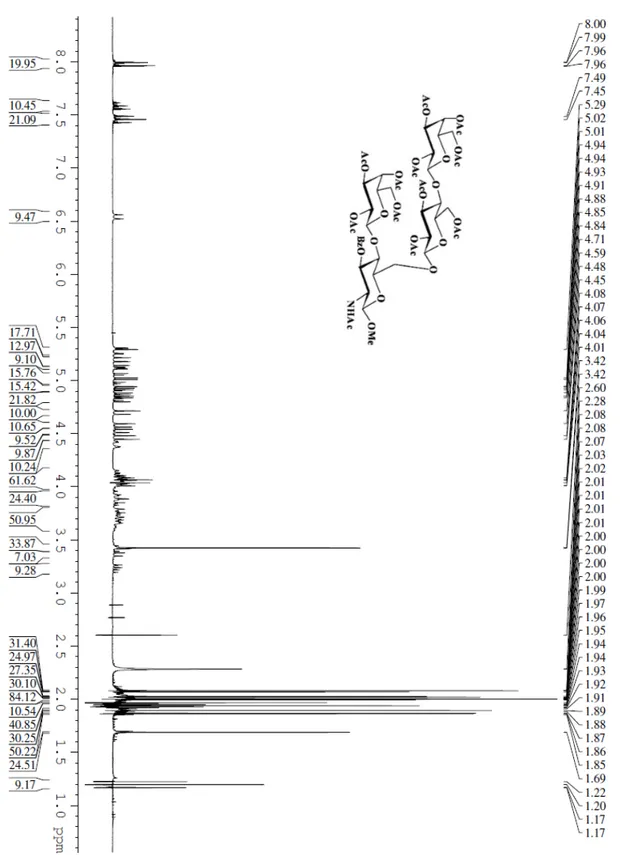
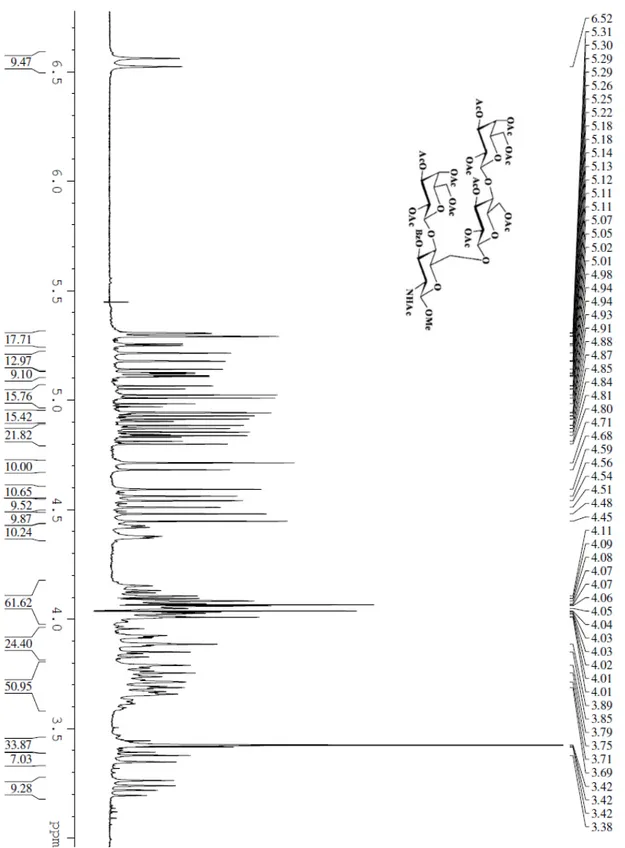
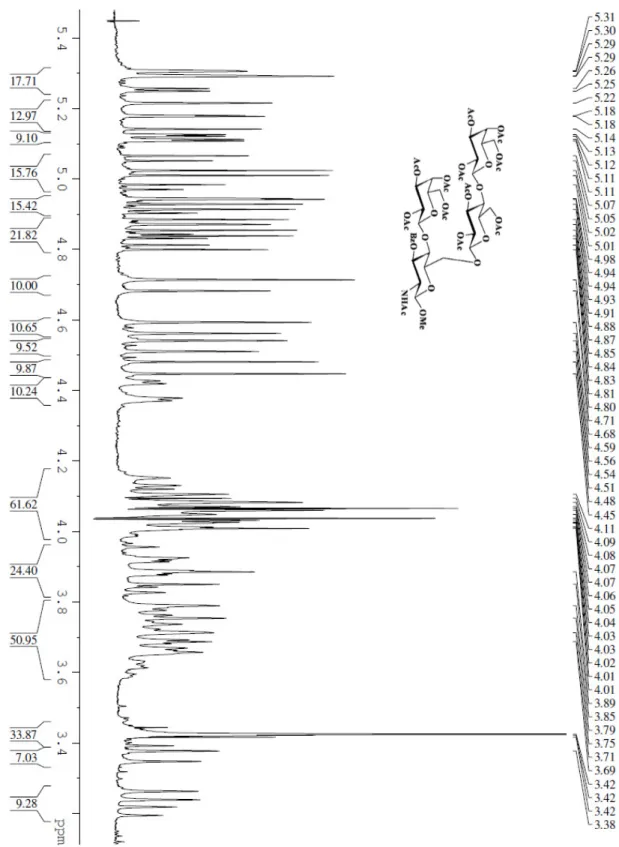
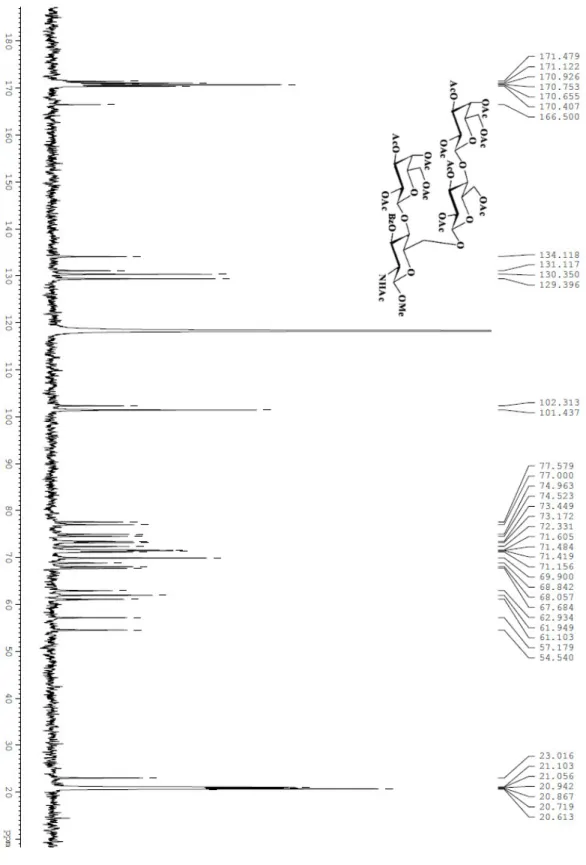
3. Parte Sperimentale
Metodiche generali: reattivi, solventi e strumentazione
I punti di fusione sono stati determinati al microscopio su apparecchio Kofler, senza ulteriore correzione. Le misure del potere ottico rotatorio sono state effettuate con un polarimetro Perkin-Elmer mod. 241, alla temperatura di 20 ± 2°C.
L’analisi NMR è stata eseguita con uno spettrometro Bruker Avance II 250 operante con radiofrequenza di 250.13 MHz per il 1H e di 62.90 MHz per il 13C. Gli spettri sono stati determinati su soluzione dei solventi riportati, impiegando come riferimento interno CD3CN nel caso della registrazione in D2O e tetrametilsilano (TMS) in tutti gli altri casi.
Nel caso della caratterizzazione spettroscopica di prodotti in miscela, le attribuzioni sono basate sulle differenze di intensità dei segnali (13C NMR) o sui valori degli integrali. La determinazione dei dati spettroscopici di tutti i composti preparati in questo lavoro di tesi è effettuata attraverso l’uso di tecniche NMR monodimensionali (1H e 13C) e bidimensionali (DEPT-135, COSY, HETCOR).
Le TLC analitiche sono state eseguite su fogli di silice supportata su alluminio di tipo DC- Alufolien Kieselgel 60 F254. La rivelazione dei composti è effettuata con una lampada
U.V. (254 nm), immergendo i fogli sia in una soluzione al 10% di H2SO4 che di acido
fosfomolibdico in EtOH e riscaldando.
La silice usata è Kieselgel 60 Merk (230-400 mesh) per cromatografie flash. I solventi utilizzati per i processi di purificazione (cromatografia su colonna, cristallizzazione, ecc.) sono usati dopo distillazione in accordo con le procedure standard.31 Le miscele dei solventi usate nelle reazioni e nelle purificazioni cromatografiche sono riportate come v/v.
Come agente essiccante delle soluzioni è stato usato MgSO4 anidro (Fluka).
La dimetilformammide (DMF) usata è quella anidra disponibile in commercio (Fluka) garantita e confezionata in bottiglie con tappo “Sure-Seal”.
I solventi anidri utilizzati nelle reazioni sono stati purificati come segue, e conservati sotto atmosfera di Argon e su setacci molecolari 4Ǻ attivati.
Il diclorometano (CH2Cl2) anidro è stato preparato dal prodotto commerciale refluito e
distillato da P2O5.
Il metanolo (MeOH) anidro è stato preparato dal prodotto commerciale refluito e distillato da CaH2 e conservato sotto atmosfera di Argon su setacci molecolari 3Ǻ attivati.
Il toluene (PhCH3) anidro è stato preparato dal rispettivo prodotto commerciale refluito
e distillato da Na metallico.
I setacci molecolari 4Ǻ e AW300 polverizzati (Aldrich) sono stati attivati per permanenza per 12 ore a 130°C a pressione ridotta (0.01 mmHg).
La resina acida Amberlyst-15 è quella commerciale lavata con MeOH.
Le reazioni che richiedono un ambiente anidro sono state condotte sotto atmosfera di Argon in pallone munito di chiusura con setto di gomma e le aggiunte successive, se necessarie, sono state effettuate con una siringa.
Preparazione del 2-acetammido-1,3,4,6-tetra-O-acetil-2-desossi-αα-ααD-glucopiranosio
(41)
In un pallone da 250 ml si sospendono 4.00 g (18.6 mmoli, 1 eq) di glucosammina cloridrato commerciale (40) in 52.5 ml di una miscela 2:1 di piridina e Ac2O e la
sospensione è lasciata in agitazione a temperatura ambiente. Dopo 20 ore l’analisi TLC (AcOEt) evidenzia la scomparsa del prodotto di partenza (Rf 0) e la formazione di un unico
prodotto a Rf 0.30. L’eliminazione dei reagenti a pressione ridotta coevaporando con
toluene (3x50 ml) fornisce un grezzo di reazione (11.5 g) che, purificato mediante flash cromatografia (esano-AcOEt 9:1), permette di isolare 41 (7.01 g, resa 97%) come solido bianco chimicamente puro all’analisi 1H e 13C NMR. La reazione è stata ripetuta nelle stesse condizioni e il derivato 41 è stato isolato puro (7.4 g, resa 82%) mediante cristallizzazione da AcOEt.
