1
SOMMARIO
INTRODUZIONE ... 3
CAPITOLO 1 - LA CARDIOPATIA ISCHEMICA: NOZIONI FISIOPATOLOGICHE E QUADRI CLINICI ... 4
1.1 La cardiopatia ischemica ... 4
1.2 Fisiopatologia ... 5
1.2.1. Regolazione del circolo coronarico ... 5
1.2.2 Il consumo miocardico di O2 (MVO2) ... 6
1.2.3 Il ruolo dell’aterosclerosi ... 8
1.2.4 Gli effetti dell’ischemia ... 11
1.3 Il ruolo della placca “vulnerabile” ... 12
1.4 Gli aspetti clinici dell’ischemia cardiaca ... 15
1.4.1 Il sintomo dolore ... 15
1.4.2 Angina Pectoris Stabile ... 17
1.4.3 Angina Pectoris Instabile ... 18
1.5 L’infarto acuto del miocardio (IMA) ... 19
1.5.1 Patogenesi dell’infarto ... 19
1.5.2 Classificazione degli infarti: N-STEMI e STEMI ... 21
1.6 Diagnostica Strumentale ... 24
1.6.1 Elettrocardiogramma ... 24
1.6.2 Marker cardiaci sierici ... 28
1.6.3 Imaging cardiaco ... 31 1.7 Complicanze dell’IMA... 33 1.7.1 Complicanze aritmiche... 33 1.7.2 Complicanze emodinamiche ... 37 1.7.3 Complicanze ischemiche ... 40 1.7.4 Altre complicanze ... 41
2
CAPITOLO 2 – LA TERAPIA RIPERFUSIVA DELL’IMA: REVISIONE DELLA
LETTERATURA ED APPROCCIO PRATICO ... 44
2.1 Le basi storiche dell’approccio riperfusivo ... 44
2.2 La terapia trombolitica ... 45
2.2.1 Meccanismo d’azione ... 45
2.2.2 l’approccio fibrinolitico endovenoso: farmaci ed evoluzione ... 46
2.3 Il confronto tra fibrinolisi e PPCI ... 53
2.4 Il “delay” nelle strategie riperfusive ... 56
2.5 la fibrinolisi extraospedaliera ... 60
2.6 La strategia riperfusiva migliore: PPCI facilitata e la “rescue PCI” ... 65
2.7 Le attuali linee guida internazionali sulle strategie riperfusive. ... 70
CAPITOLO 3 – LA REALTÀ TOSCANA DELLA RIPERFUSIONE E I DIFFERENTI APPROCCI: NETWORK E PROTOCOLLI STEMI ... 73
3.1 L’epidemiologia Toscana dell’IMA ... 73
3.1.1 Il registro IMA in Toscana ... 76
3.2 I network STEMI ... 78
3.3 La terapia fibrinolitica in Toscana: dove e come ... 79
3.3.1 Area Vasta Nord-Ovest ... 80
3.3.2 Area Vasta Centro ... 82
3.3.3 Area Vasta Sud-Est ... 83
CAPITOLO 4 – CONCLUSIONI ... 86
3
INTRODUZIONE
Le sindromi coronariche acute sono ampiamente diffuse nei paesi
industrializ-zati, e la patologia che vi sottende, l’aterosclerosi, ha un’incidenza molto alta nei paesi cosiddetti “occidentali”; l’infarto miocardico acuto rappresenta uno dei quadri estremi di questo spettro di situazioni cliniche, attestandosi tra le
prime cause di morte, specialmente in paesi come America ed Europa.
L’infarto miocardico acuto con sopraslivellamento del tratto ST è un quadro diagnostico particolare, che per la fisiopatologia che vi sottende vede nelle
strategie di riperfusione coronarica la miglior soluzione a oggi possibile per
garantire la salvezza del paziente.
Questa tesi si prefigge lo scopo di fornire una visione d’insieme sulle strategie
riperfusive, con particolare riguardo per quelle amministrabili in modo più
precoce, come la trombolisi endovenosa; per fare questo si avvale di una
revi-sione della letteratura principale riguardante l’argomento, e delle più recenti linee guida internazionali sull’infarto del miocardio. Altresì questa tesi vuole fornire una visione complessiva della realtà Toscana riguardo all’infarto e co-me tale patologia sia inquadrata e trattata dalle realtà assistenziali presenti, e in
4
CAPITOLO 1 - LA CARDIOPATIA ISCHEMICA: NOZIONI
FISIO-PATOLOGICHE E QUADRI CLINICI
1.1 La cardiopatia ischemica
Il termine cardiopatia ischemica definisce uno spettro di malattie a diversa
e-ziologia, in cui il fattore fisiopatologico unificante è rappresentato da uno
squilibrio tra la richiesta metabolica e l’apporto di ossigeno a una porzione del
miocardio. La più comune causa di cardiopatia ischemica è la malattia
atero-sclerotica di una o più arterie coronarie epicardiche, sufficiente a causare un’inadeguata perfusione del miocardio rifornito dal letto vascolare coinvolto. La cardiopatia ischemica è una delle principali cause di mortalità e disabilità e
a essa è associata una maggiore spesa sanitaria rispetto ad altre patologie
dif-fuse nei Paesi industrializzati.
Negli Stati Uniti, la cardiopatia ischemica è la malattia cronica più invalidante,
e ne sono afflitti 13 milioni di persone, più di 6 milioni soffrono di angina
pec-toris e più di 7 milioni hanno avuto un infarto miocardico. Le malattie
cardio-vascolari rappresentano ancora oggi la principale causa di morte nel nostro
pa-ese, essendo responsabili del 44% di tutti i decessi. In particolare la
cardiopa-tia ischemica è la prima causa di morte in Italia, rendendo conto del 28% di
tutte le morti. I dati del Registro Nazionale degli Eventi Coronarici e
Cerebro-vascolari mostrano un quadro sostanzialmente omogeneo in tutta Italia, con
5
donne: tra i pazienti colpiti da infarto miocardico acuto nell'età compresa fra
35 e 74 anni, 3 uomini su 10 e 4 donne su 10 muoiono entro 28 giorni dall’esordio dei sintomi, principalmente fuori dall’ospedale, prima di poter es-sere adeguatamente curati.
1.2 Fisiopatologia
Due sono i fattori che intervengono nella genesi dell’ischemia miocardica: da una parte la riduzione del flusso coronarico e dall’altra l’aumento del consumo miocardico di ossigeno (MVO2).
1.2.1. Regolazione del circolo coronarico
I vasi coronarici possono essere suddivisi in: vasi di conduttanza (grossi rami
epicardici e loro diramazioni principali) e vasi di resistenza (rami
intramiocar-dici e arteriole). Il sangue scorre attraverso le arterie coronarie secondo un
flusso fasico, in particolare durante la fase di diastole, mentre in sistole i rami
intramurali sono virtualmente occlusi dalla contrazione ventricolare; gli strati
subendocardici sono generalmente i più esposti all’ischemia, soprattutto
per-ché maggiormente esposti alla pressione telediastolica endocavitaria. A questo
proposito va ricordato che la tachicardia predispone allo sviluppo d’ischemia,
poiché, accorciando la durata della diastole, diminuisce il tempo disponibile al
flusso coronarico.
I fattori che regolano il circolo coronarico sono molteplici, e tra essi il più
6
cardiaco. Quando questa aumenta, si determinano idrolisi di ATP e
conse-guente liberazione di adenosina nell’interstizio; essa induce una
vasodilatazio-ne soprattutto a livello dei vasi di resistenza, con un conseguente aumento del
flusso coronarico proporzionale alle maggiori richieste metaboliche. L’adenosina non è la sola sostanza implicata nel processo, ma è verosimilmen-te la principale; tra gli altri elementi che contribuiscono all’omeostasi della circolazione coronarica, sono particolarmente importanti quelli di tipo
neu-roumorale, quali per esempio le interazioni simpatico-parasimpatico e le
pro-staglandine.
In condizioni basali, nel cuore l’estrazione di O2 è molto alta (circa il 70%): ne consegue che se la domanda metabolica aumenta, ad esempio per attività
fisica o stress di varia natura, l’unico meccanismo di compenso è
rappresenta-to da un proporzionale aumenrappresenta-to del flusso coronarico, che si attua attraverso la
vasodilatazione del distretto coronarico arteriolare (vasi di resistenza), e questa
capacità massima di vasodilatazione secondaria a uno stimolo metabolico è
definita riserva coronarica.
1.2.2 Il consumo miocardico di O2 (MVO2)
Il cuore è un organo aerobio e, in condizioni fisiologiche, la quantità di ener-gia richiesta per i processi metabolici basali e per l’attivazione elettrica dell’organo è minima; i principali determinanti del consumo miocardico di O2 sono invece la frequenza cardiaca, la contrattilità e la tensione di parete. È in-tuitivo che l’aumento della frequenza cardiaca aumenti il MVO2, inoltre la
ta-7
chicardia agisce anche riducendo il flusso coronarico; anche per la contrattilità
è palese che quanto questa sia maggiore, tanto più alto è il consumo di O2.
