodo SHEW (Second Harmonic Evanescent Wave); (3) il metodo Phase-Matching;
(4) il metodo Hyper-Rayleigh-Scattering [20] [21];
(5) il metodo EOAM (ElectroOptical Absorption measurements) [22] [23];
(6) il metodo EFISH (Electric Field-Induced Second Harmonic Generation) .[24][22] .
In questo lavoro di tesi si utilizzano dati sperimentali ottenuti con le tecniche EFISH e EOAM per il confronto con i risultati numerici [7] [25] [26]. Si è deciso quindi di introdurre una concisa trattazione
3. TECNICHE SPERIMENTALI 24
della tecnica EOAM, oltre ad un cenno all’analisi delle polveri, un metodo semplice e in grado di fornire risultati interessanti (che però non è stato utilizzato in questo lavoro di tesi).
3.1. Forma contratta dei tensori di suscettibilità.
Il tensore di suscettibilità SHG fornisce una relazione tra la polarizzazione di ordine II ed i campi elettrici applicati. I suoi elementi formano un tensore di III rango le cui proprietà di simmetria sono le stesse possedute dal tensore piezoelettrico. È possibile ridurre un tensore di 27 elementi in una forma contratta di soli 18: (34) Px(2)(2ω) Py(2)(2ω) Pz(2)(2ω) = 1 2
χ(2)xxx χ(2)xyy χ(2)xzz χ(2)xyz χ(2)xxz χ(2)xxy χ(2)yxx χ(2)yyy χ(2)yzz χ(2)yyz χ(2)yxz χ(2)yxy χ(2)zxx χ(2)zyy χ(2)zzz χ(2)zyz χ(2)zxz χ(2)zxy
E2 x(ω, ω) E2 y(ω, ω) E2 z(ω, ω) 2Ey(ω)Ez(ω) 2Ex(ω)Ez(ω) 2Ex(ω)Ey(ω)
Per di più, grazie alle proprietà di simmetria di Kleinmann, soltanto 10 dei suoi elementi saranno necessari per avere una completa conoscenza
L’espressione precedente è una buona descrizione per il caso più generale. La simmetria della dispo-sizione spaziale delle molecole può infatti semplificare molto questo tensore, rendendo nulle molte delle sue componenti.
3.2. Metodo delle polveri di Kurtz.
È stato scelta la trattazione di questo metodo per la sua semplicità e velocità nella valutazione di proprietà NLO del secondo ordine di un materiale. Con questa tecnica si può subito verificare se il mate-riale è o non è inattivo al fenomeno SHG. Inoltre, in caso di attività SHG, si può anche determinare se le componenti del materiale che generano radiazioni di fase concordi (componenti phase-matchable) sono più o meno numerose di quelle che invece generano onde che provocano interferenza distruttiva (componenti non phase-matchable). Si può procedere con due livelli di accuratezza differenti: qualitativo e quantita-tivo. Nell’analisi di tipo qualitativo il campione viene ridotto in un fine strato di polveri (< 0, 2 mm), depositato su un sottile vetrino e tenuto fermo con del nastro trasparente. Il campione, nell’analisi che
3. TECNICHE SPERIMENTALI 25
Figura 5. Il grafico illustra la dipendenza della risposta SHG I(2ω) rispetto alla di-mensione media delle particelle in materiali Phase-Matchabel e Non-Phase-Matchable. Le unità di misura non sono riportate perchè trattasi di un grafico qualitativo. Al di sopra del parametro < rc >, la “coherence-length”, un materiale Phase-Matchable ha una risposta SHG la cui intensità non decresce all’aumentare di < r >.
si presta maggiormente ad un livello di accuratezza di tipo quantitativo, è stavolta composto da polveri setacciate, in modo tale da avere grani di determinata dimensione (tipicamente 75 µm < ∅ < 100 µm). Il tutto viene poi deposto in una cella di silice di noto spessore.
L’apparato sperimentale consiste in Laser Q-switched la cui radiazione viene irradiata sul campione attraverso un filtro UV-¯Vis. Un secondo filtro si trova poi tra campione e fotomoltiplicatore. Grazie a questo secondo filtro è possibile registrare soltanto l’intensità della radiazione di frequenza pari 2ω dovuta all’effetto SHG del campione.