Il prodotto 41 è un solido bianco che presenta Rf 0.30 (AcOEt) e
caratteristiche chimico fisiche analoghe a quelle riportate in letteratura.171H NMR (CDCl3): δ 6.17 (d, 1H, J1,2 3.6 Hz, H-1), 5.95 (d,1H, J2,NH 9.1 Hz,
NH), 5.23 (m, 2H, H-3, H-4), 4.30 (m, 1H, H-5), 4.25 (dd, 1H, J6a, 6b 12.5 Hz, J5,6 b 4.2 Hz,
H-6b), 4.08 (m, 2H, H-2, H-6a), 2.09, 2.06, 2.05 (3s, each 3H, MeCOO), 1.94 (s, 3H, MeCON); 13C NMR (CDCl3): δ 171.4, 170.6, 170.1, 169.0, 168.6 (5 x CO), 90.5 (C-1),
70.4 (C-3), 69.7 (C-5),, 67.4 (C-4) 61.4 (C-6), 50.8 (C-2), 22.8 (MeCON), 20.7-20.5 (4 x MeCOO). I parametri NMR (1H, 13C) sono in accordo con quanto riportato in letteratura.17
Preparazione del 2-ammino-3,4,6-tri-O-acetil-2-desossi-1-O-2-N-(etan-1-il-1-ilidene)-α-D-glucopiranosio (42)
In un pallone a due colli da 250 ml, fiammeggiato e sotto atmosfera di Argon, si solubilizzano 14.0 g (36.0 mmoli, 1 eq) di 41 in 168 ml di dicloroetano (DCE) anidro e la soluzione è lasciata in agitazione magnetica a 50°C sotto atmosfera inerte. Dopo 10 minuti si aggiungono 1.21 ml ( 41.0 mmoli, 1.14 eq) di TMSOTf e la soluzione rosa è lasciata in agitazione magnetica a 50°C. Dopo 20 ore l’analisi TLC (CHCl3-MeOH 95:5) mostra la
completa scomparsa del prodotto di partenza (Rf 0.35) e la formazione di un unico prodotto
a Rf 0.43. La miscela di reazione è neutralizzata con Et3N (2 ml), trasferita in un pallone ad
un collo (250 ml) e concentrata a pressione ridotta. La purificazione flash cromatografica su gel di silice (esano-AcOEt 1:9 + 0.1% di Et3N) del grezzo (26.5 g) fornisce 2 (13.4 g,
resa 95%) chimicamente puro all’analisi NMR (1H,13C).
O OAc AcO AcO AcNH OAc
L’ossazolidina prodotto 42 è uno sciroppo giallo che presenta Rf 0.43
(CHCl3-MeOH 95:5) e caratteristiche chimico fisiche analoghe a quelle
riportate in letteratura:19a [α]
D +50 (c 1.4; CHCl3); 1H NMR (CDCl3): δ 5.99
(d, 1H, J1,2 7.4 Hz, H-1), 5.23 (t,1H, J2,3=J3,4 2.6 Hz, H-3), 4.90 (bd, 1H, J4,5 9.1 Hz, H-4),
4.12 (m, 3H, H-2, H-6a, H-6b), 3.61 (m, 1H, H-5), 2.11, 2.09, 2.08 (3s, each 3H, MeCOO);
13C NMR (CDCl
3): δ 169.7, 168.7, 168.4 (3 x C=O), 165.8 (C=N), 98.6 (C-1), 69.5 (C-3),
67.7 (C-4), 66.7 (C-5), 64.2 (C-2), 62.6 (C-6), 20.1-19.9 (3 x MeCO), 13.1 (Me). I parametri NMR (1H, 13C) sono in accordo con quanto riportato in letteratura.18b
Preparazione del 3-O-tosil-1,3-propandiolo (46a)
In un pallone da 100 ml munito di imbuto gocciolatore e agitazione meccanica sono solubilizzati 3.33 g (17.5 mmoli) di cloruro di tosile in 30 ml di una soluzione di Py-CH2Cl2 1:1 (v/v). Alla soluzione raffreddata a 0°C con un bagno di H2O-ghiaccio si
addiziona lentamente una soluzione formata da 5 ml di 1,3-propandiolo (69 m.moli) in 16 ml di miscela Py-CH2Cl2 1:1. Dopo 20 minuti, terminato il gocciolamento, si allontana il
raffreddamento, si porta la miscela a temperatura ambiente e si segue l’evolvere della reazione mediante analisi TLC (esano-AcOEt 2:3). Dopo 2 ore l’analisi TLC evidenzia la presenza del diolo in eccesso (Rf 0.10) e la formazione di un prodotto prevalente (Rf 0.31).
Si addiziona H2O (75 ml) e CH2Cl2 (75 ml), si separano le fasi e la fase acquosa è estratta
con CH2Cl2 (4x50 ml). Le fasi organiche riunite, lavate prima con HCl al 5% e poi con una
soluzione satura di NaHCO3 (70 ml), sono seccate, filtrate e concentrate a pressione ridotta.
La purificazione flash cromatografica su gel di silice (esano-AcOEt 55:45) del grezzo di reazione (3.90 g) ha permesso di isolare l’alcol 3 in resa del 58%, puro all’analisi NMR (1H, 13C).
L’alcol monotosilico 3 è uno sciroppo incolore avente Rf 0.31
(esano-AcOEt 2:3); 1H NMR (CDCl3): δ 7.76, 7.32 (sist. AA’XX’, 4H, Ar-H),
4.15 (t, 2H, J 6.1 Hz, CH2OTs), 3.68 (t, 2H, J6.0 Hz, CH2OH), 2.42 (s, 3H, MePh), 1.85
(q, 2H, CH2CH2CH2), 1.68 (s, 1H, OH); 13C NMR (CDCl3): δ 144.8, 132.8 (2xAr-C),
129.8, 127.8 (4xArCH), 67.43 (CH2OTs), 58.2 (CH2OH), 31.6 (CH2), 21.6 (MePh). I
parametri NMR (1H, 13C) sono perfettamente in accordo con quanto riportato in
letteratura.20a O OAc AcO AcO N O OTs O H
Preparazione del 3-N-t-butossicarbonil-amminopropanolo (46b)
In un pallone da 50 ml si solubilizzano 0.76 ml (10.0 mmoli, 1 eq) di 3-ammino propanolo in 6 ml di CH2Cl2, si raffredda la soluzione a 0°C e lentamente si addiziona una
soluzione costituita da 2.18 g (10.0 mmoli, 1 eq) di Boc2O in CH2Cl2 (1.5 ml) Si lascia in
agitazione a temperatura ambiente e si segue l’evolvere della reazione per TLC (Et2O).
Dopo 3 ore l’analisi TLC evidenzia la scomparsa del prodotto di partenza (Rf 0) e la
formazione di un unico prodotto a Rf 0.47. Si diluisce la miscela di reazione con 15 ml di
CH2Cl2, si aggiunge una soluzione acquosa satura di NaCl (20 ml), si separano le fasi e
quella acquosa è ulteriormente estratta con CH2Cl2 (2x20 ml). Le fasi organiche riunite,
anidrificate con MgSO4, filtrate e concentrate a pressione ridotta forniscono un grezzo che,
purificato mediante flash cromatografia su colonna di gel di silice (esano-AcOEt 1:1), permette di isolare il derivato 3a (1.56 g, resa 89%) chimicamente puro all’analisi 1H e 13C NMR.
Il composto 3a è un olio incolore che presenta Rf 0.47 (Et2O) e
caratteristiche chimico-fisiche analoghe a quelle riportate in letteratura:20b1H NMR (CDCl
3) δ: 4.78 (bs, 1H, NH), 3.66 (bs, 2H,
OCH2), 3.28 (m, 2H, CH2NH), 2.97 (bs, 1H, OH), 1.67 (m, 2H, CH2CH2CH2), 1.44 (s, 9H,
CMe3); 13C NMR (CDCl3) δ: 157.0 (C=O), 79.4 (CMe3), 59.3 (OCH2), 37.0 (CH2NH),
32.6 (CH2CH2CH2), 28.3 (CMe3). I parametri NMR (1H, 13C) sono in accordo con quanto
riportato in letteratura.20b
Preparazione del metil 2-acetamido-3,4,6-tri-O-acetil-2-desossi-ββββ-D-glucopiranoside
(47)
Metodo A: [con MeOH e CSA] - In un pallone a due colli da 50 ml, fiammeggiato e
sotto atmosfera di Argon, si solubilizzano 600 mg (1.82 mmol, 1 eq) dell’ossazolidina 42 in DCE anidro (14.5 ml), la soluzione è portata alla temperatura di 80°C e si aggiungono, in successione, 169 mg (0.73 mmoli, 0.4eq) di CSA e 0.30 ml (7.30 mmoli, 4 eq) di MeOH anidro. La miscela è lasciata in agitazione a 80°C e si segue l’evolvere della reazione per TLC (CH3Cl-MeOH 95:5). Dopo 45 minuti si evidenzia la scomparsa del prodotto di
partenza (Rf 0.43) e la formazione di un prodotto prevalente a Rf 0.37. La miscela di
reazione è diluita con CH2Cl2 (10 ml) e lavata prima con una soluzione acquosa satura di
NaHCO3 (5 ml) e poi con una soluzione acquosa satura di NaCl (5 ml). Le fasi acquose
riunite sono anidrificate con MgSO4, filtrate e concentrate a pressione ridotta. La
O
H N
H O O
purificazione flash cromatografica su colonna di gel di silice (CH3Cl-MeOH 98:2 + 0.1%
Et3N) del grezzo di reazione (540 mg) permette di isolare il glicoside 47 (379 mg, resa
58%) chimicamente puro all’analisi NMR (1H, 13C).
Metodo B: [con MeOH e BF3Et2O] In un pallone a due colli da 25 ml, fiammeggiato e
anidrificato in atmosfera di Argon, si solubilizzano 500 mg (1.52 mmoli, 1 eq) dell’ossazolidina 42 in 3.5 ml di MeOH anidro (48 eq), si raffredda la soluzione a 0°C, si aggiungono 510 µl (0.27 eq) di una miscela 1:10 di BF3Et2O e MeOH anidro e si lascia in
agitazione a temperatura ambiente seguendo l’evolvere della reazione per TLC (CH3
Cl-MeOH 95:5). Dopo 12 ore l’analisi TLC evidenzia, oltre al prodotto di partenza (Rf 0.43),
la formazione di un prodotto a Rf 0.37 e si decide di aggiungere ulteriori 19 µl (0.1 eq) di
miscela 1:10 di BF3Et2O in MeOH anidro. Dopo 7 ore a temperatura ambiente, scomparso
il prodotto di partenza (TLC), si neutralizza con Et3N (2 ml), si trasferisce la miscela di
reazione in un pallone ad un collo e si concentra a pressione ridotta. La purificazione flash cromatografica su gel di silice (esano-AcOEt 5:95 + 0.1% di Et3N) del grezzo (589 mg)
permette di isolare il metil glicoside 47 (277 mg, resa 50%) chimicamente puro all’analisi NMR (1H, 13C).
Il metil glicoside 47 è un solido bianco avente Rf 0.37 (CH3Cl-MeOH
95:5) e caratteristiche chimico fisiche analoghe a quelle riportate in letteratura:21[α]D -12 (c 1.0; CHCl3); p.f. 162°C (EtOH); 1H NMR
(CDCl3): δ 6.13 (d, 1H, J2,NH 8.3 Hz, NH), 5.23 (dd,1H, J2,3 10.0 Hz, J3,4 9.6 Hz, H-3), 5.00
(dd, 1H, J4,5 9.2 Hz, H-4), 4.55 (d, 1H, J1,2 8.1 Hz, H-1), 4.23 (dd, 1H, J5,6b 4.2 Hz, J6a,6b
12.1 Hz, H-6b), 4.07 (bd, 1H, H-6a), 3.82 (m, 1H, H-2), 3.66 (m, 1H, H-5), 3.44 (s, 3H, CH3O), 2.02 (s, 3H, MeCOO), 1.96 (s, 6H, 2 x MeCOO); 1.89 (s, 3H, MeCON); 13C NMR
(CDCl3): δ 170.7, 170.6, 170.4, 169.3 (4 x C=O), 101.4 (C-1), 72.3 (C-3), 71.5 (C-4), 68.6
(C-5), 62.0 (C-6), 56.7 (CH3O), 54.3 (C-2), 23.1 (MeCON), 20.5 (3 x MeCOO). I
parametri NMR (1H, 13C) sono in accordo con quanto riportato in letteratura.21
Preparazione del N-(t-butilossicarbonil)-amminopropil 2-acetammido-3,4,6-tri-O-acetil-2-desossi-ββ-ββ D-glucopiranoside (48)
In un pallone a due colli da 50 ml, fiammeggiato e sotto atmosfera di Argon, si attivano 200 mg di setacci molecolari 4Å, si introducono 200 mg (0.61 mmoli, 1 eq) dell’ossazolidina 42, 159.6 mg (0.91 mmoli, 1.5 eq) dell’accettore 46b, 3 ml di DCE
O OAc AcO AcO NHAc OMe
anidro, e si lascia in agitazione a temperatura ambiente per 30 minuti. Si aggiungono 11 µl (0.06 mmoli, 0.1 eq) di TMSOTf e la miscela di reazione è lasciata in agitazione a temperatura ambiente seguendo l’evolvere della reazione per TLC (AcOEt). Dopo 5 giorni si evidenzia la scomparsa del prodotto di partenza e la formazione di un prodotto prevalente a Rf 0.21. Si neutralizza la sospensione con Et3N (2 ml), si filtra su setto poroso
coperto da celite e la soluzione è concentrata a pressione ridotta. La purificazione flash cromatografica su gel di silice (esano-AcOEt 15:85 + 0.1% di Et3N) del grezzo (516 mg)
permette di isolare il glicoside 7 (178 mg, resa 58%) chimicamente puro all’analisi NMR (1H, 13C).