Il terzo fattore è la tensione della parete miocardica, che dipende direttamente da due elementi: la pressione sviluppata all’interno della camera cardiaca, de-terminata dalle resistenze all’eiezione del sangue, e il raggio medio della cavi-tà, determinato dal riempimento della stessa (cioè sarà maggiore quanto più
al-to è il rial-torno venoso o precarico). Quindi la tensione parietale dipende tanal-to
dalla pressione endocavitaria quanto dal precarico; in pratica essa aumenta sia
quando aumentano le resistenze, sia quando aumenta il ritorno venoso. Inoltre
un aumento della tensione della parete di una cavità cardiaca (postcarico) de-termina la sua ipertrofia e quindi l’aumento della massa miocardica, cui con-segue un incremento del consumo di O2.
L’instaurarsi dell’ipertrofia ha, tuttavia, anche un effetto indiretto favorevole, tendente a ridurre il consumo di O2: infatti, essa tende a ridurre il postcarico,
essendo questo inversamente proporzionale allo spessore della parete. Tuttavia quest’effetto favorevole sul consumo di O2 è annullato, di fatto, dall’aumento della massa: infatti, lo spessore della parete è proporzionale alla radice cubica
della sua massa, quindi lo spessore si accresce molto meno della massa
mio-cardica coinvolta e per questo motivo l’effetto netto dell’ipertrofia è un
au-mento del consumo di O2.
In clinica non è possibile rilevare tutte queste variabili; alcune di esse tuttavia
8
sistolica. Il prodotto tra queste due variabili (FC × PA sistolica) × 10–3 si chiama doppio prodotto ed è l’indice di MVO2 utilizzato nella pratica clinica.
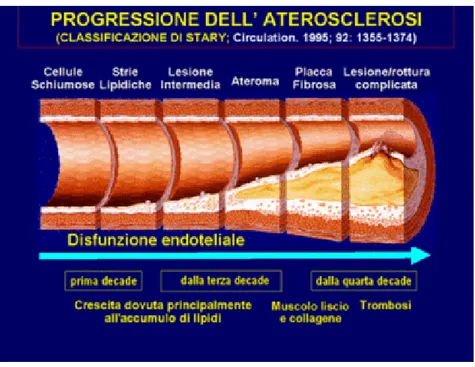
1.2.3 Il ruolo dell’aterosclerosi
Nella grande maggioranza dei casi la patologia ischemica sottende alla
presen-za di un processo aterosclerotico al livello dei vasi coronarici. Tale patologia è
condizionata dalla presenza di numerosi fattori di rischio, che agiscono in mo-do sinergico a formare un danno alle normali funzioni dell’enmo-dotelio vasale: elevati livelli plasmatici di lipoproteine LDL, bassi livelli di lipoproteine
HDL, fumo di sigaretta, ipertensione e diabete mellito sono i principali
re-sponsabili di questi fenomeni lesivi endoteliali. L’aterosclerosi coronarica
spesso inizia a svilupparsi prima dei 20 anni ed è diffusa anche tra gli adulti
asintomatici nel corso della vita.
La lesione intimale consente la deposizione di acidi grassi (nella forma di
li-poproteine LDL ossidate), che sono captati dai macrofagi tramite i loro recet-tori “scavenger”, internalizzati e digeriti. In caso di eccesso di lipidi i macro-fagi possono subire una degenerazione, divenendo cellule schiumose e
libe-rando fattori pro-infiammatori; l’accumulo di queste cellule schiumose
avvie-ne lungo liavvie-nee, che prendono il nome di strie lipidiche. Nella sede di
forma-zione di queste strie si ha il richiamo di cellule infiammatorie, come monociti
e linfociti, e il rilascio di altri fattori infiammatori e chemiotattici, con
deposi-zione di altre cellule infiammatorie, richiamo di piastrine e l’innesco di una
cascata infiammatoria. La morte di monociti e macrofagi nella sede della
9
e consentono la migrazione di cellule muscolari lisce dalla media nell’ambito dell’intima; tali cellule agiscono con fini riparativi, differenziandosi in fibro-blasti e depositando matrice extracellulare e collagene, costituendo il
cappuc-cio fibroso della placca. Si ha così la formazione della placca ateromasica vera
e propria, che istologicamente è costituita da un accumulo di cellule muscolari
lisce proliferanti, fibre connettivali e di matrice connettivale, che costituiscono
gran parte della componente superficiale (il cosiddetto cappuccio fibroso), e al
centro una deposizione di lipidi che si presentano come una massa
disorganiz-zata, alla cui formazione contribuiscono cristalli di colesterolo, residui
cellula-ri e quantità vacellula-riabili di fibcellula-rina e di altre proteine plasmatiche.
10
Le placche ateromasiche tendono a formarsi in punti in cui il flusso sanguigno
muta da laminare a turbolento, ad esempio ai punti di diramazione delle arterie
epicardiche. La presenza di una lesione aterosclerotica a questo livello
deter-mina, a valle della stenosi, una caduta di pressione che è proporzionale alla
ri-duzione del calibro vasale: il gradiente pressorio che così si crea, stimola la
di-latazione dei vasi di resistenza, allo scopo di mantenere un flusso adeguato in
condizioni basali, e questo spiega l’assenza di qualsivoglia segno clinico ed
elettrocardiografico d’ischemia anche in presenza di significativa aterosclerosi
coronarica.
Una stenosi del 50% della sezione vasale impedisce il pieno incremento del
flusso per rispondere alle aumentate richieste miocardiche; se la stenosi riduce la sezione di oltre l’80%, si ha una riduzione del flusso anche in condizioni basali, e in questa situazione l’albero coronarico è costretto a impegnare gran parte della sua “riserva” per mantenere un apporto metabolico adeguato. Se la richiesta metabolica aumenta, il circolo coronarico non è più in grado di
far fronte alle richieste, avendo esaurito la propria “riserva”, e quindi compare l’ischemia. Essa interessa inizialmente gli strati subendocardici che sono i più esposti. Un aumento del tono coronarico legato a fattori neuro umorali può
li-mitare temporaneamente la riserva coronarica, impedendo la vasodilatazione,
e in questi casi l’ischemia può comparire per richieste metaboliche inferiori;
questo spiega la variabilità della soglia ischemica che abitualmente si osserva
11
1.2.4 Gli effetti dell’ischemia
Durante gli episodi d’inadeguata perfusione la tensione di ossigeno del tessuto
miocardico si riduce e ciò può determinare disturbi transitori delle funzioni
meccaniche, biochimiche ed elettriche del miocardio.
In caso di anossia grave, gli acidi grassi non possono essere ossidati e il
gluco-sio è trasformato in lattato; il pH intracellulare si riduce, così come le riserve
miocardiche di fosfato ad alta energia (ATP e la creatina fosfato). La riduzione
delle riserve di ATP interferisce con lo scambio ionico a livello del
sarcolem-ma, con aumento del Na+ e riduzione del K+ intracellulare: l’aumento del Na+
intracellulare ha come conseguenza un incremento del Ca++ intracellulare
at-traverso un aumentato scambio Na+/Ca++. La ridotta disponibilità di ATP ab-bassa anche l’assunzione di Ca++ da parte del reticolo sarcoplasmatico e riduce l’estrusione di Ca++ dalla cellula. L’aumento del Ca++ intracellulare produce un sovraccarico di Ca++ a livello dei mitocondri e ciò deprime ulteriormente la
produzione di ATP. Il Ca++ risulta quindi avere un ruolo centrale nel circolo
vizioso che porta al danno irreversibile della cellula in caso d’ischemia
persi-stente. A livello del tessuto ischemico si producono anche radicali liberi deri-vati dall’ossigeno, cioè molecole di ossigeno con un eccesso di elettroni che le rendono chimicamente reattive; tali sostanze, per mezzo di fenomeni di
peros-sidazione, possono danneggiare la membrana cellulare e quindi contribuire al
danno ischemico.
Un fenomeno di notevole interesse risulta essere il fatto che il miocardio
12
successiva. Questo fenomeno, che è indipendente dalla neovascolarizzazione
nelle ramificazioni delle coronarie, è chiamato precondizionamento ischemico.
Questo fenomeno ha una fase precoce, che avviene entro 2 ore da un episodio
ischemico che non abbia condotto a infarto, e una tardiva, che ha luogo 24 ore
dopo e può durare da 1 a 3 giorni. Sembra che tanto la fase precoce quanto
quella tardiva dipendano, almeno in parte, dalla liberazione locale di
adenosi-na come conseguenza della degradazione di ATP nelle cellule miocardiche. In accordo con l’esistenza del precondizionamento ischemico sono le osservazio-ni riguardanti la minore gravità dell’infarto del miocardio preceduto da episodi anginosi, rispetto a quello che si verifica “a ciel sereno”.
La gravità e la durata dello squilibrio tra apporto e richiesta di ossigeno
deter-minano la reversibilità del danno, che si manifesta con alterazioni della con-trattilità (acinesie e discinesie) e dell’attività elettrica (i segni elettrocardiogra-fici dell’ischemia), nonché con alterazioni dell’emodinamica generale (scom-penso cardiaco).