Un grano di polvere sarà costituito da una certà quantità di cromofori attivi all’SHG. Aumentando la dimensione del grano aumenterà di conseguenza anche il numero di cromofori in esso presenti. Detto ciò è facile discriminare tra materiali Phase-Matchable e Non-Phase-Matchable. In figura (5) sono riportati i comportamenti di queste due tipologie di materiali. L’andamento della risposta SHG del campione permette il discrimine tra i due casi. All’aumentare delle dimensioni dei grani di polvere, in un materiale Non-Phase-Matchable abbiamo incoerenza di fase crescente tra le radiazioni generate. Ciò comporta una
3. TECNICHE SPERIMENTALI 26
interferenza distruttiva crescente (ci sono infatti sempre più cromofori) e pertanto una diminuzione della risposta SHG.
3.3. EOAM (ElectroOptical Absorption Measurements).
Il coefficiente di estinzione molare correla l’entità di assorbimento della radiazione luminosa con il numero di cromofori presenti all’interno di un campione. Operando un “controllo” sull’orientazione dei cromofori è possibile ricavare informazioni aggiuntive alla concentrazione molare. Il coefficiente di estinzione molare è infatti legato al momento di dipolo di transizione che, a sua volta, è correlato alla di-stribuzione elettronica dello stato di partenza (stato fondamentale della molecola in assenza di radiazione luminosa) e dello stato di arrivo (stato energetico eccitato indotto dalla radiazione luminosa). In questa tecnica il “controllo” è operato attraverso l’orientazione di un campo elettrico statico −→E0. Si misurano delle
differenze tra coefficienti di estinzione molare in presenza ed assenza di questo campo. Operativamente si procede pertanto alla misura del coefficiente di estinzione molare K (˜ν) in tre condizioni ambientali differenti:
(1) in assenza del campo elettrico statico esterno ⇒K (˜ν).
(2) con campo elettrico statico orientato parallelamente al vettore di polarizzazione della luce incidente, (φ = 0) ⇒ K−E→0"(˜ν)
(3) con campo elettrico statico orientato perpendicolarmente al vettore di polarizzazione della luce incidente, (φ = 90◦) ⇒K−E→0⊥(˜ν)
Il rapporto tra coefficiente di estinzione molare in presenza e assenza di −→E0è esponenzialmente correlato
al modulo del campo e all’orientazione di questo rispetto alla radiazione luminosa incidente. Nel caso in cui −→E0 sia piccolo è lecita l’espansione di Taylor di questo esponenziale:
(35) K −→ E0(φ, ˜ν) K (˜ν) = e L(φ,˜ν)−E→02 = 1 + L (φ, ˜ν) −→E 02+ . . .
Se l’assorbimento è dovuto ad una singola eccitazione elettronica, il coefficiente L (φ, ˜ν) è esprimibile attraverso una somma di contributi di forma nota e connessi ad osservabili molecolari:
3. TECNICHE SPERIMENTALI 27 dove: • r (φ) = 2−cos52(φ); • s (φ) = 3cos25(φ)−1; • t (˜ν) = 1 hc 'K(φ,˜ν) ˜ v (−1'∂K(φ,˜ν) ∂ ˜v ( φ; • u (˜ν) = 2h12c2 ' K(φ,˜ν) ˜ v (−1'∂2 K(φ,˜ν) ∂ ˜v2 ( φ;
Tramite D, E, F, G, H e I, che si ricavano attraverso un’analisi di regressione multilineare, si ottengono misure di momento di dipolo dello stato fondamentale e la variazione del momento di dipolo tra stato eccitato e stato fondamentale. Si esplicita di seguito la loro espressione più generica:
D = 1 kT ! R(1) · −→µgs"+ S(1) E =! 1 kT "2 -3 (←m→ex←gs· −→µgs) 2 − (−→µgs) 2. + 3 kT I! R(2) · −→µgs"+-!←→mex←gs· α · ←→mTex←gs " −T r(α)3 . + S(2)J F = 1 kT(−→µgs· ∆−→µex←gs) + ! R(1) · ∆−→µex←gs"+12T r(∆α) G = 1 kT (←m→ex←gs· −→µgs) (←m→ex←gs· ∆−→µex←gs) + 1 2 ! R(2) · ∆−→µex←gs"+21!←m→ex←gs· ∆α · ←m→ T ex←gs " H = ∆−→µex←gs I = (←m→ex←gs· ∆−→µex←gs)2
Il vettore ←m→ex←gs ha come elementi i versori della terna d’assi originata dalle tre componenti del momento di dipolo di transizione. I termini delle espressioni precedenti che sono piccoli e usualmente trascurati sono stati elisi graficamente. I tensori R(1) e R(2) hanno come elementi dei rapporti tra
polarizzabilità di transizione e momento di dipolo di transizione, mentre i tensori S(1)e S(2)sono di rango
superiore e coinvolgono rapporti con al numeratore l’iperpolarizzabilità di transizione. Occorre valutare questi tensori caso per caso. In un tipico sistema molecolare “push-pull”, il momento di dipolo ha una componente di entità di gran lunga superiore rispetto alle altre, diciamo essere “z” questa direzione. L’ elemento della matrice di polarizzabilità di transizione riferito a z sarà molto più importante degli altri elementi diagonali e, a maggior ragione, di quelli non diagonali. Le matrici R(1)e R(2)si semplificano
3. TECNICHE SPERIMENTALI 28
perciò nei due vettori −→R(1)e −→R(2) con le due componenti −→R(1)
z e −→R(2)z più grandi delle altre. I tensori S(1) e S(2) sono solitamente trascurati, ma in un sistema push-pull si riducono a due quantità scalari14.
I termini H ed I sono trascurati totalmente insieme ad altre componenti di D, E, F e G. Ciò che si ottiene alla fine dell’analisi di multiregressione sono delle quantità scalari, tramite cui si ricava il valore del momento di dipolo dello stato fondamentale e dello stato eccitato.
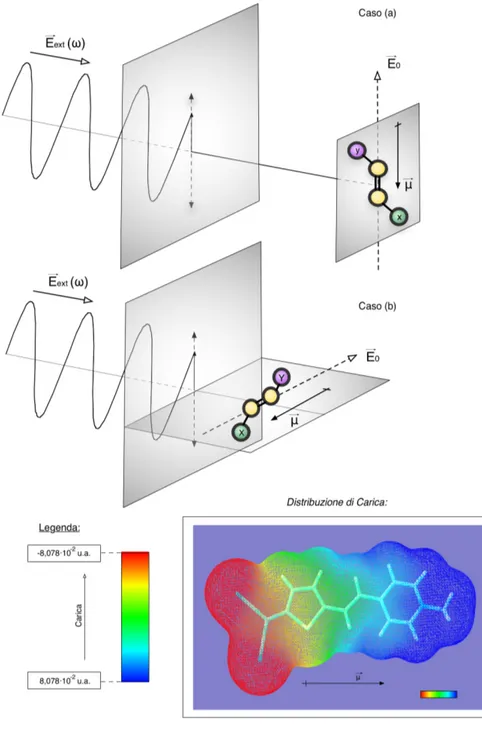
Per avere un’idea di come viene condotto l’esperimento si faccia riferimento alla figura (6) a pag.29. In figura viene simbolicamente rappresentato un sistema a trasferimento di carica. Il campo elettrico −→E0
ha un ruolo duplice: da una parte ha la funzione di orientare il dipolo della molecola lungo le sue linee di campo, e dall’altra quella di modificare attraverso un termine aggiuntivo l’Hamiltoniana del sistema isolato. Nell’espansione del momento di dipolo di transizione compaiono i tensori di polarizzabilità ed iperpolarizzabilità di transizione. Questa espansione ricorda quella del momento di dipolo (19) a pag.13:
(37) ←→µ−→E0
ex←gs= ←→µex←gs+ ←→αex←gs−→E0+
1
2!←→β ex←gs−→E0−→E0+ . . .
In assenza di campo elettrico statico si verifica assorbimento in caso di accoppiamento tra ←→µex←gse la radiazione luminosa esterna. Qualora il campo elettrico statico sia acceso, la radiazione luminosa dovrà accoppiarsi con un nuovo operatore dipolo di transizione: ←→µ→−E0
ex←gs= '
←→µex←gs+ ←→αex←gs−→E0(. Nel caso (a) riportato in figura (6) è la componente più grande di questo momento di dipolo di transizione indotto ad accoppiarsi con la radiazione luminosa −→Eext(ω). Il sistema a trasferimento di carica si comporta come fosse una vera e propria antenna molecolare, la densità elettronica viene spostata da una estremità all’altra della molecola dal campo oscillante.