Il glicoside 48 è un solido rosa avente Rf 0.21 (AcOEt),
[α]D -46.7 (c 1.0, CHCl3); p.f. 170-174°C; 1H NMR
(CDCl3): δ 6.88 (d, 1H, J2,NH 8.8 Hz, NHAc), 5.08 (dd,
1H, J2,3 10.3 Hz, J3,4 9.5 Hz, H-3), 5.02 (dd, 1H,J4,5 9.5 Hz, H-4), 5.01 (bt, 1H, NHBoc),
4.50 (d, 1H, J1,2 8.5 Hz, H-1), 4.23 (dd, 1H, J5,6b 4.6 Hz, J6a,6b 12.2 Hz, H-6b), 4.07 (dd, 1H,
J5,6a 2.0 Hz, H-6a), 3.96 (m, 1H, H-2), 3.95, 3.48 (2m, each 1H, CH2O), 3.64 (m, 1H, H-5),
3.41, 3.01 (2m, each 1H, CH2NH), 2.05 (s, 3H, MeCOO), 1.98 (s, 6H, 2 x MeCOO), 1.92
(s, 3H, MeCON), 1.70 (m, 2H, CH2), 1.41 (s, 9H, CMe3); 13C NMR (CDCl3): δ 170.5,
170.4, 170.3, 169.1 (C=O), 156.1 (OCON), 101.0 (C-1), 79.0 (CMe3), 72.9 3), 71.4
(C-5), 68.3 (C-4), 61.8 (C-6), 66.7 (CH2O), 53.7 (C-2), 36.2 (CH2NH), 29.5 (CH2), 28.0
(CMe3), 22.7 (MeCON), 20.4, 20.3, 20.2 (MeCOO). Analisi Elementare calcolata per
C22H36N2O11 (504.54): C, 52.37; H, 7.19; N, 5.55. Trovato: C, 52.39; H, 7.21; N, 5.57.
Preparazione del 3-O-tosilpropil 2-acetammido-3,4,6-tri-O-acetil-2-desossi-ββ-ββ D -glucopiranoside (49)
In un pallone a due colli da 250 ml, fiammeggiato e sotto atmosfera di Argon, si attivano 3.60 g di setacci molecolari 4Å, si addiziona una soluzione formata da 4.64 g (14.1 mmoli, 1 eq) dell’ossazolidina 42 in 56 ml di DCE anidro e la sospensione è posta in agitazione magnetica alla temperatura di 80 °C. Si addizionano, in successione, 1.64 g (7.05 mmoli, 0.5 eq) di CSA e 4.87 g (21.1 mmoli, 1.5 eq) di alcol monotosilico (46a) e la miscela di reazione è lasciata in agitazione ad 80°C sotto atmosfera inerte. Dopo 12 ore l’analisi TLC (AcOEt) evidenzia la scomparsa del prodotto di partenza (Rf 0.37), la
presenza dell’alcol 3 (Rf 0.55) e la formazione di un prodotto prevalente (visibile all’UV) a
Rf 0.40. La miscela di reazione è filtrata su strato di celite ed il filtrato, diluito con CH2Cl2
N H O O OAc AcO AcO NHAc O O
(35 ml), è lavato prima con una soluzione satura di NaHCO3 (50 ml) e successivamente
con una soluzione satura di NaCl (50 ml). La fase organica, anidrificata con MgSO4, filtrata
e concentrata a pressione ridotta fornisce un grezzo (5.90 g) che è sottoposto a purificazione flash cromatografica su gel di silice eluendo con esano-AcOEt 2:8 + 0.1% di Et3N. Si isola 5 (5.50 g, resa 69%) chimicamente puro all’analisi NMR (1H, 13C).
Il glicoside 48 è una schiuma bianca avente Rf 0.40 (AcOEt),
[α]D - 47.6 (c 1.05, CHCl3); 1H NMR (CD3CN): δ 7.88, 7.55
(sistema AA’XX’, each 2H, Ar-H), 6.52 (d, 1H, J2,NH 9.1 Hz,
NH), 5.21 (dd, 1H, J2,3 10.6 Hz, J3,4 9.4 Hz, H-3), 5.02 (dd, 1H,J4,5 10.0 Hz, H-4), 4.66 (d,
1H, J1,2 8.5 Hz, H-1), 4.29 (dd, 1H, J5,6b 5.0 Hz, J6a,6b 12.2 Hz, 6b), 4.20-4.05 (m, 3H,
H-6b, CH2OTs), 3.91-3.75 (m, 2H, H-2, H-5), 3.84 (m, 1H, CH2O), 3.63 (dt, 1H, J 6.3 Hz, J
10.4 Hz, CH2O), 2.54 (s, 3H, MePh), 2.10, 2.06, 2.03 (3s, each 3H, MeCOO), 1.94 (m, 2H,
CH2), 1.88 (s, 3H, MeCON); 13C NMR (CD3CN): δ 171.3, 171.1, 170.8, 170.5 (4 x C=O),
146.3, 133.7 (2 x Ar-C), 131.0, 128.6 (Ar-CH), 101.5 (C-1), 73.4 (C-5), 72.3 (C-4), 69.6 (C-3), 68.7 (CH2OTs), 66.1 (CH2O), 62.9 (C-6), 54.6 (C-2), 29.7 (CH2), 23.0 (MeCON),
21.6 (MePh), 20.8 (3 x MeCOO). Analisi Elementare calcolata per C24H33NO12S (559.59):
C, 51.51; H, 5.94; N, 2.50. Trovato: C, 51.53; H, 5.96; N, 2.52.
Preparazione del 3-azidopropil 2-acetammido-3,4,6-tri-O-acetil-2-desossi-ββ-ββ D
-glucopiranoside (50)
In un pallone da 250 ml si solubilizzano, in atmosfera di Argon, 5.46 g (9.7 mmoli, 1 eq) del glicoside 49, in 130 ml di DMF anidra, si addizionano 1.91 g di NaN3 (29.3 mmoli,
3 eq) e 7.22 g (19.5 mmoli, 2 eq) di TBAI e la soluzione è lasciata in agitazione a 50 °C sotto atmosfera inerte. Dopo 3 ore l’analisi TLC (AcOEt) evidenzia la scomparsa del prodotto di partenza (Rf 0.40) e la formazione di un unico prodotto (Rf 0.45) che brucia con
una colorazione arancio e non visibile all’UV. Si concentra a pressione ridotta, si ripartisce il residuo fra CH2Cl2 (50 ml) e H2O (50 ml), si separano le fasi e quella acquosa è
ulteriormente estratta con CH2Cl2 (3x30 ml). Le fasi organiche riunite, anidrificate con
MgSO4, filtrate e concentrate a pressione ridotta forniscono un grezzo che viene purificato
mediante flash cromatografia su gel di silice eluendo con esano-AcOEt 2:8 + 0.1% di Et3N.
Si isola l’azide 6 (4.14 g, resa 98%) chimicamente pura all’analisi NMR (1H, 13C).
L’azide 50 è un solido giallino avente Rf 0.45 (AcOEt), [α]D-0.96
(c 1.04, CHCl ); p.f. 115-117 °C (sul cromatografato); 1H NMR OTs O O OAc AcO AcO NHAc O O OAc AcO N
(CD3CN): δ 6.50 (d, 1H, J2,NH 9.6 Hz, NH), 5.15 (dd, 1H, J2,3 10.6 Hz, J3,4 9.5 Hz, H-3),
4.93 (dd, 1H,J4,5 10.0 Hz, H-4), 4.62 (d, 1H, J 1,2 8.5 Hz, H-1), 4.21 (dd, 1H, J5,6b 5.0 Hz,
J6a,6b 12.2 Hz, H-6b), 4.05 (dd, 1H, J5,6a 2.5 Hz, H-6a), 3.85-3.72 (m, 2H, H-2, H-5), 3.85
(dt, 1H, J 10.3 Hz, 6.0 Hz, CH2O), 3.57 (dt, 1H, J 6.2 Hz, J 10.3 Hz, CH2O), 3.35 (t, 2H, J
6.7 Hz, CH2N3), 2.02, 1.97, 1.94, (3s, each 3H, MeCOO), 1.86 (m, 2H, CH2), 1.83 (s, 3H,
MeCON); 13C NMR (CD3CN): δ 171.3, 171.1, 170.8, 170.6 (4 x C=O), 101.6 (C-1), 73.4
(C-3), 72.3 (C-5), 69.7 (C-4), 67.3 (CH2O), 62.9 (C-6), 54.7 (C-2), 48.8 (CH2N3), 29.5
(CH2), 23.1 (MeCON), 20.8 (3 x MeCOO). Analisi Elementare Calcalata per C17H26N4O9
(430.42): C, 47.44; H, 6.09; N, 13.02. Trovato: C, 47.46; H, 6.11; N, 13.04.
Preparazione del 3-azidopropil 2-acetammido-2-desossi-4,6-O-isopropilidene-ββ-ββ D -glucopiranoside (54)
In un pallone da 100 ml si solubilizzano 1.12 g (2.61 mmoli, 1 eq) di 50 in 8 ml di MeOH, si raffredda la soluzione a 0°C, si aggiungono 1.3 ml di una soluzione di MeONa in MeOH 0.33 M e si lascia la miscela di reazione in agitazione a 0°C. Dopo 1.5 ore l’analisi TLC (CH3Cl-MeOH 8:2) mostra la scomparsa del prodotto di partenza (Rf 0.90) la
formazione di un unico prodotto a Rf 0.21. La soluzione è neutralizza con resina acida
Amberlyst (H+), filtrata e concentrata a pressione ridotta. Si ottiene un grezzo (794 mg, resa quantitativa) che all’analisi NMR (1H e 13C) risulta costituito esclusivamente dal triolo
52 che viene utilizzato nella reazione successiva senza ulteriori purificazioni.
Il triolo 52 è un solido bianco avente Rf 0.21 (CH3Cl-MeOH 8:2),
[α]D-11.0 (c 1.03, MeOH); p.f. 170-172 °C (grezzo). Lo spettro 1H NMR (CD
3CN) consente di individuare solo alcuni segnali
diagnostici a δ 8.03 (d, 1H, J2,NH 9.0 Hz, NH), 4.38 (d, 1H, J1,2 8.3 Hz, H-1), 3.35 (t, 2H, J
6.8 Hz, CH2N3), 1.90 (s, 3H, MeCON); 1.82 (m, 2H, CH2); 13C NMR (CD3CN): δ 173.6
(C=O), 102.5 (C-1), 77.5, 75.6, 71.8 (C-3, C-5, C-4), 67.0 (CH2O), 62.5(C-6), 57.0 (C-2),
49.0 (CH2N3), 29.8 (CH2), 23.0 (MeCON).