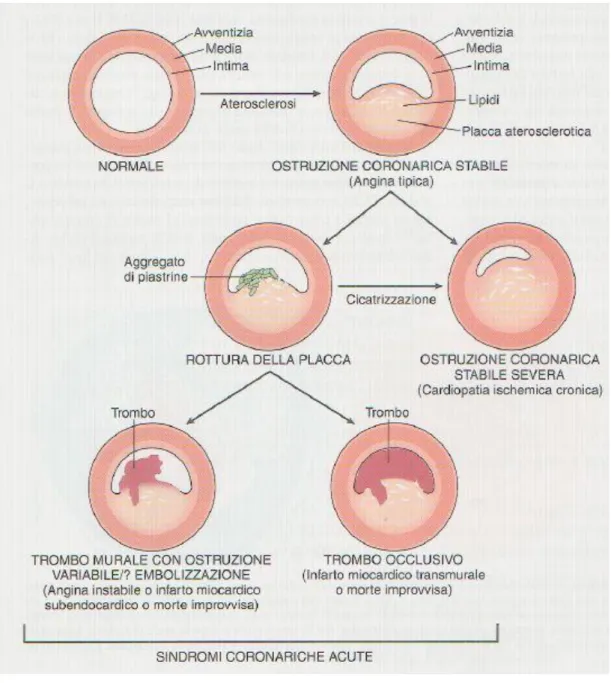
1.3 Il ruolo della placca “vulnerabile”
Le variazioni morfologiche delle placche aterosclerotiche dipendono dalla
di-versa combinazione delle componenti che le costituiscono, soprattutto al
livel-lo del cappuccio fibroso. La placca ateromasica, aumentando di volume, tende
a protrudere nel lume; nel suo pieno sviluppo, essa può andare incontro a una
13
che possono sopraggiungere su una placca sono l’ulcerazione, la trombosi, l’emorragia e le calcificazioni (nell’ambito della placca stessa), e le lesioni a-neurismatiche della parete, per atrofia della tonaca media e della sua parte
ela-stica. Le più temibili, ai fini della trattazione della cardiopatia ischemica, sono sicuramente l’ulcerazione con formazione di trombosi, le quali si estrinsecano soprattutto nelle cosiddette “placche vulnerabili”, in cui la componente fibrosa è molto ridotta, o con una componente calcifica abbondante che le rende
parti-colarmente fragili al traumatismo prodotto dal flusso ematico. La rottura della
lamina fibromuscolare della placca (il cappuccio fibroso) consente ai fattori
della coagulazione di entrare in contatto col fattore tissutale, proteina
pro-coagulante altamente trombogenica, che viene espresso dalle cellule schiumo-se derivanti dai macrofagi nel “core” lipidico della placca. Questa reazione in-nesca la cascata della coagulazione e porta alla formazione di un trombo, che
può limitarsi nella sua estensione alla placca stessa o estendersi oltre la stessa
determinando una riduzione del lume ulteriore a quella già causata dalla
plac-ca stessa.
Se il trombo che si viene a formare è di tipo non occlusivo o transitorio, la
rot-tura della placca può essere asintomatica o dar luogo a sintomi ischemici epi-sodici quali l’angina a riposo; i trombi occlusivi persistenti danno spesso ori-gine a infarto del miocardio, soprattutto in assenza di un circolo collaterale
ben sviluppato, che si forma tipicamente nelle aterosclerosi coronariche di
grado severo con un flusso cronicamente ridotto. La tipologia della placca è
an-14
dando a modificare nella sua evoluzione i quadri sintomatologici e quindi le
sindromi cliniche che possono affliggere il paziente.
Una placca fissurata, alla risoluzione del trombo, andrà incontro a
cicatrizza-zione, con conseguente ulteriore riduzione del calibro vasale, peggioramento dell’ostruzione e quindi della cardiopatia ischemica di base.
Figura 2 – I possibili esiti della rottura di placca “vulnerabile” (da Robbins e Cotran, “le basi patologiche delle malattie” 7ma ed., 2006.)
15
1.4 Gli aspetti clinici dell’ischemia cardiaca
Secondo quanto precedentemente detto, la cardiopatia ischemica viene divisa
in varie sindromi. La differenziazione è basata soprattutto sui dati clinici, in
primis il dolore e i suoi connotati (tipologia, insorgenza, durata,
localizzazio-ne, irradiazioni); a questo e ad altri rilievi obiettivi, come frequenza cardiaca,
pressione arteriosa ecc., sono associati dati elettrocardiografici e di laboratorio
capaci di individuare, se presenti, i segni di necrosi cellulare tipici dell’infarto
miocardico acuto.
1.4.1 Il sintomo dolore
Il dolore anginoso classico è caratteristico e d’importanza cruciale al rilievo
anamnestico: un dolore oppressivo o costrittivo retrosternale, indicato dal
pa-ziente ponendo la mano aperta sulla regione sternale, irradiato al collo, alle spalle, alla superficie ulnare dell’arto superiore sinistro (ma anche del destro), di durata variabile in base alla sindrome clinica (pochi minuti nell’ischemia, oltre 30 minuti nell’infarto).
Per le sue peculiarità contribuisce in modo essenziale al sospetto diagnostico
di cardiopatia ischemica, ma vista la grande variabilità di presentazioni è im-portante raccogliere un’anamnesi prossima accurata, seguendo uno schema che tenga presente le seguenti variabili:
Qualità del dolore: oppressione o pesantezza, bruciore, tensione, senso di affanno, sensazione di dolore profondo e non superficiale, inizio e
16
cessazione graduali. Il dolore non è influenzato dagli atti respiratori,
dalla posizione del corpo né dalla pressione della parete toracica.
Localizzazione: abitualmente il dolore è retro sternale mediano. Esso ha molteplici irradiazioni (superficie ulnare dell’arto superiore sinistro o raramente del destro, spalle, mandibola, epigastrio, regione
interscapo-lo-vertebrale). Raramente si presenta solo nelle sedi d’irradiazione
sen-za essere presente in sede retrosternale.
Durata: da 1-2 a 20 min. Naturalmente durate diverse hanno significato diverso: quanto più prolungato è il dolore, tanto più grave è la
situazio-ne.
Fattori precipitanti: esercizio fisico, temperatura rigida, periodo post-prandiale, rapporto sessuale, emozione.
Risposta alla trinitrina: regressione graduale del dolore dopo 1-5 min dall’assunzione sublinguale.
17
Nella valutazione del paziente anginoso è importante tenere presente il grado
di limitazione funzionale indotto dalla malattia, poiché questo è un indice, per
quanto grossolano, della gravità del danno anatomico. A tale riguardo la
Ca-nadian Heart Society raccomanda la seguente classificazione:
I. L’angina è provocata solo da sforzo fisico molto intenso e inusuale per
il soggetto; tutte le attività ordinarie possono essere svolte senza
pro-blemi.
II. È presente una modesta limitazione dell’attività fisica; il soggetto ha
angina se deve fare, per esempio, due rampe di scale, particolarmente in
condizioni sfavorevoli (dopo un pasto o in una giornata fredda).
III. Il paziente è decisamente limitato nella sua attività fisica ed è
sintoma-tico per sforzi lievi.
IV. Qualunque tipo di sforzo provoca angina, che può essere presente
an-che a riposo.
1.4.2 Angina Pectoris Stabile
Questa sindrome clinica episodica è dovuta all’ischemia miocardica
transito-ria. Il tipico paziente è un uomo oltre i 50 anni o una donna oltre il 60 anni,
che lamenta disturbi al torace, descritti come il dolore toracico classico;
l’angina di solito ha un andamento crescente e decrescente, dura dai 2 ai 5
mi-nuti e si può irradiare nelle sedi tipiche. Tale sindrome è indotta da sforzi fisi-ci o da emozioni, e la soglia per la comparsa dell’angina è fissa, fisi-cioè si ripre-senta a ogni carico di lavoro uguale o superiore a quello che ha scatenato il
18
paziente assume nitroderivati sublinguali. Al sintomo dolore si può associare
dispnea, tachicardia con palpitazioni, astenia.
Dal punto di vista diagnostico si hanno modesti segni elettrocardiografici, che
scompaiono al recedere della sintomatologia e devono perciò essere indagati con un test da stimolo, come l’elettrocardiogramma sotto sforzo (che permette tra l’altro di stabilire la soglia ischemica del paziente); non si hanno alterazioni di laboratorio significative, con una costante negatività degli indici di
miocar-dionecrosi.
1.4.3 Angina Pectoris Instabile
L’angina instabile è definita come un disturbo ischemico con almeno una delle seguenti tre caratteristiche: 1) compare a riposo o a minimi sforzi, e dura oltre
10 minuti; 2) è intensa e di recente insorgenza; 3) si verifica con una
sintoma-tologia in crescendo.
Il paziente tipico presenta un quadro molto simile a quello dell’angina stabile,
e si differenzia da questa per i criteri sopracitati, oltre che per i segni
elettro-cardiografici che si fanno più specifici: sottoslivellamento del tratto ST di
al-meno 0,1mV, inversione delle onde T (di solito consensuali come direzione al
complesso QRS) o alterazione della loro morfologia, diventando più rigide (dette “a punta”). Questi segni elettrocardiografici correlano con la sintomato-logia, ma non con le indagini di laboratorio, che evidenziano una completa
19
1.5 L’infarto acuto del miocardio (IMA)
L’infarto miocardico è dovuto a un’ischemia acuta che persiste sufficiente-mente a lungo da provocare la necrosi cellulare. Esso, pertanto, è
caratterizza-to da un danno anacaratterizza-tomico irreversibile del miocardio, diversamente da quancaratterizza-to accade nell’angina in cui l’alterazione è reversibile; tale situazione si manife-sta con disturbi della meccanica, della conduzione e dell’emodinamica che
ca-ratterizzano i sintomi, i segni e le complicanze di tale patologia.