La media della somma delle due rimanenti componenti del momento di dipolo di transizione, quelle di entità più piccola, saranno poi ricavate con l’ulteriore esperimento in cui il campo elettrico statico è orientato perpendicolarmente al vettore di polarizzazione della luce incidente, caso (b) della figura (6). I due casi (a) e (b) rappresentano le due orientazioni, con φ = 0 e φ = 90◦, tra −→E
ext(ω) e −→E0.
14Ad esempio, nel caso di un sistema push-pull con dipolo prevalentemente orientato lungo la direzione z, il tensoreS(1)¨ si riduce ad un solo elemento di entità non nulla:
• S(1)→ S(1)
3. TECNICHE SPERIMENTALI 29
Figura 6. Interazione tra la radiazione ed un sistema molecolare a trasferimento di carica. Le estremità della molecola, donatore ed accettore, sono indicate rispettivamente con un cerchietto viola contrassegnato da una “Y” ed un cerchietto verde contrassegnato da una “X”. Si distinguono due casi: (a) campo elettrico statico −→E0 parallelo alla
radia-zione incidente −→Eext(ω), (b) campo elettrico statico −→E0 perpendicolare alla radiazione
incidente −→Eext(ω). In basso viene poi riportata, a titolo d’esempio, la distribuzione elet-tronica ottenuta con analisi di popolazione NBO di una delle molecole oggetto di questo lavoro di tesi.
3. TECNICHE SPERIMENTALI 30
Le polarizzabilità ed iperpolarizzabilità statiche, α0e β0, si calcolano ad un livello di approssimazione
TLM15utilizzando le osservabili misurate spettroscopicamente e le seguenti equazioni:
(38) α0= ←→µ2 ex←gsλex←gs hc (39) β0= 6←→µ2 ex←gs∆µex←gsλ2ex←gs (hc)2
15TLM è un acronimo di Two State Model (Modello a due stati). Questa approssimazione viene trattata nella parte III di
Parte 3
4. Descrizione QM dei sistemi molecolari in soluzione
Si consideri valida l’approssimazione di “Born Oppenheimer”. Separando la funzione d’onda in una parte nucleare ed una parte elettronica si costruisce una Hamiltoniana effettiva che dipenderà dalle coordinate di tutti quanti gli elettroni {q1· · · qi· · · qn} e parametricamente dalle coordinante dei nuclei {Q1· · · Qi· · · QN}.
L’idea di base è quella di introdurre un termine aggiuntivo ˆVint all’Hamiltoniana del sistema mole-colare isolato. Così facendo tale sistema diventerà il “soluto” in virtù di questo termine addizionale che tiene conto della interazione del solvente sul soluto. L’operatore ˆVint dipenderà sia dalla distribuzione elettronica e nucleare del soluto che dalla distribuzione delle molecole di solvente (che indico con il termine ρsolvente).
(40) Hˆsoluto({qi}; {Qi}) = ˆH(0)soluto({qi}; {Qi}) + ˆVint({qi}, {Qi}, ρsolvente)
(41) Hˆsoluto({qi}; {Qi}) | ψ({qi}; {Qi}) # = E({Qi}) | ψ({qi}; {Qi}) #
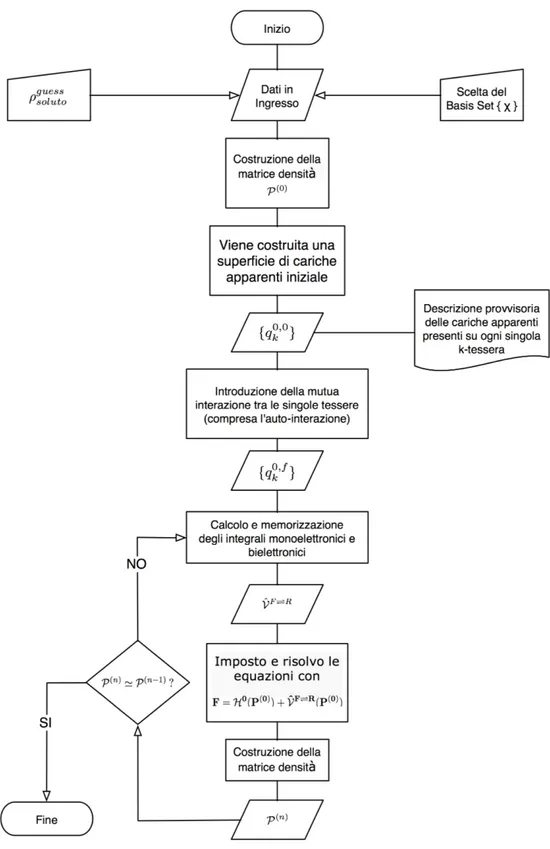