In un pallone da 50 ml, si sciolgono, sotto atmosfera di Argon, 794 mg di 52 grezzo, (2.61 mmoli, 1 eq) in 10 ml di DMF anidra, raffreddata la soluzione a 0°C, si addizionano 0.75 ml (7.83 mmoli, 3 eq) di 2-metossipropene (2-MP) distillato di fresco, 60.6 mg (0.261 mmoli, 0.1 eq) di CSA e la soluzione è lasciata in agitazione a temperatura ambiente e in atmosfera inerte. Dopo 1 ora l’analisi TLC (CHCl3-MeOH 9:1) evidenzia la presenza del
prodotto di partenza a (Rf 0.09) e la formazione di un unico prodotto a Rf 0.35, si raffredda
O O OH O H O H NHAc N3
la soluzione a 0°C e si addizionano ulteriori 100 µl (0.25 eq) di 2-metossipropene (2-MP). Dopo 3 ore, evidenziata (TLC) la completa conversione del prodotto di partenza nel derivato a Rf 0.35, si neutralizza la miscela di reazione con Et3N (0.5 ml) e si concentra la
soluzione a pressione ridotta. Il grezzo (1.10 g) è purificato mediante flash cromatografia su gel di silice (CHCl3-MeOH 94:6 + 0.1% di Et3N) e si isola il derivato 54 (793 mg, resa
88% calcolata da 6) chimicamente puro all’analisi NMR (1H, 13C).
Il composto 54 è una schiuma bianca avente Rf 0.35 (CHCl3
-MeOH 9:1), [α]D-46.5 (c 1.11, CHCl3); 1H NMR (CD3CN): δ
6.61 (d, 1H, J2,NH 9.2 Hz, NH), 4.40 (d, 1H, J1,2 8.3 Hz, H-1),
3.84-3.68 [m, 4H, H6a, H6b, CH2O (1H), OH-3], 3.63 (dd, 1H, J2,3 8.7 Hz, J3,4 9.5 Hz, H-3),
3.57-3.40 [m, 3H, H-2, H-4, CH2O (1H)], 3.34 (t, 2H, J6.7 Hz, CH2N3), 3.18 (dt, 1H, J4,5
9.8 Hz, J5,6a=J5,6b 5.6 Hz, H-5), 1.90 (s, 3H, MeCO), 1.76 (m, 2H, CH2), 1.46, 1.34 (2s,
each 3H, Me2C); 13C NMR (CD3CN): δ 171.6 (C=O), 102.6 (C-1), 100.2 (CMe2), 74.8
(C-4), 73.1 (C-3), 68.1 (C-5), 67.1 (CH2O), 62.6 (C-6), 57.6 (C-2), 48.8 (CH2N3), 29.6 (CH2),
29.4, 19.4 (Me2C), 23.3 (MeCO).Analisi Elementare calcolato per C14H24N4O6 (344.37): C,
48.83; H, 7.02; N, 16.27. Trovato: C, 48.86; H, 7.05; N, 16.29.
Preparazione del 3-azidopropil 2-acetamido-3-O-benzil-2-desossi-4,6-O-isopropilidene-ββ-ββ D-glucopiranoside (56)
In un pallone da 25 ml si solubilizzano 300 mg (0.87 mmoli, 1 eq) di 54 in 6 ml di THF umido, si aggiungono 5.75 mg (0.022 mmoli, 0.025 eq) di 18-crown-6, si raffredda la soluzione a 0°C, si aggiungono 195 mg (3.48 mmoli, 4 eq) di KOH polverizzato, si lascia in agitazione per trenta minuti, si addizionano 0.21 ml (1.74 mmoli, 12 eq) di BnBr e la sospensione è lasciata in agitazione a temperatura ambiente. Dopo 4 ore l’analisi TLC (AcOEt) evidenzia la completa scomparsa del prodotto di partenza (Rf 0.14) e la
formazione di un unico prodotto, visibile all’UV, a Rf 0.57. Si raffredda la sospensione con
un bagno di acqua-ghiaccio, si aggiunge lentamente MeOH (2 ml) e si eliminano i solventi a pressione ridotta. Il residuo è ripartito fra CH2Cl2 (5 ml) e H2O (5 ml), si separano le fasi
e quella acquosa è estratta con CH2Cl2 (3x10 ml). Le fasi organiche riunite, anidrificate
con MgSO4, filtrate e concentrate a pressione ridotta forniscono un grezzo (434 mg) che,
purificato mediante flash cromatografia su gel di silice (esano-AcOEt 1:1 + 0.1% di Et3N),
fornisce il 3-O-benzil derivato 56 (275 mg, resa 75%) chimicamente puro all’analisi 1H e
13C NMR. O O O O O H NHAc N3
In una ripetizione della sequenza desacetilazione-isopropilidenazione-benzilazione, il 3-O-benzil derivato 56 è stato ottenuto, senza purificazioni intermedie, dal tri-O-acetil derivato 50 in resa complessiva del 58%.
Il prodotto 56 è un solido bianco avente Rf 0.23 (esano-AcOEt
1:1); p.f. 138-141°C (cromatografato); [α]D +18.5 (c 1.0,
CHCl3); 1H NMR (CD3CN): δ 7.37-7.24 (m, 5H, Ar-H), 6.52
(d, 1H, J2,NH 9.4 Hz, NH), 4.74, 4.57 (sistema AB, 2H, JA,B 12.0 Hz, CH2Ph), 4.43 (d, 1H,
J1,2 8.4 Hz, H-1), 3.84 (dt, 1H, J5.7 Hz, J10.3 Hz, CH2O), 3.81-3.66 (m, 3H, H-4, H6a
,H6b), 3.73 (m, 1H, J2,3 8.8 Hz, H-2), 3.52 (dd, 1H, J3,4 10.3 Hz, H-3), 3.51 (dt, 1H, J6.3
Hz, J10.3 Hz, CH2O), 3.33 (t, 2H, J6.7 Hz, CH2N3), 3.22 (dt, 1H, J 4,5 9.8 Hz, J5,6a = J 5,6b
5.5 Hz, H-5), 1.85 (s, 3H, MeCO), 1.77 (m, 2H, CH2), 1.49, 1.37 (2s, each 3H, Me2C); 13C
NMR (CD3CN): δ 170.7 (C=O), 140.0 (Ar-C), 129.1-128.3 (Ar-CH), 102.9 (C-1), 100.1
(CMe2), 79.9 (C-3), 75.3 (C-4), 74.2 (CH2Ph), 67.8 (C-5), 67.0 (CH2O), 62.8 (C-6), 55.7
(C-2), 48.7 (CH2N3), 29.5 (CH2), 29.3, 19.5 (Me2C), 23.3 (MeCO). Analisi Elementare
calcolato per C21H30N4O6 (434.50): C, 58.05; H, 6.96; N, 12.89. Trovato: C, 58.08; H, 6.99;
N, 12.92.
Preparazione del 3-azidopropil 2-acetammido-3-O-acetil-2-desossi-4,6-O-isopropilidene-ββ-ββ D-glucopiranoside (57)
In un pallone da 25 ml si solubilizzano 360 mg (1.04 mmoli, 1 eq) di 54 in 2 ml di piridina, si aggiungono 1 ml di Ac2O (1.9 mmoli, 2 eq) e la soluzione è lasciata in
agitazione a temperatura ambiente. Dopo 12 ore l’analisi TLC (CHCl3-MeOH 9:1) mostra
la scomparsa del prodotto di partenza (Rf 0.35) e la formazione di un unico prodotto a Rf
0.70. Dopo eliminazione dei reagenti mediante co-evaporazione con toluene (3x15 ml) a pressione ridotta si ottiene un grezzo (420 mg) che viene purificato mediante flash cromatografia su colonna di gel di silice (esano-AcOEt 4:6 + 0.1% Et3N). Si isola il
3-O-acetil derivato 57 (388 mg, resa 97%) chimicamente puro all’analisi 1H e 13C NMR.
Il prodotto 57 è un solido giallino avente Rf 0.25 (esano-AcOEt
3:7); p.f. 160-164°C (cromatografato); [α]D -52.1 (c 1.0, CHCl3); 1H NMR (CD3CN): δ 6.47 (d, 1H, J2,NH 9.5 Hz, NH), 5.00 (dd, 1H, J2,3 9.4 Hz, J3,4 10.3 Hz, H-3), 4.54 (d, 1H, J1,2 8.5 Hz, H-1), 3.90-3.70 (m, 4H, H-2, H-4, H6a ,H6b), 3.82 (m, 1H, CH2O), 3.53 (dt, 1H, J 6.3 Hz, J 10.2 Hz, CH2O), 3.35 (m, 1H, H-5), 3.33 (t, 2H, J6.7 Hz, CH2N3), 1.96 (s, 3H, MeCOO), 1.82 (s, 3H, O O O O AcO NHAc N3 O O O O NHAc N3 BnO
MeCON), 1.78 (m, 2H, CH2), 1.44, 1.30 (2s, each 3H, Me2C); 13C NMR (CD3CN): δ 171.2,
170.7 (2 x CO), 102.4 (C-1), 100.4 (CMe2), 73.1 (C-3), 72.6 (C-4), 68.0 (C-5), 67.2
(CH2O), 62.6 (C-6), 55.2 (C-2), 48.7 (CH2N3), 29.5 (CH2), 29.3, 19.4 (Me2C), 23.1
(MeCON), 20.9 (MeCOO). Analisi Elementare calcolato per C16H26N4O7 (386.41): C,
49.73; H, 6.78; N, 14.50. Trovato: C, 49.75; H, 6.80; N, 14.52.
Preparazione del 3-azidopropil 2-acetammido-3-O-benzoil-2-desossi-4,6-O-isopropilidene-ββ-ββ D-glucopiranoside (58)
In un pallone da 25 ml, si solubilizzano 411.7 mg (1.20 mmoli, 1 eq) di 54 in 4.5 ml di piridina anidra, si raffredda a 0°C, si addizionano 0.43 ml (3.71 mmoli, 3.1 eq) di BzCl e la soluzione è lasciata in agitazione magnetica a 0°C. Dopo 1.5 ore l’analisi TLC (AcOEt) rivela la completa trasformazione del prodotto di partenza (Rf 0.14) in un unico prodotto,
visibile all’UV, a Rf 0.62. Si aggiunge ghiaccio, la miscela di reazione è lasciata in
agitazione per 15 minuti, si diluisce con CHCl3 (10 ml), si separano le fasi e quella
organica è lavata prima con una soluzione diluita (1M) di HCl (5 ml) e successivamente con una soluzione satura di NaHCO3 (5 ml). Le fasi organiche riunite, seccate su MgSO4,
filtrate e concentrate a pressione ridotta forniscono un grezzo (620 mg) che viene purificato per flash cromatografia su gel di silice eluendo con esano-AcOEt 1:1 + 0.1% di Et3N. Si isola il 3-O-benzoil derivato 58 (520 mg, resa 96%) chimicamente puro all’analisi 1H e 13C NMR.
Il prodotto 58 è una schiuma bianca avente Rf 0.27
(esano-AcOEt 1:1), [α]D -31.2 (c 0.99, CHCl3); 1H NMR (CDCl3): δ 7.98 (m, 2H, Ar-H), 7.65-7.20 (m, 3H, Ar-H), 7.04 (d, 1H, J2,NH 7.5 Hz, NH), 5.51 (t, 1H, J2,3 =J3,4 10.1 Hz, H-3), 4.60 (d, 1H, J1,2 8.6 Hz, H-1), 4.48 (m, 1H, H-2,), 4.10-3.60 (m, 5H, H-4, H6a ,H6b, CH2O), 3.50 (m, 1H, H-5), 3.35 (t, 2H, J 6.8 Hz, CH2N3), 1.83 (s, 3H, MeCON), 1.74 (m, 2H, CH2), 1.47, 1.33 (2s, each 3H, Me2C); 13C NMR (CDCl
3): δ 170.4 (MeCO), 167.0 (PhCO), 133.4, 129.7, 128.5 (Ar-CH), 129.0
(Ar-C), 101.9 (C-1), 99.6 (CMe2), 73.2 (C-3), 71.7 (C-4), 66.7 (C-5), 65.9 (CH2O), 61.8
(C-6), 53.6 (C-2), 47.7 (CH2N3), 28.7 (CH2), 28.6, 18.7 (Me2C), 23.0 (MeCON). Analisi
Elementare calcolato per C21H28N4O7 (448.48): C, 56.24; H, 6.29; N, 12.49. Trovato: C,
56.25; H, 6.30; N, 12.50. O O O O NHAc N3 BzO
Preparazione del metil 2-acetammido-3-O-benzoil-2-desossi-4,6-O-isopropilidene-ββββ-D
-glucopiranoside (62)
Una soluzione del metil derivato 47 (340 mg, 0.94 mmoli, 1 eq) in 2.5 ml di MeOH è stata trattata con MeONa in MeOH (0.47 ml) secondo quanto descritto nella preparazione del triolo 52. La reazione è risultata completa in 1 ora e l’analisi TLC (CHCl3-MeOH 8:2)
ha rivelato la completa scomparsa del materiale di partenza e la formazione di un unico spot a Rf 0.18. L’analisi NMR (1H e 13C) del grezzo (193 mg, resa 87%) evidenzia la
presenza esclusiva del triolo 51, che può essere utilizzato nella reazione successiva senza ulteriori purificazioni.