1.5.1 Patogenesi dell’infarto
La rottura di una placca vulnerabile è l’evento più comune alla base dell’IMA: l’esposizione del fattore tissutale induce l’attivazione della coagulazione, con l’attivazione dei fattori VII e X, causando la conversione della protrombina in trombina, che a sua volta converte il fibrinogeno in fibrina; tale processo è
au-toalimentante, e porta alla formazione di un trombo occludente il lume. L’occlusione coronarica è il fattore patogenetico più importante della necrosi miocardica, ma non è l’unico: a esso se ne possono associare altri, che contri-buiscono a far precipitare la necrosi o almeno a renderla più estesa. Alla
pre-senza di un’ischemia miocardica da improvvisa riduzione del flusso in un di-stretto coronarico, si verifica un’aumentata increzione di catecolamine: questo fatto ha ripercussioni negative a livello miocardico. Infatti, un aumento delle
catecolamine circolanti provoca un incremento della frequenza cardiaca e un
aumento delle resistenze periferiche; inoltre esso stimola un aumento della contrattilità miocardica nelle zone non direttamente interessate dall’ischemia. Tutti questi eventi comportano un sensibile incremento del consumo
miocar-20
dico di O2 e possono innescare un circolo vizioso che aggrava e prolunga l’ischemia miocardica sino a far precipitare la necrosi. Se la necrosi è già in at-to, questa sequela di eventi fa sì che essa, alla fine, risulti più estesa.
Perciò, a definire la storia naturale di un infarto nelle sue due modalità di
pre-sentazione, concorrono 3 fattori: 1) la rottura di una placca vulnerabile e la presenza di trombi; 2) l’ostruzione dinamica, data dalla costrizione dei vasi co-ronarici; 3) l’aumento delle richieste metaboliche del miocardio o un minore apporto di ossigeno dal flusso coronarico.
Nella maggior parte degli infarti transmurali (in questi situazione la parete
miocardica è interessata per tutto il suo spessore) siamo di fronte ad un’occlusione completa: essa è dovuta nella stragrande maggioranza dei casi a una trombosi, e con molta minor frequenza è dovuta a uno spasmo coronarico.
Entrambi gli eventi si verificano preferenzialmente, per non dire
esclusiva-mente, a livello di lesioni aterosclerotiche. Trombosi e spasmo possono
coesi-stere e anzi influenzarsi vicendevolmente; basta pensare al ruolo delle
piastri-ne piastri-nella gepiastri-nesi del trombo e al fatto che esse liberano sostanze vasoattive,
co-me il trombossano A2, che possiede una potente azione vasocostrittrice.
Nel caso d’infarto intramurale (in questa situazione la parete non è interessata per tutto il suo spessore ma solo nei suoi strati subendocardici) il rilievo più frequente è quello di un’occlusione subtotale o di un’occlusione totale in pre-senza di circolo collaterale, su cui possono sommarsi fenomeni vasospastici transitori che azzerano temporaneamente l’apporto di sangue al tessuto
cardia-21
co. Il danno si manifesta al tessuto subendocardico sia perché è irrorato dai
rami più terminali del letto coronarico, sia perché a questo livello la pressione
intracavitaria è elevata (soprattutto in diastole, momento in cui ricordiamo si
ha la maggior parte del flusso coronarico), e questa contrasta la perfusione
co-ronarica mantenendo questi tessuti in uno stato d’ischemia cronica.
Figura 4 – La progressione dell’infarto: da sinistra, ischemia, infarto subendocardico o intramurale, infar-to transmurale (da Robbins e Cotran, “le basi painfar-tologiche delle malattie” 7ma ed., 2006.)
La sede di un infarto è in rapporto alla coronaria occlusa e la sua estensione dipende dal punto in cui l’occlusione è avvenuta (quanto più prossimale l’ostruzione tanto più estesa la necrosi). L’estensione della necrosi dipende anche dalla presenza o assenza di circoli collaterali.
1.5.2 Classificazione degli infarti: N-STEMI e STEMI
In base ai reperti elettrocardiografici che otteniamo dai tracciati di pazienti con IMA, possiamo non solo avere un’idea della sede di lesione, ma anche della sua estensione intramurale; tale differenza, cambiando molto la prognosi sia
22
per la fase acuta sia per le complicanze, ha portato a classificare l’infarto
se-condo questi rilievi.
Si parla di Infarto del Miocardio senza sopraslivellamento del tratto ST
(N-STEMI) in presenza di fenomeni ischemici acuti in cui si abbia la
positivizza-zione degli enzimi di miocardionecrosi. Clinicamente abbiamo un quadro di
angina pectoris, caratterizzato da dolore precordiale classico, dispnea, cute
fredda e pallida, tachicardia sinusale, terzo e/o quarto tono cardiaco, rantoli
al-le basi polmonari e talvolta ipotensione.
All’ECG si avrà quindi un quadro caratterizzato da segni tipici dell’ischemia cardiaca (sottoslivellamento del tratto ST di almeno 0,1mV, inversione dell’onda T con morfologia “a punta”, assenza di onde Q di necrosi) con un aumento degli indici di miocardionecrosi. Questi infarti generalmente sono di
tipo intramurale (con preferenziale localizzazione subendocardica), cosa che
correla con una gravità minore del quadro elettrocardiografico e
laboratoristi-co rispetto agli infarti transmurali, e laboratoristi-con prognosi generale e laboratoristi-complicanze
me-no severe.
L’Infarto del Miocardio con sopraslivellamento del tratto ST (STEMI) è più comunemente di tipo transmurale, ed è caratterizzato da una sintomatologia
diversa e più grave: il paziente lamenta un dolore profondo e viscerale, di tipo opprimente “a morsa”, simile a quello dell’angina pectoris ma che si verifica comunemente a riposo (sono state osservate variazioni circadiane con una
risve-23
glio). Il dolore è più intenso e dura più a lungo, di regola supera i 30 min e non scompare con la cessazione dell’attività fisica, a differenza dell’angina. La lo-calizzazione è precordiale, ma spesso anche subxifoidea/epigastrica, e ciò
por-ta il paziente a rifiupor-tare la possibilità di un atpor-tacco cardiaco e a considerare una
patologia digestiva, idea avvalorata dai frequenti sintomi da attivazione auto-nomica che accompagnano questo tipo d’infarti (sudorazione, nausea, vomito). Il quadro è caratterizzato inoltre da profonda astenia, e una sintomatologia an-siosa fino alla sensazione di “morte imminente”, oltre che da tutti i sintomi già visti nel NSTEMI.
Figura 5 – Le variazioni del tratto ST caratterizzanti le definizioni di STEMI e NSTEMI (da www.thrombosisadviser.com.)
In passato la presenza di onde Q anomale era considerata un segno di IMA
24
onde Q. Accurati studi di correlazione tra ECG e quadri patologici hanno
indi-cato tuttavia che infarti transmurali possono manifestarsi senza onde Q e che
infarti sub endocardici possono alcune volte essere associati a onde Q; per
questo motivo gli infarti sono attualmente indicati come “Q” e “ non Q” in
ag-giunta alla dicitura STEMI e NSTEMI.
1.6 Diagnostica Strumentale
Al fine di una rapida e corretta diagnosi di IMA, e per una corretta scelta
tera-peutica, il medico può affidarsi a molteplici strumenti diagnostici, invasivi e
non, che forniscono un quadro clinico completo e dirimente.
Attualmente la diagnosi di IMA e la successiva classificazione in NSTEMI e
STEMI si basa, oltre che sui reperti obiettivi, su esami non cruenti, come l’elettrocardiografia, gli esami di laboratorio e l’ecocardiografia, e su esami cruenti, come l’angiografia coronarica.
1.6.1 Elettrocardiogramma
I segni elettrocardiografici di IMA non sono stabili: la necrosi miocardica è un evento dinamico che evolve nel tempo e l’elettrocardiogramma testimonia, con la propria evoluzione, le successive alterazioni fisiopatologiche a livello
miocardico. In caso d’infarto, le variazioni delle fasi di depolarizzazione
(QRS) sono spesso accompagnate da anomalie di ripolarizzazione (ST-T);
abi-tualmente si distinguono quattro stadi di evoluzione elettrocardiografica dell’infarto acuto del miocardio:
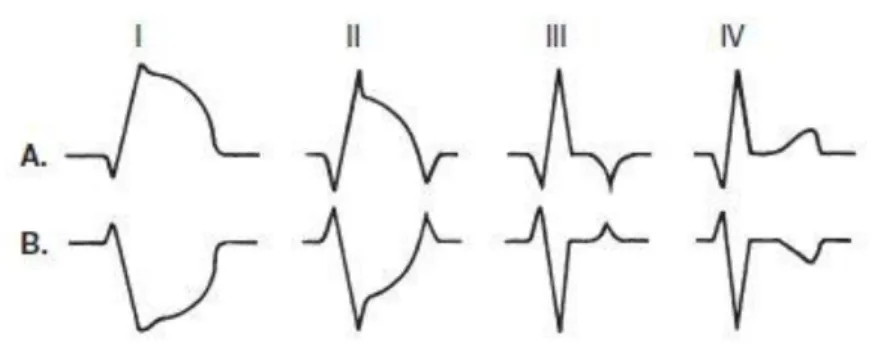
25 I stadio. È caratterizzato da iniziali segni di necrosi (piccola onda Q) e marcati
segni di lesione subepicardica (sopraslivellamento del tratto ST). In questa
fa-se il sopraslivellamento è talmente marcato che tende a inglobare l’onda T e quindi maschera l’ischemia subepicardica (onda T negativa) della zona perin-fartuale. Quest’aspetto è rilevabile nelle derivazioni prospicienti la necrosi; in quelle opposte si rilevano segni speculari: particolarmente comune una lesione
subendocardica (sottoslivellamento di ST) in sede inferiore, nel caso di necrosi
anteriore, oppure una lesione subendocardica antero - settale in caso di necrosi
inferiore. Le alterazioni elettrocardiografiche del I stadio sono caratteristiche delle prime ore dall’esordio dei sintomi.