Tutti i modelli continui sono stati elaborati cercando di ottenere un buon compromesso tra una accurata descrizione per il sistema soluto e l’esigenza di semplificare l’intorno chimico. L’ambiente intorno al “soluto” è costituito da un grande insieme di molecole, il “solvente”, e viene descritto come fosse un mezzo dielettrico continuo dotato di una certa costante dielettrica [27][28]. Questo approccio, tipico dei modelli di “focalizzazione” (focused models), permette di focalizzare l’attenzione su una piccola parte del sistema, nel nostro caso il soluto. E’ importante sottolineare che nel gergo tecnico PCM il termine “soluto” ha un significato generalizzato: con esso si indica soltanto una porzione del sistema che viene spesso descritta rigorosamente con metodi QM ab-initio. Si può considerare soluto anche una piccola imperfezione di un cristallo o il sito attivo di un enzima.
4. DESCRIZIONE QM DEI SISTEMI MOLECOLARI IN SOLUZIONE 33
L’Hamiltoniana del sistema totale, ˆHF R({f}, {r}), si suddivide in due parti: con ˆHF({f}) si descrive la porzione “soluto” (l’apice F sta per Focused), mentre con ˆHR({r}) si tiene conto di tutto ciò che compone il sistema e non è soluto (l’apice R sta per “Rest”) .
(42) HˆF R({f}, {r}) = ˆ
HF({f}) + ˆ
HR({r}) + ˆ
HF⇔R({f}, {r})
Con {f} ed {r} si indica l’insieme dei gradi di libertà della parte F ed R, mentre l’ultimo termine a secondo membro dell’espressione precedente rappresenta il contributo di mutua interazione tra le due parti. Non si è interessati ad una dettagliata descrizione del sistema “R”, ma soltanto ad una buona descrizione delle interazioni. Si elimina quindi il termine ˆHR({r}) e si ottiene una Hamiltoniana efficace:
(43) HˆF Ref f({f}, {r}) = ˆHF({f}) + ˆHF⇔R({f}, {r})
Un ulteriore e più consistente approssimazione consiste nella pressoché completa eliminazione dei gra-di gra-di libertà {r}. A tal uopo si introduce una funzione gra-di risposta del solvente in luogo della gra-dipendenza di ˆHF⇔R({f}, {r}) dal set di coordinate {r}. L’operatore che approssimerà l’ultimo termine dell’espres-sione precedente avrà sempre una dipendenza dall’intero set di coordinate {f} del soluto, ma presenterà il non indifferente vantaggio di dipendere ulteriormente da un massimo di due vettori posizione −→red−→r) attraverso una dipendenza funzionale da Q(−→r ,−→r)), la funzione di risposta del solvente.
(44) HˆF Ref f({f}, {r}) = ˆHF({f}) + ˆVF⇔R[{f}, Q(−→r , − → r))]
L’interazione tra soluto e solvente è composta da contributi di vario genere, idealmente tutti presenti all’interno della funzione di risposta Q(−→r ,−→r)). Limitandoci ai soli contributi elettrostatici, la funzione risposta è analoga a quella ottenibile da un dielettrico una volta acceso un campo elettrico esterno −→E.
4. DESCRIZIONE QM DEI SISTEMI MOLECOLARI IN SOLUZIONE 34
In elettrostatica classica la funzione di polarizzazione −→P di un mezzo dielettrico immerso in una zona dove è presente un campo elettrico è proporzionale al campo elettrico stesso:
(45) −→P = -− 1
4π −→E
Con −→P si descrive la risposta del dielettrico al campo elettrico esterno, per ottenerla si è reso necessario introdurre il concetto di permettività dielettrica -. Importante è capire che includendo la permettività dielettrica - all’interno della funzione risposta del solvente si ha la possibilità di descrivere le interazioni elettrostatiche soluto-solvente eliminando pressoché completamente tutti quanti i gradi di libertà {r}.