Il triolo 51 è una schiuma bianca e presenta Rf 0.18 (CH3Cl-MeOH 8:2),
[α]D-11.0 (c 1.03, MeOH); Lo spettro 1H NMR (CD3CN) consente di
individuare solo alcuni segnali diagnostici a δ 4.38 (d, 1H, J1,2 7.9 Hz,
H-1), 3.29 (s, 3H, OMe), 1.81 (s, 3H, MeCON); 13C NMR (CD
3OD): δ 173.8 (C=O), 103.5
(C-1), 77.9, 76.2, 72.0 (C-3, C-5, C-4), 62.7 (C-6), 57.2, 56.9 (C-2, OMe), 22.9 (MeCON). L’isopropilidenazione di 51 grezzo (193 mg, 0.82 mmoli, 1 eq) è condotta in DMF (3 ml) con 2-metossipropene (0.24 ml, 2.46 mmoli, 3 eq) e CSA (19.1 mg, 0.082 mmoli, 0.1 eq) seguendo la procedura descritta nella preparazione di 54. Dopo 4.5 ore l’analisi TLC (CHCl3-MeOH 8:2) evidenzia la completa scomparsa del prodotto di partenza (Rf 0.18) e
la formazione di un unico prodotto a Rf 0.47. Dopo trattamento della miscela di reazione si
ottiene un grezzo (260 mg,) che all’analisi NMR (1H e 13C) risulta costituito prevalentemente dal derivato 53, che è stato utilizzato nella reazione successiva senza ulteriori purificazioni.
Il prodotto 53 è un solido bianco che presenta Rf 0.47 (CHCl3-MeOH
8:2) e caratteristiche chimico fisiche analoghe a quelle riportate in letteratura:23 13C NMR (CDCl3): δ 172.1 (C=O), 103.1 (C-1), 99.2
(CMe2), 73.8 (C-4), 71.7 (C-3), 66.7 (C-5), 61.7 (C-6), 56.8, 56.5 (C-2, OMe), 18.7
(Me2C), 22.9 (MeCO). I parametri NMR (1H, 13C) sono in accordo con quanto riportato in
letteratura.23
Una soluzione di 53 grezzo (260 mg) in piridina anidra (3 ml) è stata trattata con BzCl (290 µl, 2.54 mmoli 3.1eq) secondo quanto descritto nella preparazione di 58. La reazione risulta completa in 3 ore e l’analisi TLC (CHCl3-MeOH 9:1) ha rivelato la completa
scomparsa del materiale di partenza (Rf 0.23) e la formazione di un unico spot a Rf 0.56.
Dopo trattamento e purificazione flash cromatografica su gel di silice (esano-AcOEt 25:75
O O O NHAc HO OMe O OH O H O H NHAc OMe
+ 0.1% di Et3N) del grezzo di reazione (511 mg) si isola il 3-O-benzoil derivato 62 (520
mg, resa 60% calcolata da 47) chimicamente puro all’analisi 1H e 13C NMR.
Il derivato 62 è una schiuma bianca avente Rf 0.23 (esano-AcOEt
25:75), [α]D-27.5 (c 1.1, CHCl3); 1H NMR (CD3CN): δ 7.98 (m, 2H,
Ar-H), 7.63 (m, 1H, Ar-H), 7.48 (m, 2H, Ar-H), 6.56 (d, 1H, J2,NH 9.7
Hz, NH), 5.27 (dd, 1H, J2,3 10.2 Hz, J3,4 9.4 Hz, H-3), 4.56 (d, 1H, J1,2 8.5 Hz, H-1), 4.01
(dd,1H, H-2), 3.96 (dd, 1H, J4,5 9.7 Hz, H-4), 3.95-3.79 (m, 2H,H6a, H6b), 3.42 (m, 1H,
H-5), 3.42 (s, 3H, OMe), 1.70 (s, 3H, MeCO), 1.44, 1.27 (2s, each 3H, Me2C); 13C NMR
(CD3CN): δ 170.8 (CO), 166.8 (PhCO), 134.3-129.5 (Ar-CH), 130.8 (Ar-C), 103.3 (C-1),
100.4 (CMe2), 74.1 (C-3), 72.7 (C-4), 67.9 (C-5), 62.6 (C-6), 57.3 (OMe), 55.2 (C-2), 29.3,
19.4 (Me2C), 23.0 (MeCO). Analisi Elementare calcolato per C19H25NO7 (379.41): C,
60.15; H, 6.64; N, 3.69. Trovato: C, 60.18; H, 6.67; N, 3.72.
Preparazione del 3-azidopropil 2-acetammido-3-O-acetil-2-desossi-ββββ-D
-glucopiranoside (60)
In un pallone da 25 ml, si solubilizzano 252 mg (0.65 mmoli, 1 eq) di 57 in 5 ml di AcOH acquoso al 70% e la miscela di reazione è lasciata in agitazione a 40°C. Dopo 40 minuti l’analisi TLC (esano-AcOEt 3:7) evidenzia la scomparsa del prodotto di partenza (Rf 0.23) e la formazione di un prodotto a Rf 0. La soluzione è stata concentrata a pressione
ridotta e ripetutamente coevaporata con toluene (5x20 ml). Il grezzo (155 mg) viene purificato attraverso flash cromatografia su gel di silice eluendo con CHCl3-MeOH 9:1 +
0.1% di Et3N. Si isola 60 (154 mg, resa 68%) chimicamente puro all’analisi NMR(1H e 13C).
Il prodotto 60 è un solido bianco avente Rf 0.27 (CHCl3-MeOH
9:1), p.f. 143-146 °C (cromatografato), [α]D -78.0 (c 0.60 MeOH); 1H NMR (CDCl 3-CD3CN): δ 7.84 (d, 1H, J2,NH 9.2 Hz, NH), 4.96 (dd, 1H, J2,3 10.4 Hz, J3,4 9.3 Hz, H-3), 4.39 (d, 1H, J1,2 8.4 Hz, 1), 3.85-3.68 [m, 4H, H-2, H-6a, H-6b, CH2O (1H)], 3.56 (t, 1H, J4,5 9.3 Hz, H-4), 3.41 [dt, 1H, J6.0 Hz, J10.1 Hz, CH2O (1H)], 3.35 (m, 3H, H-5, OH-4, OH-6), 3.17 (t, 2H, J6.7 Hz, CH2N3), 1.86, 1.83
(2s, each 3H, MeCON, MeCOO), 1.66 (m, 2H, CH2). 13C NMR (CDCl3-CD3OD): δ 171.4,
171.3 (2 x CO), 100.8 (C-1), 75.7, 75.3 (C-3, C-5), 68.1 (C-4), 65.8 (CH2O), 61.0 (C-6),
53.7 (C-2), 49.1 (CH2N3), 28.4 (CH2), 22.4 (MeCON), 20.4 (MeCOO).Analisi Elementare
O O O NHAc BzO OMe O H OH O O AcO NHAc N3
calcolato per C13H22N4O7 (346.34): C, 45.08; H, 6.40; N, 16.18. Trovato: C, 45.10; H, 6.42;
N, 16.20.
Preparazione del 3-azidopropil 2-acetammido-3-O-benzil-2-desossi-ββ-ββ D -glucopiranoside (59)
Una soluzione di 56 (657 mg, 1.51 mmoli, 1 eq) in AcOH acquoso 70% (16 ml) è lasciata in agitazione a 40°C secondo quanto descritto per la preparazione di 60. Dopo 40 minuti l’analisi TLC (AcOEt) evidenzia la scomparsa del prodotto di partenza (Rf 0.54) e
la formazione di un unico prodotto a Rf 0.15. Dopo trattamento della miscela di reazione e
purificazione flash cromatografica su gel di silice (AcOEt + 0.1% di Et3N) del grezzo (660
mg) si isola 59 (590 mg, resa 99%) chimicamente puro all’analisi 1H e 13C NMR.
Il diolo 59 è un solido bianco avente Rf 0.15 (AcOEt), [α]D +1.68
(c 0.95, MeOH); p.f. 153-158°C (cromatografato); 1H NMR (CD3CN): δ 7.37-7.24 (m, 5H, Ar-H), 6.60 (d, 1H, J2,NH 9.1 Hz,
NH), 4.79, 4.63 (sistema AB, 2H, JA,B 11.3 Hz, CH2Ph), 4.40 (d, 1H, J1,2 8.4 Hz, H-1), 3.84
(dt, 1H, 5.8 Hz, J10.2 Hz, CH2O), 3.75-3.60 (m, 4H, H-2, H6a, H6b, OH-4), 3.53 (dt, 1H, J
6.3 Hz, J10.2 Hz, CH2O), 3.45 (m, 2H, H-3, H-4), 3.28 (ddd, 1H, J 4,5 9.3 Hz, J 5,6a 3.0Hz,
J 5,6b 5.5 Hz, H-5), 3.35 (t, 2H, J6.8 Hz, CH2N3), 3.1 (bt, 1H, OH-6), 1.84 (s, 3H, MeCO),
1.77 (m, 2H, CH2); 13C NMR (CD3CN): δ 170.9 (CO), 140.1 (Ar-C), 129.1, 128.8, 128.3
(Ar-CH), 102.2 (C-1), 83.4 (C-3), 76.9 (C-5), 74.9 (CH2Ph), 71.8 (C-4), 66.7 (CH2O), 62.7
(C-6), 55.5 (C-2), 48.8 (CH2N3), 29.6 (CH2), 23.3 (MeCO). Analisi Elementare calcolato
per C18H26N4O6 (394.43): C, 54.81; H, 6.64; N, 14.20. Trovato: C, 54.83; H, 6.66; N,
14.22.
Preparazione del 3-azidopropil 2-acetammido-3-O-benzoil-2-desossi-ββ-ββ D -glucopiranoside (61)
Una soluzione di 58 (490 mg, 1.09 mmoli, 1 eq) in AcOH acquoso 70% (5 ml) è lasciata in agitazione a 40°C secondo quanto descritto per la preparazione di 60. Dopo 1 ora l’analisi TLC (esano-AcOEt 2:8) evidenzia la scomparsa del prodotto di partenza (Rf
0.48) e la formazione di un unico prodotto a Rf 0.15. Dopo trattamento della miscela di
reazione e purificazione flash cromatografica su gel di silice (AcOEt + 0.1% di Et3N) del
grezzo (465 mg) si isola 62 (411 mg, resa 92%) chimicamente puro all’analisi NMR (1H,
13C). O H OH O O NHAc N3 BnO
In una ripetizione della reazione condotta utilizzando il grezzo proveniente dalla reazione di benzoilazione di 54 (1.57 g, 4.56 mmoli) e contenente 58, dopo purificazione cromatografica si isola 61 (1.50 g) in resa dell’81% calcolata a partire da 54.