II stadio. In questa fase la lesione subepicardica (sopraslivellamento del tratto
ST) si attenua; i segni di necrosi divengono più evidenti (l’onda Q si
appro-fondisce) e compare l’ischemia subepicardica (si rende evidente una T
negati-va). Questo stadio è quindi caratterizzato dalla simultanea presenza di necrosi,
lesione e ischemia. La durata di questo stadio è molto variabile: queste
altera-zioni elettrocardiografiche possono durare da poche ore a parecchi giorni. La
persistenza per lungo tempo della lesione subepicardica (sopraslivellamento
del tratto ST), particolarmente comune negli infarti anteriori, può essere
e-spressione di un aneurisma del ventricolo sinistro nella sede della necrosi.
III stadio. Questa fase è caratterizzata dalla scomparsa della lesione
subepi-cardica (sopraslivellamento del tratto ST) e dall’approfondimento dei segni d’ischemia subepicardica (approfondimento delle onde T negative). I segni e-lettrocardiografici sono quindi quelli di necrosi e ischemia. In molti casi
que-26
sto stadio si prolunga nel tempo, tanto da rimanere testimonianza definitiva dell’infarto pregresso.
IV stadio. In alcuni casi, dopo alcune settimane o anche più precocemente, si
può assistere alla progressiva riduzione dei segni d’ischemia subepicardica
(onde T negative) e a volte anche alla normalizzazione della ripolarizzazione. In questi casi l’unico segno elettrocardiografico del precedente infarto è un’onda Q di necrosi.
Figura 6 - Rappresentazione schematica dell’evoluzione elettrocardiografica in 4 stadi dell’infarto miocar-dico. A. Aspetto nelle derivazioni prospicienti la zona necrotica; B. aspetto nelle derivazioni opposte alla
zona necrotica (aspetto speculare) (da Rugarli, “medicina interna sistematica”, 2005)
Poiché in alcune derivazioni la presenza di onda Q è fisiologica, è necessario delineare alcune caratteristiche tipiche dell’onda Q patologica, indicativa di necrosi: durata superiore a 0,04 sec; presenza frequente di uncinature o
irrego-larità nella sua branca discendente iniziale; profondità variabile nelle diverse
derivazioni. Quest’ultimo punto richiede le seguenti precisazioni:
In aVL > 2 mm e > 25% della R;
27
In D2 e aVF > 2 mm e > 25% della R;
In V5 e V6 > 2 mm e > 15% della R;
In D3 la diagnosi di Q patologica è spesso difficile; la diagnosi sarà più certa alla presenza delle seguenti caratteristiche: a) presenza simultanea
di Q in D2 e aVF; b) assenza di riduzione della profondità durante
in-spirazione forzata; c) profondità > 6 mm.
L’ECG non solo consente la diagnosi d’infarto, ma anche la sua localizzazione approssimativa e il giudizio sulla sua estensione. Va tuttavia ricordato che la
terminologia elettrocardiografica non rispecchia in modo assolutamente fedele
la localizzazione anatomica: questo modo di classificare l’IMA resta utile per
la definizione dei vari tipi d’infarto cardiaco, ma è inadeguata per individuare a livello di quale arteria coronarica si sia verificata l’occlusione. In linea di massima si può dire che l’infarto anteriore interessi la coronaria sinistra ante-riore discendente, quello laterale la circonflessa e quello infeante-riore, nell’80% dei casi, la coronaria destra e, nei rimanenti, la coronaria sinistra circonflessa.
Studi più accurati condotti analizzando le alterazioni che si hanno
precoce-mente a carico del tratto ST (che sono un indicatore migliore dell’onda Q che
può mancare negli infarti intramurali) hanno consentito analisi più precise.
Nel caso degli infarti anteriori, un’elevazione del tratto ST in V1, V2, V3 e aVL con un sottoslivellamento di più di 1 mm in aVF indica un’occlusione prossi-male della coronaria sinistra anteriore discendente; l’elevazione di ST in V1,
28
tratto, indica un’occlusione di questa stessa arteria distalmente all’origine del primo ramo diagonale.
Per quanto riguarda gli infarti inferiori, un’elevazione del tratto ST in D3 di entità maggiore che in D2, e con sottoslivellamento di questo tratto di più di 1
mm in D1 e aVL, indica un’occlusione della coronaria destra; invece, un’elevazione di ST in D3 non superiore rispetto a D2 e con ST isoelettrico o elevato in aVL indica un’occlusione della coronaria sinistra circonflessa.
L’evoluzione nel tempo delle alterazioni del tratto ST è anche un ottimo indice della riperfusione che consegue a un infarto del miocardio. Infatti, si pensa che
quanto più rapidamente il tratto ST si riporti sull’isoelettrica tanto migliore è la riperfusione del miocardio che segue l’infarto. Una rapida riduzione di più del 70% dell’elevazione del tratto ST nelle derivazioni dove era più alterato è
associata con il decorso più favorevole.
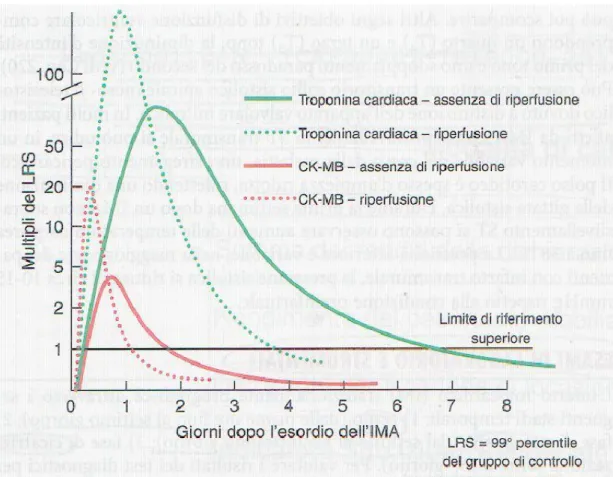
1.6.2 Marker cardiaci sierici
Determinate proteine, denominate marker di miocardionecrosi, sono rilasciate
nel sangue dal tessuto miocardio necrotico dopo un IMA, sia N-STEMI sia
STEMI. La velocità di rilascio di tali marcatori dipende dalla loro
localizza-zione nei miociti, dal loro peso molecolare e dal flusso refluo dalla zona di ne-crosi: l’andamento nel tempo del rilascio di questi marker riveste una grande importanza diagnostica, ma contemporaneamente le strategie di riperfusione
29
ematochimici arrivino dal laboratorio. Importante è anche la quantità totale di proteina rilasciata, che correla alle dimensioni dell’infarto.
Le troponine miocardio-specifiche T e I (TnT e TnI) sono diverse come
se-quenza rispetto a quelle della muscolatura striata scheletrica, e sono dosate
tramite test quantitativi basati su anticorpi monoclonali altamente specifici;
queste due isoforme delle troponine non sono normalmente presenti nel
san-gue dei soggetti sani, e aumentano dopo uno STEMI fino a 20 volte il limite
superiore della norma. TnT e TnI sono i marcatori sierici principali per
identi-ficare gli IMA; le loro specificità e sensibilità, maggiori degli altri marker,
permettono di distinguere l’angina instabile dal NSTEMI, e di identificare
le-sioni molto piccole (sotto il range di sensibilità degli altri marcatori), inoltre
permettono di distinguere la lesione se di origine muscolo-scheletrica e non
cardiaca. I livelli di TnT e TnI restano elevati per 7-10 giorni dopo STEMI.
L’enzima creatin fosfochinasi (CK) nel sangue aumenta rapidamente dopo
IMA, entro 4-8 ore, per normalizzarsi dopo 48-72 ore. Questo marker è molto
meno specifico per l’infarto del miocardio, infatti subisce elevazioni anche
dopo malattia o trauma muscolo-scheletrico (anche iatrogeno, come le iniezio-ni intramuscolo). L’isoenzima MB della creatin fosfochinasi (CK-MB) ha il vantaggio di non essere presente nel sangue in concentrazioni significative nei
tessuti diversi dal miocardio ed è quindi molto più specifico ai fini della
dia-gnosi di IMA, ma anche questa isoforma ha una specificità minore delle
30
Figura 7 – Movimento dei marker di miocardionecrosi solitamente utilizzati nello STEMI (da Harrison, “principi di medicina interna” 17ma ed., 2009.)
In passato sono stati usati anche altri indici di miocardionecrosi, con
sensibili-tà e specificisensibili-tà minori di troponine e CK-MB, quali mioglobina (MB),
transa-minasi glutammico - ossalacetica (SGOT) e lattico - deidrogenasi (LDH), in
seguito abbandonati per la loro inferiorità diagnostica.