Una generalizzazione del concetto di “Focused Model” è rappresentato dal “Layering”. Con “Layering” si intende la divisione del sistema in regioni concentriche. Ogni “Layer” rappresenta una parte di sistema trattata a differente livello di accuratezza. Nel più semplice schema di “Layering” si hanno due sole regioni: una può essere ad esempio rappresentata dal soluto descritto a livello QM, mentre l’altra può essere rappresentato dal solvente descritto a livello continuo.
Adesso si elencano tutte le caratteristiche del modello continuo di base, modello che viene preso come punto di partenza, da cui ci si discosterà sia introducendo ulteriori rifiniture e sia trascurando alcuni termini:
• il soluto deve essere descritto omogeneamente ad un livello QM;
• le interazioni soluto-solvente sono limitate a quelle di natura elettrostatica; • il sistema modello è una soluzione molto diluita e con un solo soluto;
• viene presa in considerazione solo la distribuzione elettronica dello stato fondamentale del soluto; • il solvente è isotropo e all’equilibrio con temperatura e pressione fissata.
In tutti quanti i modelli continui notevole importanza è rivestita dal concetto di cavità. Il soluto viene idealmente inserito all’interno di una cavità vuota all’interno del mezzo continuo. Critiche sono la di-mensione e la forma della cavità, tanto che molti modelli differiscono proprio per questo. L’ideale è avere una cavità che approssima al meglio lo spazio fisico occupato dal soluto. Un ottimo compromesso tra velocità di calcolo e accuratezza si basa sulla definizione di cavità come sovrapposizione di sfere atomiche con raggi vicini a quelli di van der Walls (uno dei set di raggi di vdW più utilizzato è quello definito da Bondi[29]). Per la costruzione della cavità uno dei metodi attualmente implementato nei moderni
5. SCF HARTREE-FOCK (METODO HF) 35
programmi di calcolo è conosciuto come GEPOL [30] [31] [32].
Prima di entrare nel dettaglio dei modelli continui di solvatazione occorre presentare una breve descrizione dei metodi QM usati in questo lavoro di tesi per la determinazione di energia e proprietà molecolari. Il formalismo del metodo Hatree-Fock (HF) viene presentato in maniera più estesa rispetto alla Teoria del Funzionale Densità (DFT) ed al Metodo Møller-Plesset (metodo MP). Con il metodo Hartree-Fock si esemplificano infatti sia il modello PCM che il metodo della risposta lineare.
5. SCF Hartree-Fock (Metodo HF)
Dato un insieme di k orbitali spaziali ortonormali {ψi} è possibile costruire un insieme di 2k spin-orbitali {χi} moltiplicando ciascuna funzione ψi per le due funzioni di spin α(ω) e β(ω). Questi spin-orbitali dipendono dalle coordinate spaziali r e dai valori di spin indicati con ω:
(46) {ψi(r)α(ω)} i = 1, 2, . . . , k
{ψi(r)β(ω)} i = 1, 2, . . . , k
Lo stato |ψ# di un sistema si esprime attraverso un determinante di Slater al fine di ottenere funzioni d’onda che soddisfino il principio di esclusione di Pauli. Un determinante di Slater è generalmente indicato con un vettore riga contente tutti gli elementi diagonali di una matrice di Slater, ed in caso di N elettroni si ha: (47) |ψ# = |χ1(1)χ2(2) . . . χ2k(N)# = $ 1 N ! %1 2 Det K K K K K K K K K K K K K χ1(1) χ2(1) . . . χ2k(1) χ1(2) χ2(2) . . . χ2k(2) ... ... ... ... χ1(N) χ2(N) . . . χ2k(N) K K K K K K K K K K K K K
L’energia di |ψ# è ricavabile come valore di aspettazione dell’operatore ˆH, l’operatore Hamiltoniana.
(48) E0= $ψ| ˆH |ψ# = N / i $χi| ˆh |χi# + 1 2 N / i N / j $χiχj| |χiχj# = N / i hi+1 2 N / i N / j (Jij− Kij)
5. SCF HARTREE-FOCK (METODO HF) 36 dove: • hi=Lχ∗i(1) ' −1 2∇2(1) − <N uclei Q 1 rQ1 ( χi(1)dr(1) ;
• l’ultimo termine è l’energia di interazione elettronica Coulombiana e di Scambio.