Il diolo 61 è una schiuma bianca avente Rf 0.29 (AcOEt), [α]D
-3.54 (c 1.0, MeOH); 1H NMR (CD
3CN): δ 8.00 (m, 2H, Ar-H),
7.61 (m, 1H, Ar-H), 7.48 (m, 2H, Ar-H), 6.98 (d, 1H, J2,NH 8.5 Hz,
NH), 5.23 (dd, 1H, J2,3 9.4 Hz, H-3), 4.62 (d, 1H, J1,2 8.5 Hz, H-1), 4.38 (bt, 1H, OH-6),
3.96 (dd, 1H, H-2), 3.90-3.51 (m, 6H, H-4, H6a, H6b, OH-4, CH2O), 3.45 (ddd, 1H, J4,5 10.3
Hz, J 5,6a 2.6 Hz, J 5,6b 6.8 Hz,H-5), 4.38 (bt, 1H, OH-6), 3.35 (t, 2H, J6.8 Hz, CH2N3),
1.79 (m, 2H, CH2), 1.70 (s, 3H, MeCO); 13C NMR (CD3CN): δ 171.3 (MeCO), 167.2
(PhCO), 130.9 (Ar-C), 134.1, 130.4, 129.4 (Ar-CH), 101.6 (C-1), 77.5 (C-3), 76.9 (C-5), 69.5 (C-4), 66.9 (CH2O), 62.3 (C-6), 54.7 (C-2), 48.8 (CH2N3), 29.5 (CH2), 23.0 (MeCO).
Analisi Elementare calcolato per C18H24N4O7 (408.41): C, 52.94; H, 5.92; N, 13.72.
Trovato: C, 52.96; H, 5.94; N, 13.74.
Preparazione del metil 2-acetammido-3-O-benzoil-2-desossi-ββββ-D-glucopiranoside (63)
Una soluzione di 622 (202 mg, 0.533 mmoli, 1 eq) in AcOH acquoso 70% (3 ml) è lasciata in agitazione a 40°C secondo quanto descritto per la preparazione di 60. Dopo 1 ora l’analisi TLC (CHCl3-MeOH 9:1) mostra la scomparsa del prodotto di partenza (Rf
0.72) e la formazione di un unico prodotto a Rf 0.27. Dopo trattamento della reazione e
purificazione flash cromatografica su gel di silice (CHCl3-MeOH 9:1 + 0.1% di Et3N) del
grezzo (220 mg) si isola 63 (174 mg, resa 96%) chimicamente puro all’analisi NMR (1H,
13C).
Il diolo 63 è una schiuma bianca avente Rf 0.27 (CHCl3-MeOH 9:1),
[α]D -44.1 (c 1.0; CHCl3); e caratteristiche chimico fisiche analoghe a
quelle riportate in letteratura:24 [α]D -47 (c 0.5; CHCl3); p.f. 100-102°C
(dec).
Preparazione del 3-azidopropil 2-acetammido-3-O-benzil-2-desossi-6-O-p-metossibenzil-ββββ-D-glucopiranoside (65)
In un pallone da 100 ml si solubilizzano 200 mg (0.51 mmoli, 1 eq) del diolo 59 in toluene (40 ml), si aggiungono 152 mg (0.61 mmoli, 1.2 eq) di Bu2SnO e la soluzione è
O H OH O O NHAc N3 BzO O H OH O NHAc BzO OMe
lasciata a riflusso del solvente (bagno esterno 140°C) in presenza di un’apparecchiatura munita di colonna Dean-Stark per allontanare l’acqua che si forma durante la reazione. Dopo 12 ore, la soluzione è raffreddata a 80°C e si aggiungono 197 mg (0.61 mmoli, 1.2 eq) di TBAB e 104 µl (0.76 mmoli, 1.5 eq) di PMBCl e si lascia in agitazione seguendo l’evolvere della reazione mediante analisi TLC (CHCl3-MeOH 9:1). Dopo 1 ora, non
osservando (TLC) nessuna evoluzione della reazione, si aggiungono 208 µl (1.52 mmoli, 3 eq) di PMBCl e dopo 3 ore si osserva sia il prodotto di partenza (Rf 0.32) che la
formazione di un prodotto a Rf 0.46. Si aggiungono altri 208 µl di PMBCl, (1.52 mmoli, 3
eq) e dopo 2 ore si osserva la quasi completa scomparsa del materiale di partenza. Si aggiunge Et3N (2 ml), si concentra a pressione ridotta e la purificazione flash
cromatografica (esano-AcOEt 6:4 + 0.1% di Et3N) del grezzo (1.5 g) fornisce 65 (160 mg,
resa 65%) chimicamente puro all’analisi NMR (1H, 13C).
Il prodotto 65 è una schiuma bianca avente Rf 0.44 (AcOEt); 1H
NMR (CD3CN): δ 7.38-7.25 (m, 7H, Ar-H), 6.91 (m, 2H, Ar-H),
6.52 (d, 1H, J2,NH 9.4 Hz, NH), 4.78, 4.63 (sistema AB, 2H, JA,B
11.3 Hz, CH2Ph), 4.47 (s, 2H, CH2PMB), 4.38 (d, 1H, J1,2 8.4 Hz, H-1), 3.81 (dt, 1H, 5.9
Hz, J10.3 Hz, CH2O), 3.78-3.71 (m, 2H, H6a, H6b), 3.77 (s, 3H, OMe), 3.65 (m, 2H, H-2,
OH-4), 3.52 (dt, 1H, J 5.9 Hz, J10.3 Hz, CH2O), 3.46-3.39 (m, 3H, H-3, H-4, H-5), 3.34
(t, 2H, J6.8 Hz, CH2N3), 1.84 (s, 3H, MeCO), 1.77 (m, 2H, CH2); 13C NMR (CD3CN): δ
170.7 (CO), 160.1 (PMB C), 140.1 (C), 131.6 (PMB C), 130.2, 114.5 (PMB Ar-CH), 129.1, 128.8, 128.3 (Ar-CH), 102.2 (C-1), 83.5 (C-3), 76.1 (C-5), 74.9 (CH2Ph), 73.4
(CH2PMB), 71.9 (C-4), 70.4 (C-6), 66.8 (CH2O), 55.8 (OMe), 55.4 (C-2), 48.9 (CH2N3),
29.6 (CH2), 23.3 (MeCO). Analisi Elementare calcolato per C26H34N4O7 (514.58): C, 60.69;
H, 6.66; N, 10.89. Trovato: C, 60.72; H, 6.69; N, 10.92.
Preparazione del 3-azidopropil 2-acetammido-3-O-acetil-6-O-t-butildimetilsilil-2-desossi-ββββ-D-glucopiranoside (67)
Metodo A: [con TBDMSCl e imidazolo] - In un pallone da 25 ml si solubilizzano 277
mg (0.80 mmoli, 1 eq) di diolo 60 in 8.3 ml di DMF, si raffredda la soluzione a 0°C, si addizionano 109 mg (1.60 mmoli, 2 eq) di imidazolo e 133 mg (0.88 mmoli, 1.1 eq) di TBDMSCl e la miscela di reazione è lasciata in agitazione a temperatura ambiente. Dopo 1.5 ore, evidenziata (TLC, AcOEt) la presenza del prodotto di partenza (Rf 0.12) insieme
ad un prodotto a Rf 0.33, si raffredda a 0°C e si aggiungono ulteriori 74 mg (1.4 eq) di
O H OPMB O O NHAc N3 BnO
imidazolo e 84 mg (0.7 eq) di TBDMSCl e si lascia in agitazione a temperatura ambiente. Dopo 1.5 ore l’analisi TLC mostra una non completa conversione del materiale di partenza nel prodotto a Rf 0.33 e la miscela di reazione è diluita con Et2O (20 ml) e lavata con una
soluzione satura di NaCl (20 ml). Si separano le fasi e quella acquosa è estratta prima con Et2O (3 x 20 ml) e poi con AcOEt (3 x 20 ml). Le fasi organiche riunite, anidrificate con
MgSO4, filtrate ed evaporate a pressione ridotta forniscono un grezzo (290 mg) che,
purificato mediante flash cromatografia su gel di silice (AcOEt + 0.1% di Et3N), permette
di recuperare il diolo 60 non reagito (96 mg, resa 34%) e il prodotto 67 (211 mg, resa 48%) chimicamente puro all’analisi 1H e 13C NMR.
Metodo B: [con TBDMSCl e Py] - In un pallone da 25 ml si solubilizzano 138 mg
(0.399 mmoli, 1 eq) di 60 in 1.5 ml di piridina, si addizionano 120 mg (0.798 mmoli, 1 eq) di TBDMSCl e la miscela di reazione è lasciata in agitazione a temperatura ambiente. Dopo 3.5 ore l’analisi TLC (CHCl3-MeOH 9:1) evidenzia la scomparsa del prodotto di
partenza (Rf 0.31) e la formazione di un prodotto prevalente a Rf 0.46. Si diluisce con
CHCl3 (10 ml), si lava prima con una soluzione satura di NaHCO3 (5 ml) e poi con una
soluzione satura di NaCl (5 ml), si separano le fasi e quella acquosa è estratta prima con CHCl3 (3x20 ml). Le fasi organiche riunite, anidrificate con MgSO4, filtrate ed evaporate a
pressione ridotta forniscono un grezzo (120 mg) che, purificato mediante flash cromatografia su colonna di gel di silice (AcOEt + 0.1% di Et3N), permette di recuperare
67 (51.4 mg, resa 28%) chimicamente puro all’analisi 1H e 13C NMR.
Il prodotto 67 è una schiuma bianca avente Rf 0.33 (AcOEt), [α]D
-50.4 (c 0.70, CHCl3); 1H NMR (CD3CN): δ 6.56 (d, 1H, J2,NH 9.4
Hz, NH), 4.88 (dd, 1H, J2,3 10.6 Hz, J3,4 9.0 Hz, H-3), 4.46 (d, 1H,
J1,2 8.5 Hz, H-1), 3.89 (dd, 1H, J5,6a 2.4 Hz, J6a,6b 11.3 Hz, H-6a), 3.83 [m, 3H, H-6b,OH-4,
CH2O (1H)], 3.67 (ddd, 1H, H-2), 3.58 [m, 2H, H-4, CH2O (1H)], 3.33 (m, 1H,H-5), 3.33
(t, 2H, J6.4 Hz, CH2N3), 2.00 (s, 3H, MeCOO), 1.81 (s, 3H, MeCON), 1.79 (m, 2H, CH2),
0.90 [s, 9H, SiCMe3], 0.08, 0.07 (2s, each 3H, Me2Si); 13C NMR (CD3CN): δ 171.65,
171.02 (2 x CO), 101.6 (C-1), 77.0 (C-5), 76.7 (C-3), 69.3 (C-4), 66.9 (CH2O), 63.5 (C-6),
54.6 (C-2), 48.8 (CH2N3), 29.5 (CH2), 26.2 (SiCMe3), 23.1 (MeCON), 21.1 (MeCOO),
18.9 [SiCMe3], -5.05, -5.14 (Me2Si). Analisi elementare calcolata per C19H36N4O7Si
(460.61): C, 49.55; H, 7.88; N, 12.16. Trovato: C, 49.57; H, 7.90; N, 12.18. O H OTBDMS O O NHAc N3 AcO
Preparazione del 3-azidopropil 2-acetammido-3-O-benzoil-6-O-t-butildimetilsilil-2-desossi-ββββ-D-glucopiranoside (68)
Metodo A: [con TBDMSCl e imidazolo] – Una soluzione di 61 (388 mg, 0.95 mmoli, 1
eq) in DMF (10 ml) è trattata con imidazolo (155 mg, 2.27 mmoli, 2.4 eq) e TBDMSCl (187 mg, 1.24 mmoli, 1.3 eq) secondo quanto descritto per la preparazione di 67 (Metodo A). Dopo 3 ore l’analisi TLC (AcOEt) evidenzia, oltre alla presenza del prodotto di partenza (Rf 0.29), la formazione di un prodotto a Rf 0.76. Dopo trattamento della miscela
di reazione e purificazione flash cromatografica su gel di silice (prima esano-AcOEt 2:8 + 0.1% di Et3N poi esano-AcOEt 1:9 + 0.1% di Et3N) del grezzo (450 mg) si recupera il
diolo non reagito 61 (201 mg, resa 52%) e il derivato 68 (182 mg, resa 37%) chimicamente puro all’analisi NMR (1H, 13C).