Importanti, ma di marginale rilevanza diagnostica, sono quelle reazioni defini-te “non specifiche” all’infarto miocardico acuto: si ha una leucocitosi polimor-fonucleata entro poche ore, e che persiste per 3-7 giorni fino a valori di 12.000
31
che aumenta più lentamente, con un picco nella prima settimana dopo IMA e
che resta elevata per 1-2 settimane.
1.6.3 Imaging cardiaco
Nei pazienti con infarto miocardico l’ecocardiogramma bidimensionale mette costantemente in evidenza zone di alterata cinesi: il rilievo di zone
ipocineti-che o acinetiipocineti-che permette non solo di confermare la diagnosi d’infarto, ma
an-che di definire la sede in misura molto più precisa di quanto non consenta l’elettrocardiogramma. Oltre all’alterata cinesi, la zona infartuale presenta an-che un minor spessore rispetto alle zone di miocardio non necrotico. La stima della funzione ventricolare sinistra, effettuata con l’ecocardiogramma tramite il calcolo della frazione d’eiezione (EF), si correla bene alla stima di questo valore ottenuta per via angiografica, ed è un utile elemento per formulare un
giudizio prognostico.
Se l’ecocardiografia è affidabile nello scoprire e localizzare un infarto, meno precisa è nel quantizzarne l’estensione, poiché non è in grado di distinguere con esattezza tra tessuto necrotico e tessuto ischemico: entrambi, infatti, si contraggono in modo anomalo e quindi spesso l’ecocardiografia può sovrasti-mare la reale estensione della necrosi. Oltre a questo, l’ecocardiografia non è
in grado di distinguere, se usato da solo, uno STEMI da una pregressa
cicatri-ce miocardica da infarto, perché entrambe le situazioni si presentano con un’alterata cinesi. Per questo l’ecocardiografia si dimostra utile come esame complementare all’ECG e al rilievo dei marker chimici.
32
Quando l’ECG non è diagnostico, la precoce osservazione della presenza o as-senza di alterazioni della cinesi parietale all’ecocardiogramma può aiutare a decidere se il paziente debba o no ricevere una terapia di riperfusione; l’esame
è quindi particolarmente utile nella diagnosi precoce, al letto del malato, e per
la diagnosi della maggior parte delle complicanze meccaniche dell’infarto:
a-neurisma, difetto interventricolare, malfunzionamento o rottura di un muscolo
papillare, rottura della parete libera con tamponamento cardiaco. Inoltre è la
tecnica più semplice e affidabile per diagnosticare un versamento pericardico o l’eventuale presenza di trombi ventricolari.
Infine, con l’ecocardiografia si può rilevare un’alterata motilità delle pareti del ventricolo destro, una sua eventuale dilatazione e una riduzione della sua
fra-zione di eiefra-zione; questi sono tutti elementi che consentono di diagnosticare
con sicurezza un infarto ventricolare destro, la cui diagnosi secondo criteri
cli-nici ed elettrocardiografici può essere spesso difficile se non impossibile.
È doveroso citare l’imaging con radioisotopi come la scintigrafia miocardica
con 201tallio o 99mtecnezio. Queste tecniche sono utili per valutare con
preci-sione i parametri morfologici e funzionali di un IMA, ma spesso risultano
in-daginose nella situazione di emergenza imposta dall’infarto; oltre a questo, tali tecniche non riescono a distinguere, come l’ecografia, una lesione cronica miocardica da un infarto in atto, e quindi difettano in specificità per IMA.
33
1.7 Complicanze dell’IMA
Le complicanze di un infarto possono essere suddivise in tre grandi gruppi:
complicanze aritmiche; complicanze emodinamiche, come la compromissione
della funzione di pompa, le rotture (del setto, del muscolo papillare, della
pa-rete libera), l’infarto del ventricolo destro e l’aneurisma; complicanze
ische-miche per estensione della necrosi, o per l’angina precoce postinfartuale. A
queste se ne aggiungono altre non inquadrabili unitariamente, come i
fenome-ni trombo-embolici, la pericardite epistenocardica e la sindrome di Dressler. È
importante tenere presente che molte di queste complicanze possono
coesiste-re e complicacoesiste-re simultaneamente o in tempi successivi l’evoluzione di un
me-desimo infarto.
1.7.1 Complicanze aritmiche
Nella fase acuta di un infarto possono insorgere pressoché tutte le aritmie; in
questo contesto esse assumono un significato particolarmente grave perché
possono influire negativamente sulla funzione di pompa del cuore, che è già
compromessa e possono portare a un’estensione della necrosi sia attraverso una ridotta perfusione coronarica, sia attraverso l’aumento del consumo di O2; possono infine, anche le meno pericolose, evolvere rapidamente verso le
arit-mie maggiori, come la fibrillazione ventricolare o il blocco atrioventricolare totale e l’asistolia.
Le extrasistoli ventricolari compaiono probabilmente nel 100% dei casi d’infarto: quando hanno alcune caratteristiche particolari (frequenza superiore a 10/min, bigeminismo e trigeminismo, presenza di coppie, precocità con
fe-34
nomeno R/T, cioè l’extrasistole insorge in corrispondenza dell’onda T del complesso precedente, multifocalità) possono evolvere rapidamente verso la
tachicardia ventricolare e verso la fibrillazione ventricolare.
La tachicardia ventricolare insorge quando tre o più extrasistoli ventricolari si
presentano in rapida successione; possono presentarsi come brevi salve di
e-xtrasistoli, oppure essere persistenti. Nel caso delle brevi salve, spesso non si
hanno conseguenze emodinamiche ma il rischio di fibrillazione ventricolare è
molto elevato. Nel caso di tachicardia ventricolare persistente (frequenza tra
150 e 200/min) si verifica una riduzione marcata della portata cardiaca cui si
associa ipotensione, ipoperfusione coronarica, con aggravamento del
fenome-no ischemico. Si instaura un circolo vizioso che rapidamente aggrava il quadro
clinico; in alcuni casi l’esordio della tachicardia ventricolare si accompagna a
sincope per la brusca riduzione della portata a livello cerebrale. Una forma
particolare di tachicardia ventricolare è il cosiddetto ritmo idioventricolare
ac-celerato: in questo caso la frequenza della tachicardia è molto più bassa
(90-100/min) e le conseguenze emodinamiche sono minori, il rischio di evoluzione
verso la fibrillazione è molto modesto; spesso quest’aritmia insorge alla
pre-senza di una bradicardia sinusale.
Nella fibrillazione ventricolare i ventricoli si contraggono con una frequenza
tra 400 e 600/min, in modo desincronizzato; il cuore è praticamente fermo e si
instaura il quadro dell’arresto circolatorio, con danno cerebrale irreversibile
nel giro di pochi minuti. La fibrillazione ventricolare è suddivisa dal punto di
35
nel primo caso essa insorge in pazienti per il resto non complicati e con una
buona situazione emodinamica; la seconda complica i quadri di grave deficit
emodinamico e spesso ne rappresenta l’episodio terminale.
È di fondamentale importanza ricordare che la quasi totalità delle morti
preo-spedaliere in caso d’infarto sono dovute a fibrillazione ventricolare primitiva;
essa, quindi, non è legata alla gravità della necrosi dal punto di vista
emodi-namico. Tuttavia, poiché la maggior latenza nei tempi di ricovero di un infarto
è legata al ritardo con cui il paziente avverte il medico, o comunque richiede un’assistenza, la soluzione di questo problema si basa fondamentalmente su una corretta educazione sanitaria che informi la gente sui sintomi d’allarme di un attacco coronarico.
Le aritmie sopraventricolari, come la comparsa di flutter o fibrillazione atriale,
complicano il 5-10% degli infarti miocardici. La loro insorgenza è spesso
e-spressione di una compromissione funzionale del ventricolo sinistro con
au-mento della pressione telediastolica e conseguente dilatazione dell’atrio
sini-stro; probabilmente è proprio la dilatazione atriale il meccanismo più comune d’innesco di queste aritmie. Pur essendo molto meno gravi delle aritmie ven-tricolari maggiori, esse richiedono una diagnosi precoce e un trattamento im-mediato per due ragioni: l’elevata frequenza ventricolare, che caratterizza que-ste aritmie nella maggior parte dei casi, comporta un aumento del consumo
miocardico di O2 e quindi tende ad aggravare l’ischemia e può comportare
36
questi casi con funzione ventricolare sinistra già depressa) comporta una
ridu-zione della portata cardiaca e quindi peggiora il quadro emodinamico.
La bradicardia sinusale complica più frequentemente le fasi iniziali di un
in-farto inferiore ed è causata da un ipertono vagale. La sua importanza è legata
al fatto che può ridurre (se di grado elevato) la portata cardiaca; inoltre spesso
la fibrillazione ventricolare insorge improvvisamente proprio in presenza di
bradicardia spiccata.