Si è interessati ad un singolo determinante che sia la migliore approssimazione possibile dello stato fondamentale di un sistema descritto dall’ Hamiltoniana elettronica ˆH. Per fare questo occorre che gli spin-orbitali siano tali da ottenere un determinante che corrisponda ad uno stato energetico di minimo (principio variazionale). Per ricavare l’energia di un signolo determinante si procede ad una risoluzione numerica dell’equazione di Schrödinger con il metodo dei campi autocoerenti (SCF) ideato da D. R. Hartree e perfezionato poi da V. Fock e J. C. Slater con l’introduzione degli effetti dello scambio fra gli elettroni [33][6][34].
All’interno di questa approssimazione il termine Coulombiano diventa:
(49) Jij = M M χ∗i(1)χ∗j(2) 1 r12 χi(1)χj(2)dr(1)dr(2)
Anche per l’operatore di scambio si ha una procedimento analogo, la differenza sostanziale è che quest’ultimo non ha una interpretazione ricavabile dai concetti di fisica classica, ma come conseguenza del principio di antisimmetria:
(50) Kij = M M χ∗i(1)χ∗j(2) 1 r12χi (2)χj(1)dr(1)dr(2)
L’energia del sistema ha una dipendenza funzionale dalla funzione d’onda sistema. L’applicazione del metodo variazionale (e cioè la minimizzazione dell’energia (48) rispetto agli spin-orbitali) porta alla semplificazione del problema iniziale della determinazione di una funzione d’onda di N elettroni in quello di N problemi monoelettronici del tipo:
5. SCF HARTREE-FOCK (METODO HF) 37
dove l’operatore di Fock è definito come:
(52) ˆh(1) + N / j ˆ Jj(1) − N / j ˆ Kj(1) = ˆf (1)
La risoluzione algebrica passa attraverso uno sviluppo della parte spaziale degli spin-orbitali utiliz-zando un insieme di M funzioni di base {φM}:
(53) χi=
M / u=1
Cuiφu= C1iφ1+ C2iφ2+ C3iφ4+ · · · + CM iφM
Sostituendo tale sviluppo in equazione (51), moltiplicando per ognuna delle M funzioni φ∗ v(1) ed integrando si ottiene il set di equazioni Roothan:
(54) M / u=1 Cui$φv(1)| ˆfi(1) |φu(1)# = -i M / u=1 Cui$φv(1) |φu(1)#
Alla fine di questa procedura si ottiene un set di equazioni che in notazione compatta assumono il seguente aspetto:
(55) FC =
SC-dove:
• la matrice F è somma di contributi monoelettronici e bielettronici Fvu= $φv| ˆf|φu#; • la matrice S è la matrice di sovrapposizione di elementi Svu= $φv |φu#.
La densità di carica si esprime come:
(56) ρ(r) =
N /
i
5. SCF HARTREE-FOCK (METODO HF) 38
che per un sistema a guscio chiuso diventa:
(57) ρ(r) = 2
N/2 / i
|ψi(r)|2
Inserendo l’espansione (53) in (58) si ottiene l’espressione della matrice densità P:
(58) ρ(r) = M / u M / v 2 N/2 / i CuiC∗vi φuφ∗v= M / u M / v [Puv] φuφ∗v Avendo introdotto la matrice P gli elementi della matrice F diventano::
(59) Flm = $φl| ˆh |φm# + N / u N / v PuvGlmuv dove: Glmuv = 2 M M φ∗l(1)φ∗u(2) 1 r12 φv(2)φv(2)φm(1)dr(1)dr(2)−2 M M φ∗l(1)φ∗u(2) 1 r12 φv(2)φm(2)φv(1)dr(1)dr(2)
Il primo termine a secondo membro di (59) resta fisso nel corso delle iterazioni, mentre il secondo termine a secondo membro viene aggiornato ad ogni ciclo poiché ad ogni iterazione viene aggiornata la matrice densità P.