Metodo B: [con TBDMSCl e Py] - Una soluzione di 61 (1.54 g, 3.77 mmoli, 1 eq) in
piridina (28 ml) è trattata con TBDMSCl (1.14 g, 7.54 mmoli, 2 eq) secondo quanto descritto per la preparazione di 67 (Metodo B). Dopo 2 ore l’analisi TLC (AcOEt) evidenzia la completa scomparsa del prodotto di partenza (Rf 0.29) e la formazione di un
unico prodotto a Rf 0.76. Dopo trattamento della miscela di reazione e purificazione flash
cromatografica su gel di silice (esano-AcOEt 1:1 + 0.1% di Et3N) del grezzo (2.07 g) si
isola 68 (1.83 g, resa 93%) chimicamente puro all’analisi NMR (1H, 13C).
Il prodotto 68 è una schiuma bianca avente Rf 0.76 (AcOEt), [α]D
-11.2 (c 1.0, CHCl3); 1H NMR (CD3CN): δ 8.00 (m, 2H, Ar-H),
7.61 (m, 1H, Ar-H), 7.49 (m, 2H, Ar-H), 6.73 (d, 1H, J2,NH 9.6 Hz,
NH), 5.19 (dd, 1H, J2,3 10.6 Hz, J3,4 9.0 Hz, H-3), 4.54 (d, 1H, J1,2 8.5 Hz, H-1), 3.98-3.78
(m, 4H, H-2, H-6a, H-6b, OH-4), 3.69 (bt, 1H, J4,5 9.5 Hz, H-4), 3.41 (ddd, 1H, J5,6a 2.4 Hz,
J5,6b 4.27 Hz,H-5), 3.89 (m, 1H, CH2O), 3.57 (dt, 1H, J6.2 Hz, J10.2 Hz, CH2O), 3.35 (t,
2H, J6.8 Hz, CH2N3), 1.82 (m, 2H, CH2), 1.69 (s, 3H, MeCO), 0.91 (s, 9H, SiCMe3), 0.10,
0.08 (2s, each 3H, Me2Si); 13C NMR (CD3CN): δ 171.0 (MeCO), 167.2 (PhCO), 134.1,
130.4, 129.4 (Ar-CH), 131.0 (Ar-C), 101.6 (C-1), 77.6 (C-3), 77.1 (C-5), 69.4 (C-4), 66.9 (CH2O), 63.5 (C-6), 54.7 (C-2), 48.8 (CH2N3), 29.5 (CH2), 26.2 (CMe3), 23.0 (MeCO),
18.9 (SiCMe3), -5.02, -5.09 (Me2Si). Analisi Elementare calcolato per C24H38N4O7Si
(522.68): C, 55.15; H, 7.33; N, 10.72. Trovato: C, 55.18; H, 7.36; N, 10.75. O H OTBDMS O O NHAc N3 BzO
Preparazione del 3-azidopropil 2-acetammido-3-O-benzil-6-O-t-butildimetilsilil-2-desossi-ββββ-D-glucopiranoside (66)
Una soluzione di 59 (590 mg, 1.49 mmoli, 1 eq) in piridina (11 ml) è trattata con TBDMSCl (451 mg, 2.99 mmoli, 2 eq) secondo quanto descritto nella preparazione di 67 (Metodo B). Dopo 2 ore l’analisi TLC (AcOEt) evidenzia, oltre alla presenza di una piccola quantità del diolo di partenza (Rf 0.15), la formazione di un unico prodotto a Rf
0.58. Si addizionano ulteriori 150 mg (1.0 mmoli) di TBDMSCl e, dopo 2 ore, l’analisi TLC mostra la completa scomparsa del prodotto di partenza. Dopo trattamento della miscela di reazione e purificazione flash cromatografica su gel di silice (esano-AcOEt 1:1 + 0.1% di Et3N) del grezzo (747 mg) si isola il derivato 66 (681 mg, resa 89%)
chimicamente puro all’analisi NMR (1H, 13C).
Il prodotto 66 è una schiuma bianca avente Rf 0.25 (esano-AcOEt
1:1), [α]D+11.2 (c 1.3, CHCl3); 1H NMR (CD3CN): δ 7.40-7.24
(m, 5H, Ar-H), 6.72 (m, 1H, NH), 4.80, 4.64 (sistema AB, 2H, JA,B 11.3 Hz, CH2Ph), 4.37 (d, 1H, J1,2 8.4 Hz, H-1), 3.90 (dd, 1H, J5,6b 2.5 Hz, H-6b), 3.84
(m, 1H, CH2O), 3.82-3.65 (m, 3H, H-2, H-6a, OH-4), 3.54-3.44 (m, 2H, H-3, H-4), 3.45
(m, 1H, CH2O), 3.26 (ddd, 1H, J5,6a 5.3 Hz, H-5), 3.33 (t, 2H, J6.8 Hz, CH2N3), 1.75 (m,
2H, CH2), 1.85 (s, 3H, MeCO), 0.91 (s, 9H, SiCMe3), 0.09 (s, 6H, Me2Si); 13C NMR
(CD3CN): δ 170.9 (CO), 140.0 (Ar-C), 129.1, 128.7, 128.3 (Ar-CH), 102.1 1), 83.6
(C-3), 77.1 (C-5), 71.6 (C-4), 66.6 (CH2O), 63.8 (C-6), 55.4 (C-2), 48.8 (CH2N3), 29.6 (CH2),
26.2 (CMe3), 23.4 (MeCO),18.9 (SiCMe3), -5.0, -5.1 (Me2Si).Analisi Elementare calcolata
per C24H40N4O6Si (508.70): C, 56.67; H, 7.93; N, 11.01. Trovato: C, 56.69; H, 7.95; N,
11.03.
Preparazione del metil 2-acetammido-3-O-benzoil-6-O-t-butildimetilsilil-2-desossi-βββ
β-D-glucopiranoside (69)
Una soluzione di 63 (159 mg, 0.468 mmoli, 1 eq) in piridina (3.5 ml) è trattata con TBDMSCl (141 mg, 0.94 mmoli, 2 eq) secondo quanto descritto nella preparazione di 67 (Metodo B). Dopo 2 ore l’analisi TLC (CHCl3-MeOH 9:1) evidenzia, oltre alla presenza di
una piccola quantità del diolo di partenza (Rf 0.27), la formazione di un unico prodotto a Rf
0.70. Si addizionano ulteriori 36 mg (0.24 mmoli, 0.5 eq) di TBDMSCl e, dopo 3 ore, l’analisi TLC mostra la completa scomparsa del prodotto di partenza. Dopo trattamento della miscela di reazione e purificazione flash cromatografica su gel di silice (esano-AcOEt
O H OTBDMS O O NHAc N3 BnO
35:65 + 0.1 % di Et3N) del grezzo (197 mg) si isola 69 (172 mg, resa 81%) chimicamente
puro all’analisi NMR (1H, 13C).
Il prodotto 69 è un solido bianco avente Rf 0.70 (CHCl3-MeOH 9:1), p.f.
165-172°C (cromatografato); [α]D -18.8 (c 1.0, CHCl3); 1H NMR
(CD3CN): δ 8.00 (m, 2H, H), 7.61 (m, 1H, H), 7.48 (m, 2H,
Ar-H), 6.58 (d, 1H, J2,NH 9.6 Hz, NH), 5.17 (dd, 1H, J2,3 10.6 Hz, J3,4 9.0 Hz, H-3), 4.48 (d, 1H,
J1,2 8.5 Hz, H-1), 3.94 (dd, 1H, J5,6b 2.5 Hz, J6a,6b 11.3 Hz,H-6b), 3.90 (dd, 1H, J5,6a 4.7 Hz,
H-6a), 3.89 (m, 1H, H-2), 3.82 (d, 1H, J4,OH 5.2 Hz,OH-4), 3.70 (ddd, 1H, J4,5 9.7 Hz,
H-4), 3.43 (ds, 3H, OMe), 3.42 (ddd, 1H, H-5), 1.67 (s, 3H, MeCO), 0.91 (s, 9H, SiCMe3),
0.11, 0.10 (2s, each 3H, Me2Si); 13C NMR (CD3CN): δ 170.8 (MeCO), 167.2 (PhCO),
134.1, 130.4, 129.4 (Ar-CH), 131.1 (Ar-C), 102.5 (C-1), 77.8 (C-3), 77.1 (C-5), 69.4 (C-4), 63.5 (C-6), 56.9 (OMe), 54.6 (C-2), 26.2 (CMe3), 23.0 (MeCO), 18.9 (SiCMe3), -5.0, -5.1
(Me2Si). Analisi Elementare calcolato per C22H35NO7Si (453.61): C, 58.25; H, 7.78; N,
3.09. Trovato: C, 55.29; H, 7.81; N, 3.12.
Preparazione del 2,3,4,6-tetra-O-acetil-αα-ααD-glucopiranosil tricloroacetimmidato (22)
In un pallone da 250 ml si solubilizzano 830.8 mg (9.22 mmoli, 1.2 eq) di NH2NH2⋅AcOH in DMF (74.4 ml), si aggiungono 3.0 g (7.68 mmoli, 1 eq) di
1,2,3,4,6-penta-O-acetil-α,β-D-glucopiranosio commerciale (71) e la miscela di reazione è lasciata in agitazione in atmosfera inerte a 60 °C. Dopo 2 ore, evidenziata (TLC, esano-AcOEt 3:7) la presenza del prodotto di partenza (Rf 0.58) e la formazione di un unico prodotto a Rf 0.45,
si addizionano, in successione, quattro porzioni di NH2NH2⋅AcOH (ciascuna di 830 mg,
1.2 eq) e dopo 5 ore dall’inizio della reazione si osserva (TLC) la completa conversione del materiale di partenza nel prodotto a Rf minore. Si elimina il solvente a pressione ridotta, si
ripartisce il residuo fra CH2Cl2 (40 ml) e una soluzione acquosa di NaCl al 5% (40 ml), si
separano le fasi e quella acquosa è ulteriormente estratta con CH2Cl2 (3x30 ml). Le fasi
organiche riunite, anidrificate con MgSO4, filtrate e concentrate a pressione ridotta
forniscono un grezzo (3.37 g) che è purificato attraverso flash cromatografia su gel di silice eluendo con esano-AcOEt 1:1. Si isola il derivato 72 (2.39 g, resa 89%) che all’analisi NMR (1H, 13C) si presenta come una miscela di anomeri 72α e 72β in rapporto 4:1
calcolato sulla base delle altezze relative ai segnali dei C-1 a δ 89.9 e 95.1 rispettivamente.
O H OTBDMS O NHAc BzO OMe
La miscela di anomeri 72α,β è una schiuma bianca e presenta 13C NMR (CDCl3): δ 170.4, 170.2, 170.1, 169.9 (CO, α,βα,βα,β), 95.1 (C-1, βα,β βββ), 89.9 (C-1, α α α α), 70.3, 70.1 (C-3, C-5, ββββ), 68.1, 67.8 (C-2, C-4, ββββ), 68,0, 67.7, 66.9, 65.4 (C-2, C-3, C-4, C-5, αααα), 63.3 (C-6, αααα), 61.1 (C-6, ββββ), 20.3-20.1 (MeCO, α,βα,βα,βα,β).
In un pallone da 50 ml si solubilizzano 1.32 g (3.79 mmoli, 1 eq) di 72 in 10 ml di CH2Cl2 anidro, si addizionano 2.0 ml (22.8 mmoli, 6 eq) di CCl3CN, si raffredda la
soluzione a 0 °C e si aggiungono 31 µl (0.205 mmoli, 0.054 eq) di DBU. Si lascia in agitazione a temperatura ambiente. Dopo 2.5 ore l’analisi TLC (esano-AcOEt 1:1) evidenzia la scomparsa del prodotto di partenza (Rf 0.24) e la formazione di un unico
prodotto a Rf 0.47. Si eliminano i solventi a pressione ridotta ed il grezzo (1.86 g) viene
purificato mediante flash cromatografia su silice eluendo con esano-AcOEt 7:3 + 0.1% di Et3N. Si isola il derivato 22 (1.70 g) in resa del 91%.
Il composto 22 è uno schiuma bianca avente Rf 0.47 (esano:AcOEt 1:1) e
caratteristiche chimico fisiche analoghe a quelle riportate in letteratura:12 [α]D +115 (c 1.0, CHCl3); 13C NMR (CDCl3): δ 170.0, 169.8, 169.7,
169.6 (4 x CO), 160.5 (C=N), 93.2 (C-1), 90.5 (CCl3), 68,7, 67.2, 67.1,
66.6 (C-2, C-3, C-4, C-5), 61.0 (C-6), 20.7-20.3 (4xMeCO).