La comparsa di blocchi atrioventricolari, da un punto di vista
elettrofisiologi-co, ma anche da un punto di vista clinico pratielettrofisiologi-co, è utile che sia distinta in base
al fatto che essi si verifichino per interessamento delle strutture del tessuto di
conduzione sopra il fascio di His (blocchi soprahissiani) o per alterazioni a
li-vello del fascio di His e della sua suddivisione nelle branche (blocchi hissiani
o sottohissiani). I blocchi soprahissiani complicano più spesso un infarto
infe-riore, ma raramente sono espressione di una lesione necrotica interessante
di-rettamente il tessuto di conduzione; più spesso essi sono dovuti a ischemia o a
edema perinfartuale e pertanto sono transitori. All’ECG si presentano come
blocchi di I grado che possono evolvere verso un blocco di II grado tipo
Mo-bitz 1 (con fenomeno di Luciani-Wenckebach) e quindi verso un blocco di
grado elevato, sino al blocco atrioventricolare totale. Abitualmente, la
progres-sione verso i gradi maggiori di blocco è lenta, i complessi ventricolari sono
so-litamente stretti. I blocchi sottohissiani si manifestano inizialmente con un blocco di conduzione intraventricolare, che all’ECG si manifesta come blocco di branca. Gli aspetti elettrocardiografici più frequenti sono l’emiblocco
ante-37
riore sinistro, il blocco di branca sinistro, l’associazione di emiblocco anteriore
sinistro con blocco di branca destra e infine l’associazione di blocco di branca
destra con emiblocco posteriore sinistro. Il P-Q può essere normale oppure
può essere presente un blocco A-V di I grado. Questi quadri sono espressione
di un interessamento diretto del tessuto di conduzione da parte della necrosi; si
associano a infarto anteriore e sono più comuni negli infarti particolarmente
estesi. La loro pericolosità sta nel fatto che possono improvvisamente evolvere
in modo brusco verso il blocco totale o addirittura verso l’asistolia. La
progno-si è sfavorevole progno-sia per la concomitante estenprogno-sione della necroprogno-si progno-sia per il
ri-schio d’improvvisa comparsa del blocco A-V totale.
1.7.2 Complicanze emodinamiche
Shock cardiogeno e scompenso sono le complicanze più gravi, caratterizzate
dalla prognosi peggiore e da limitate possibilità terapeutiche; entrambe
conse-guono a una grave depressione della funzione di pompa del ventricolo sinistro.
Esistono vari gradi di passaggio dal quadro dello scompenso a quello dello
shock, secondo che prevalga un aumento della pressione tele diastolica del
ventricolo sinistro o una riduzione della portata cardiaca; nei casi più gravi
so-no presenti entrambi i feso-nomeni.Clinicamente, l’aumento della pressione tele-diastolica del ventricolo sinistro si manifesta con un aumento della pressione a
livello dei capillari polmonari. Quando questa pressione supera il valore di 15
mmHg, può iniziare la congestione vascolare polmonare (rilevabile al Rx
tora-ce); quando supera i 20 mmHg, può iniziare la trasudazione negli alveoli e
38
quadro di edema polmonare acuto. Viceversa, la riduzione della portata
car-diaca è caratterizzata clinicamente da ipotensione (PA sistolica inferiore a 90
mmHg) con segni d’ipoperfusione periferica quali: oliguria, confusione
men-tale, cute ipotermica. Queste manifestazioni cliniche di shock conclamato non si manifestano abitualmente sino a che l’indice cardiaco (cioè la portata car-diaca per m2 di superficie corporea) non scende sotto il valore di 2,2
l/min/m2. Un quadro particolare di shock, con prognosi molto più favorevole
rispetto a quella dello shock cardiogeno, è lo shock indotto da ipovolemia.
La perforazione del setto interventricolare avviene quando la necrosi interessa
il setto interventricolare, con una rottura nella sua porzione muscolare. Il
qua-dro emodinamico è caratterizzato da un brusco aumento della portata a livello
del circolo polmonare per la presenza di shunt sinistro - destro acuto; la
porta-ta sistemica si riduce e il ventricolo sinistro è sottoposto a un improvviso
so-vraccarico di volume. Ne consegue rapidamente un quadro di scompenso e/o
di shock. Clinicamente la diagnosi può essere posta in base alla comparsa di
un soffio olosistolico abitualmente d’intensità elevata, massimo al mesocardio
e a volte irradiato anche verso destra, associato a un fremito; il secondo tono
può apparire ampiamente sdoppiato. La diagnosi può essere confermata con l’ausilio del cateterismo cardiaco destro, dell’ecocardiografia bidimensionale e dell’angiocardiografia radioisotopica.
Una rottura del muscolo papillare avviene quando lo interessa direttamente
con due evenienze possibili: il malfunzionamento del muscolo, con
ri-39
gurgito mitralico massivo. La prima evenienza è abbastanza comune ed è
dia-gnosticabile clinicamente per la comparsa di un soffio meso-telesistolico alla
punta; il grado di compromissione emodinamica è in funzione dell’entità del
rigurgito, ma il fenomeno può essere transitorio; la seconda evenienza è per
rara e si diagnostica per la comparsa di un soffio olosistolico alla punta, che si
associa a un quadro di scompenso acuto dovuto all’improvviso sovraccarico di
volume del ventricolo sinistro.
Temibile complicanza degli infarti transmurali è la rottura della parete libera.
Circa il 5% degli infarti transmurali si complica con la formazione di una
breccia a livello della parete libera del ventricolo sinistro. La complicanza è
più frequente nelle persone anziane e nel sesso femminile; essa abitualmente
compare tra la 3ª e la 10ª giornata dall’infarto. I segni clinici sono quelli del
tamponamento cardiaco acuto. Un caratteristico aspetto è quello della disso-ciazione elettromeccanica: mentre il polso è completamente assente, l’attività elettrica del cuore si mantiene e all’ECG si rileva la persistenza di complessi
ventricolari; questa complicanza è rapidamente mortale.
L’infarto ventricolare destr colpisce circa un terzo degli infarti inferiori; questa localizzazione passa spesso inosservata per la scarsità dei segni clinici ed
elet-trocardiografici; se l’interessamento del ventricolo destro è esteso, si possono
manifestare segni di scompenso destro (congestione giugulare ed epatica) con
o senza ipotensione. L’ecocardiografia bidimensionale e la ventricolografia
radioisotopica hanno permesso di formulare questa diagnosi con molta
40
Un aneurisma ventricolare può comparire in circa il 12-15% dei pazienti
so-pravvissuti a un infarto. Esso è rappresentato da una zona della parete
ventri-colare che non solo rimane acinetica, ma diviene discinetica, cioè si estroflette
durante la sistole. Le zone più frequentemente interessate sono la punta e la
parete anteriore del ventricolo sinistro. Le conseguenze di un aneurisma
ven-tricolare possono essere di tre tipi: a) la sua presenza compromette la regolare
dinamica di contrazione e riduce ulteriormente l’EF; ne può conseguire un
quadro di scompenso; b) in presenza di un aneurisma sono più frequenti le
a-ritmie ventricolari, ad esempio la tachicardia ventricolare, persistenti dopo la fase acuta; c) all’interno dell’aneurisma si formano più facilmente trombi mu-rali che possono dare origine a episodi embolici sistemici. All’obiettività si
ri-leva un itto solri-levante o un evidente impulso sistolico precordiale; quasi
sem-pre è sem-presente un terzo tono. All’ECG un segno comune è la persistenza di un
sopraslivellamento del tratto ST nella sede della necrosi, anche a distanza di
tempo dalla fase acuta. L’ecocardiografia e la ventricolografia radioisotopica
consentono la conferma della diagnosi di aneurisma, la valutazione della sua
estensione e la sua localizzazione; con l’ecocardiografia è possibile anche
evi-denziare la presenza di trombi al livello della parete discinesica, e valutarne l’estensione.
1.7.3 Complicanze ischemiche
La maggior parte dei pazienti non lamenta dolori anginosi nell’immediato
pe-riodo postinfartuale. Tuttavia, alcuni di essi possono presentare dolori
que-41
sti sintomi può essere premonitrice di una recidiva e di un’estensione dell’area
infartuata.
L’angina precoce postinfartuale ha assunto negli ultimi anni particolare
rile-vanza in rapporto all’effettuazione in fase acuta della terapia trombolitica. Se
con la trombolisi si ottiene un’efficace riperfusione, ne consegue una limita-zione dell’area necrotica, ma simultaneamente viene a crearsi una zona di miocardio “a rischio” costituita dalla zona di miocardio che è stato salvato
dal-la trombolisi, ma che è esposto al danno ischemico. Questo rischio è legato sia
alla possibile riocclusione trombotica della coronaria, sia alla persistenza di
una grave stenosi residua, su cui il trombo occlusivo si era precedentemente
formato. Per questi motivi, anche se non si manifestano sintomi di angina, è
importante valutare, abitualmente prima della dimissione, la presenza di un’eventuale ischemia residua. Ciò si attua eseguendo un test da sforzo sotto-massimale, se necessario associato alla scintigrafia miocardica con 201Tl; tutti i
pazienti con ischemia residua, sia essa spontanea o indotta dal test da sforzo,
hanno indicazione a coronarografia per valutare l’eventuale necessità di una
rivascolarizzazione miocardica con by-pass o angioplastica coronarica.
1.7.4 Altre complicanze
In caso d’infarto miocardico transmurale è possibile rilevare, dal punto di vista
anatomopatologico, la presenza di una pericardite localizzata alla regione che
sovrasta la necrosi miocardica; tale patologia è detta pericardite
epistenocardi-ca, e si presenta nel 50% dei casi di IMA. Questa complicanza si verifica
42
al 16% dei pazienti con infarto miocardico si rilevano sfregamenti pericardici,
ma questa percentuale tuttavia sottostima la reale incidenza della pericardite,
perché spesso gli sfregamenti sono fugaci e quindi sfuggono all’osservazione.