Alla fine di questo processo interativo si ottengono M orbitali spaziali dotati ciascuno di una certa energia orbitalica -a. I primi N/2 saranno utilizzati per costruire il determinante che chiameremo
K KψHF
0
6. TEORIA DEL FUNZIONALE DENSITÀ (DFT) 39
Operando sul determinanteKKψHF
0
N
con l’Hamiltoniana ˆH del sistema si ottiene l’energia HF:
(60) HˆKKψHF0 N= EHF 0 K KψHF 0 N (61) E0HF = 2 N/2 / a -a− N/2 / a N/2 / b (2Jab− Kab)
6. Teoria del funzionale densità (DFT)
La teoria del funzionale densità si basa sui due teoremi di Hohenerg e Kohn: l’energia elettronica dello stato fondamentale è determinato dalla densità di carica [35]. L’approccio basato sulla densità di carica rispetto a quello basato sulla funzione d’onda ha dei vantaggi quando aumenta la complessità del sistema in esame. La densità elettronica dipende da solo tre coordinate indipendentemente dalle dimensioni del sistema in esame. Anche se la corrispondenza tra densità di carica ed energia di stato fondamentale esiste, tuttavia non si conosce la forma del funzionale che connette queste due quantità.
Supponendo di conoscere la forma di questo funzionale si può ricondurre l’approccio DFT ad un siste-ma di equazioni che ricordano l’approccio Hartree-Fock. Le equazioni KS16da risolvere sono riconducibili
alla (51): (62) − 1 2∇21− N uclei/ Q ZQ e 2 rQ1 + M ρ(r 2) r12 dr2+ VχC(r1) K KϕKS i (r1)N= -iKKϕKSi (r1)N dove:
• i primi due termini sono un analogo dell’operatore monoelettronico ˆh del metodo Hartree Fock; • il termineL ρ(r2)
r12 dr2tiene conto dell’interazione Coulombiana;
• il termine VχC è il potenziale di scambio correlazione, la derivata funzionale dell’energia di correlazione e di scambio EχC ;
6. TEORIA DEL FUNZIONALE DENSITÀ (DFT) 40
• KKϕKS i
N
.è ll’orbitale di Kohn-Sham (KS) la cui relativa energia orbitalica è -i. L’equivalente della densità di carica definita in (58) è:
(63) ρ(r) = 2 N/2 / i=1 K KϕKS i (r1)KK 2
Il set di equazioni Kohn-Sham si risolve in modalità autocoerente, analogamente al set di equazioni di Roothaan (55). Si parte con un densità di carica ρguess. Una volta stabilito il funzionale EχC da utilizzare si opera una derivazione rispetto alla densità di carica iniziale per ottenere VχC(r). A questo punto si risolve il sistema di equazioni KS per ottenere un nuovo set {ϕKS
i } di orbitali KS con i quali costruire una nuova densità di carica ρ)(r). Si reitera il processo fino a quando densità di carica ed energia di scambio-correlazione non convergano entro un certo intervallo di tolleranza. Analogamente a quanto fatto per il metodo Hartree-Fock, gli orbitali KS possono essere espressi come espansione in serie di un insieme di funzioni di base e la risoluzione delle equazioni (62) si riduce a trovare i coefficienti di tale espansione.
Una volta nota la distribuzione di carica finale ρf inale(r) si ricava l’energia come funzionale della densità di carica: (64) E[ρ] =−1 2 n / i M ϕ∗i(r1)∇2ϕ(r1)dr1− N uc/ Q M ZQρ(r 1) rQ1 dr1+ 1 2 M ρ(r 1)ρ(r2) r12 dr1dr2+ EχC[ρ]
Punto cruciale è la scelta della forma del funzionale EχC. All’interno di questo lavoro di tesi si è fatto uso del funzionale EB3LY P
χC e di una sua evoluzione per i sistemi a trasferimento di carica chiamata CAM-B3LYP illustrata nella parte IV di questo elaborato di tesi.
(65) EχCB3LY P = AEχDirac+ (1 − A)EχCHF + BEχBecke+ (1 − C)ECV W N+ CECLY P dove:
• A=0,2 B=0,72 e C=0,19 sono parametri di fitting ottenuti in fase di sviluppo del funzionale; • il funzionale locale EDirac
χ [ρ] = −CxL ρ
4 3(r)dr;
• il funzionale non locale EBecke88 χ [ρ] = 12 L ρ(r)-β χ[ρ, ∇ρ, r] dove -βχ[ρ, ∇ρ, r] = −βρ 1 3 |∇ρ| 2 ρ83(1+6βsinh−1x) e β = 0, 0042 è un parametro di regressione ottenuto mediante fitting su sei gas nobili;