Preparazione del 4-O-(2,3,4,6-tetra-O-acetil-ββ-ββ D-galattopiranosil)-2,3,6-tri-O-acetil-ααα α-D-glucopiranosil tricloroacetimidato (20)
Una soluzione di peracetil lattosio 73 (3.00 g, 4.42 mmoli, 1 eq) in DMF anidra (60 ml) è trattata con NH2NH2⋅AcOH (714 mg, 7.93 mmoli, 1.8 eq) secondo quanto descritto nella
preparazione di 22. Dopo 4 ore l’analisi TLC (esano-AcOEt 2:8) evidenzia la scomparsa del prodotto di partenza (Rf 0.56) e la formazione di un unico prodotto a Rf 0.44. Dopo
trattamento della miscela di reazione e purificazione flash cromatografica su gel di silice (esano-AcOEt 9:1) del grezzo (3.0 g) si isola 74 (1.80 g, resa 64%) che, all’analisi NMR (1H, 13C) si presenta come una miscela di anomeri 74α e 74β in rapporto 75:25 calcolato sulla base delle altezze dei segnali C-1 a δ 89.6 e 94.7 rispettivamente.
La miscela di anomeri 74α,β è una schiuma bianca e presenta
13C NMR (CDCl 3): δ 171.2, 170.4, 170.2, 170.1, 169.9, 169.6, 168.9 (CO, α,βα,βα,βα,β), 100.7 (C-1’, α,βα,βα,βα,β), 94.7 (C-1, ββββ), 89.6 (C-1, αααα), 76.1, 72.8, 72.5, 72.3, 71.2, 70.8, 70.3, 69.4, 68.8, 67.7, 66.4 (2, 3, 4, 5, 2’, C-OAc OAc OH O OAc AcO OAc OAc O O AcO AcO NH CCl3 O OAc OH O OAc AcO OAc OAc O OAc AcO
Una soluzione di 74 (498 mg, 0.78 mmoli, 1 eq) in CH2Cl2 anidro (3 ml) è trattata con
CCl3CN (0.47 ml, 4.71 mmoli, 6 eq) e 60 µl (0.0424 mmoli, 0.054 eq) di una soluzione
1:10 di DBU-CH2Cl2 anidro. Dopo 1.5 ore l’analisi TLC (esano-AcOEt 3:7) evidenzia la
scomparsa del prodotto di partenza (Rf 0.31) e la formazione di un unico prodotto a Rf
0.52. Dopo trattamento della miscela di reazione e purificazione flash cromatografica su gel di silice (esano-AcOEt 6:4) del grezzo (658 mg) si isola 20 (567 mg, resa 93%) puro all’analisi NMR (1H, 13C).
Il composto 20 è una schiuma bianca avente Rf 0.52
(esano-AcOEt 3:7) e caratteristiche chimico fisiche analoghe a quelle riportate in letteratura:12 [α]D+95 (c 1.0, CHCl3); 1H
NMR (CDCl3): δ 170.1-168.9 (CO), 160.7 (C=N), 101.0
(C-1’), 92.6 (C-1), 90.4 (CCl3), 75.7, 70.9, 70.7, 70.4, 69.7, 69.3, 68.8, 66.4 (2, 3, 4,
C-5, C-2’, C-3’, C-4’, C-5’), 61.2, 60.5 (C-6, C-6’), 20.6-20.3 (MeCO).
Preparazione del tiofenil 2,3,4,6-tetra-O-acetil-αα-ααD-glucopiranoside (75)
In un pallone da 25 ml si solubilizzano 901 mg (3.31 mmoli) di tiofenil β-D-galattopiranoside, presente in laboratorio,in 30 ml di una miscela 2:1 di piridina e Ac2O, e
la sospensione è lasciata in agitazione a temperatura ambiente. Dopo 12 ore l’analisi TLC (AcOEt) evidenzia la scomparsa del prodotto di partenza (Rf 0.10) e la formazione di un
unico prodotto a Rf 0.74. L’eliminazione dei reagenti a pressione ridotta co-evaporando
con toluene (3x50 ml) fornisce un grezzo di reazione (1.69 g) che, purificato mediante flash cromatografia (esano-AcOEt 65:35), permette di isolare 75 (1.60 g, resa 91%) chimicamente puro all’analisi 1H e 13C NMR.
Il composto 75 è una schiuma bianca avente Rf 0.74 (AcOEt) e presenta 1H NMR (CDCl
3): δ 7.56 (m, 2H, Ar-H), 7.07 (m, 3H, Ar-H), 5.46 (bd,
1H, J3,4 3.3 Hz, H-4), 5.30 (t, 1H, J2,3 = J1,1 9.9 Hz, 2), 5.10 (dd, 1H,
H-3), 4.91 (d, 1H, H-1), 4.32 (dd, 1H, J5,6b 7.1 Hz, J6a,6b 11.3 Hz,H-6b), 4.14 (dd, 1H, J5,6a 6.0
Hz, H-6a), 4.01 (m, 1H, H-5), 2.15, 2.11, 2.00, 1.99 (4s, each 3H, 4xMeCO);13C NMR (CDCl3): δ 170.2, 170.0, 169.9, 169.2 (4xMeCO), 132.3 C), 132.2, 128.9, 128.0
(Ar-CH), 86.4 (C-1), 74.2, 71.8, 67.1, 67.0 (C-2, C-3, C-4, C-5), 61.5 (C-6), 21.3-20.5 (4xMeCO). OAc OAc SPh O OAc AcO O NH CCl3 O OAc O AcO AcO OAc OAc O OAc AcO
Procedure generali di glicosidazione
.
Metodo A: [con TMSOTf] - In un pallone a due colli, fiammeggiato e sotto atmosfera
di Argon, si solubilizzano l’opportuno glicosil accettore (1 eq) e glicosil donatore (1.5-2.0 eq) in toluene anidro, la soluzione è portata a secco alla pompa meccanica munita di una trappola raffreddata a -78°C e lasciata sotto vuoto per 40 minuti in modo da eliminare eventuali tracce di H2O attraverso formazione di un azeotropo. Il residuo è solubilizzato in
CH2Cl2 anidro e addizionato di setacci molecolari AW300 attivati. La sospensione
risultante è lasciata in atmosfera di Argon ed in agitazione a temperatura ambiente. Dopo 30 minuti si raffredda all’opportuna temperatura (-30 °C) e si addiziona una soluzione 1:10 di TMSOTf in CH2Cl2 contenente 0.1-0.5 eq di TMSOTf. Si lascia in agitazione facendo
andare la miscela a temperatura ambiente seguendo l’evolvere della reazione mediante analisi TLC. Si neutralizza la miscela di reazione con Et3N, si filtra su strato di celite, il
filtrato si concentra a pressione ridotta ed il grezzo è sottoposto a purificazione flash cromatografica eluendo con l’opportuna miscela di solvente.
Metodo B: [con BF3⋅Et2O] - In un pallone a due colli, fiammeggiato e sotto atmosfera
di Argon, si solubilizzano l’opportuno glicosil accettore (1 eq) e glicosil donatore (5 eq) in CH2Cl2 anidro, si addizionano 2 eq di BF3⋅Et2O e si lascia in agitazione a 40°C seguendo
l’evolvere della reazione mediante analisi TLC. Si neutralizza la miscela di reazione con Et3N, si concentra a pressione ridotta ed il grezzo è sottoposto a purificazione flash
cromatografica eluendo con l’opportuna miscela di solvente.
Metodo C: [con NIS-AgOTf] – In un pallone a due colli, fiammeggiato e sotto
atmosfera di Argon, dopo attivazione dei setacci molecolari AW300 (500 mg) si addiziona una soluzione formata dall’opportuno glicosil accettore (1 eq) e dal glicosil donatore (1.5-2.0 eq) in CH2Cl2 anidro e la soluzione è lasciata in agitazione a temperatura ambiente.
Dopo 30 minuti, si raffredda a -30 °C, si aggiungono in successione NIS (2 eq) e AgOTf (0.6 eq). La miscela di reazione viene lasciata in agitazione a temperatura ambiente seguendo l’evolvere della reazione mediante analisi TLC. Si filtra la miscela di reazione su strato di celite lavando ripetutamente il filtro con CH2Cl2 e il filtrato è lavato prima con
una soluzione acquosa di Na2S2O3 al 10% e successivamente con una soluzione satura di
NaHCO3. Si separano le fasi e quelle acquose sono estratte con CH2Cl2 (3x20 ml). Le fasi
reazione è sottoposto a purificazione flash cromatografica eluendo con l’opportuna miscela di solvente
Preparazione del 3-azidopropil 4-O-(2,3,4,6-tetra-O-acetil-ββββ-D -galattopiranosil)-2-acetammido-3-O-benzil-2-desossi-6-O-p-metossibenzil-ββββ-D-glucopiranoside (76a)
Prova 1: [con TMSOTf, t.a.] – La reazione di glicosidazione è effettuata secondo
quanto descritto nella procedura generale (Metodo A), conducendo la reazione a temperatura ambiente ed utilizzando 200 mg di setacci molecolari AW300 attivati, 123.8 mg (0.25 mmoli, 1.77 eq) di tricloroacetoimidato 22, 73.0 mg (0.142 mmoli, 1 eq) di glicosil accettore 65, CH2Cl2 anidro (1.5 ml) e 30 µl (0.00142 mmoli, 0.01 eq) di una
soluzione 1:10 di TMSOTf–CH2Cl2 (50 µl di TMSOTf e si porta a 500 µl con CH2Cl2
anidro). Dopo 12 h l’analisi TLC (esano-AcOEt 1:1) rivela la scomparsa del donatore 22 (Rf 0.47), la presenza di tracce dell’accettore 65 (Rf 0.13) e la presenza di due macchie
visibili all’UV a Rf 0.25 e 0.33. Dopo trattamento della reazione il grezzo è sottoposto ad
acetilazione convenzionale con Ac2O (2 ml) e piridina (4 ml) per 12 ore. Dopo
co-evaporazione con toluene a pressione ridotta si ottiene un residuo (255 mg) che all’analisi TLC (esano-AcOEt 7:3) risulta costituito dal penta-O-acetil-galattopiranosio 71 (Rf 0.47),
dall’accettore acetilato 80a (Rf 0.35) e da un terzo prodotto (Rf 0.21), visibile all’UV, che
non ha subito variazione di Rf durante l’acetilazione. La purificazione flash cromatografica
eluendo prima con esano-AcOEt 1:1 + Et3N e successivamente esano-AcOEt 7:3 + Et3N
consente di isolare 71 (58 mg, resa 60%), 80a (57 mg, resa 72%) e una miscela (16 mg) che all’analisi NMR risulta costituita dal disaccaride 76a e dall’ortoestere 78a in rapporto 8:2.
Prova 2: [con TMSOTf, prima a -30°C e poi t.a.] – Una sospensione formata da setacci
molecolari 4Å attivati (200 mg), il donatore 22 (84.6 mg, 0.17 mmoli, 1.8 eq), l’accettore
65 (50 mg, 0.097 mmoli, 1 eq) e CH2Cl2 anidro (1 ml) è trattata, alla temperatura di -30°C,
con 20 µl di una soluzione 1:10 di TMSOTf–CH2Cl2 (0.01 eq) come descritto nella
procedura generale (Metodo A). Dopo 3 ore l’analisi TLC (esano-AcOEt 6:4) evidenzia la presenza sia di 22 (Rf 0.47) sia di 65 (Rf 0.13) e tracce di una macchia a Rf 0.31. Si
addizionano ulteriori 20 µl di una soluzione 1:10 di TMSOTf–CH2Cl2 e la sospensione è
lasciata andare lentamente a temperatura ambiente. Dopo una notte l’analisi TLC evidenzia una situazione analoga alla precedente se non per un aumento dell’intensità della macchia a Rf 0.31. Si raffredda a -20°C, si addizionano ulteriori 20 µl di soluzione 1:10 di