Oltre agli sfregamenti, la caratteristica clinica più importante è il dolore
tora-cico, che spesso pone un problema di diagnosi differenziale con un dolore
i-schemico, specie se non si rilevano sfregamenti; tuttavia, le caratteristiche del
dolore pericardico (che aumenta con l’inspirazione, si riduce con l’assunzione
della posizione seduta e abitualmente ha localizzazione e proiezioni diverse di
quello ischemico) consentono la differenziazione. L’ECG può mostrare una
ripresa della lesione subepicardica e/o un’accentuazione dell’ischemia; nel
ca-so di una reazione pericarditica localizzata, Rx torace ed ecocardiogramma
non sono diagnostici, ma lo diventano, specie il secondo, alla presenza di un
versamento pericardico diffuso.
La sindrome di Dressler, o pericardite postinfartuale, che è in sostanza un caso
particolare di pericardite postpericardiotomica, è caratterizzata da febbre e da
dolore pleuropericardico; si ritiene che la sua genesi sia dovuta a un
meccani-smo autoimmune. Essa insorge da 1 a 6, talora a 12 settimane dopo un infarto.
Il quadro clinico è dominato dalla febbre, che può raggiungere anche i 40 °C, e
dal dolore con le caratteristiche della pericardite ed eventualmente della
pleu-rite; la presenza di sfregamenti pericardici è costante, ma spesso fugace e per questo essi possono sfuggire all’esame obiettivo. L’ECG può mostrare i segni distintivi della pericardite; in questo caso essi sono più facilmente
43
della necrosi sono stabilizzati; l’Rx torace e l’ecocardiogramma possono
di-mostrare un versamento pleurico e pericardico.
L’infarto miocardico è una situazione favorevole all’insorgenza di embolia polmonare e sistemica. L’embolia è più frequente negli infarti estesi,
compli-cati da shock e scompenso; l’embolia polmonare è un reperto relativamente
frequente in sede autoptica; a livello clinico, la sua insorgenza sfugge spesso all’osservazione se l’embolia è di modesta entità. L’embolia sistemica può conseguire alla formazione di trombi murali a livello del ventricolo sinistro;
essa è più frequente nell’infarto anteriore e della punta, specie in presenza di
aneurisma. Con l’ecocardiografia, trombi murali sono stati rilevati in oltre un
44
CAPITOLO 2 – LA TERAPIA RIPERFUSIVA DELL’IMA:
REVISIO-NE DELLA LETTERATURA ED APPROCCIO PRATICO
2.1 Le basi storiche dell’approccio riperfusivo
Negli ultimi 20 anni si sono compiuti notevoli progressi nella gestione dei pa-zienti con IMA, e in particolare dell’infarto STEMI, soprattutto a seguito del riconoscimento del ruolo patogenetico dei fenomeni trombotici associati alle
placche aterosclerotiche come trigger d’infarto del miocardio.1 Un’enfasi
sempre maggiore è stata posta sulla terapia riperfusiva, il cui ruolo è di
ripri-stinare precocemente la pervietà coronarica.
I primi metodi di riperfusione erano basati su un approccio invasivo: Rentrop, sul finire degli anni ‘70, è stato il primo a eseguire una riperfusione invasiva con ricanalizzazione meccanica, durante una complicazione di angiografia
co-ronarica; in seguito lui stesso ha aggiunto la fibrinolisi intracoronarica alla
ri-canalizzazione meccanica, formando la base per il trattamento trombolitico
in-tracoronarico dello STEMI.2
Nel 1980 le prime prove di uso del farmaco streptochinasi, direttamente al
li-vello intracoronarico, hanno mostrato una riduzione delle dimensioni
dell'in-farto, nonché una diminuzione della mortalità3; da questi si è passati a
succes-sivi trials in cui la fibrinolisi era eseguita per via endovenosa, poiché
l'angio-grafia coronarica di emergenza in quel periodo sembrava poco praticabile4, 5.
Si è passati in seguito ad una prima serie di studi aventi lo scopo di combinare
endove-45
nosa prima, per guadagnare tempo, ed in seguito una rapida angioplastica
co-ronarica; Purtroppo, questi tentativi di strategia farmacoinvasiva e meccanica
combinata hanno prodotto risultati clinici poveri rispetto alla fibrinolisi per via
endovenosa come trattamento esclusivo, e sono stati rapidamente
abbandona-ti6.
2.2 La terapia trombolitica
2.2.1 Meccanismo d’azione
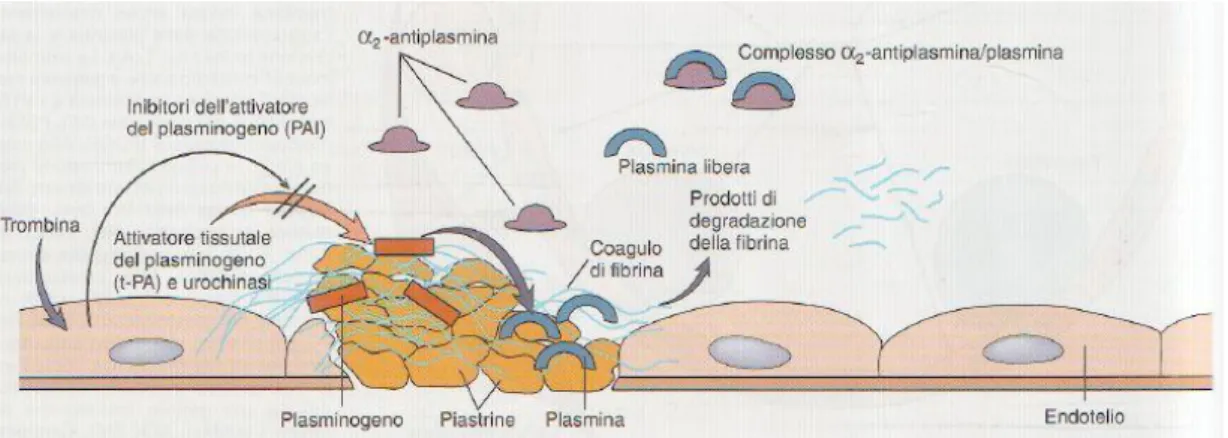
Tutti i farmaci di questo tipo agiscono convertendo il plasminogeno, un proen-zima circolante, in plasmina, l’enproen-zima attivo; questa degrada la matrice di fi-brina del trombo, formando prodotti di degradazione solubili della fifi-brina. La fibrinolisi endogena è regolata a due livelli. Gli inibitori dell’attivatore del plasminogeno (PAI) prevengono l’eccessiva attivazione del plasminogeno, re-golando l’attività dell’attivatore tissutale del plasminogeno (t-PA) e di quello di tipo urochinasico (u-PA). Una volta formata la plasmina, essa è regolata dai suoi inibitori, il più importante dei quali è l’α2-antiplasmina, cui consegue che, per dosi farmacologiche di attivatori del plasminogeno, la concentrazione di plasmina così formata sia superiore a quella dell’α2-antiplasmina.
Il sistema fibrinolitico endogeno è in grado di indirizzare la formazione di
pla-smina specificatamente sulla fibrina del coagulo: plasminogeno e t-PA legano
la fibrina e formano un complesso ternario in grado di promuovere l’attivazione del plasminogeno; al contrario della plasmina libera, quella
atti-46
vata in questo modo è protetta dall’inattivazione, e la fibrina così degradata at-tiva a cascata altro plasminogeno e t-PA, in un feedback positivo.
La plasmina svincolata dalla sua regolazione, cioè non legata alla fibrina, può
degradare anche il fibrinogeno e altri fattori della coagulazione, determinando
uno stato litico sistemico, che è alla base del rischio emorragico di questi
far-maci. Lo sviluppo farmacologico si è quindi indirizzato su sostanze in grado di
attivare il plasminogeno legato alla fibrina (dette fibrino - specifiche) rispetto
ad altre attivanti il plasminogeno libero.
Figura 8 – Rappresentazione schematica del sistema fibrinolitico (da Robbins e Cotran, “le basi patologi-che delle malattie” 7ma ed., 2006.)
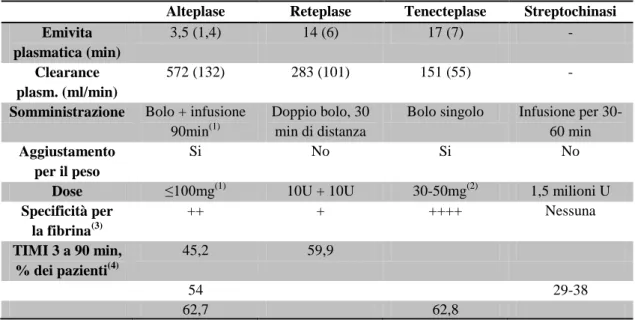
2.2.2 l’approccio fibrinolitico endovenoso: farmaci ed evoluzione
Dal punto di vista dell’approccio farmacologico allo STEMI, l’attenzione in seguito a questi primi studi si è spostata verso farmaci fibrinolitici che
consen-tissero una maggior maneggevolezza, sia in termini di sicurezza sia di
effica-cia; il farmaco storico nell’ambito dei fibrinolitici è la streptochinasi (SK), una