3. MATERIALI E METODI
3.1 Trattamento degli animali
Ratti Wistar sani di 50 giorni, provenienti dalla stessa nidiata e cresciuti in ambiente a temperatura costante di 22°C, sono stati trattati giornalmente con iniezioni intraperitoneali di Acetil-L-Carnitina (ALC, Sigma Tau Laboratories, Pomezia, Italy; 100 mg/kg di peso corporeo), per 21 giorni. Ratti di controllo sono stati trattati con equivalenti volumi della stessa soluzione.
Gli animali sono stati sacrificati tramite decapitazione. I loro cervelli sono stati isolati in condizioni sterili a bassa temperatura e, dopo asportazione del cervelletto, sono stati immediatamente congelati in azoto liquido e conservati a –80°C.
3.2 Estrazione dell’RNA
L’RNA totale è stato isolato dal cervello degli animali di controllo e trattati, ottimizzando la metodologia descritta da Chirwing et al. (1979), come descritto da Traina et al., (2004). Per minimizzare la contaminazione da RNAsi, sono state utilizzate soluzioni trattate con DEPC (dietilpirocarbonato). I campioni sono stati omogeneizzati con un omogenizzatore Ultra-Turrax, immediatamente dopo essere stati immersi nel tampone GITC. L’omogenato è
stato stratificato su un cuscinetto di CsCl 5,7M. Il gradiente è stato centrifugato in ultracentrifuga Beckman L8-70M con rotore Kontron TY65 a 40.000 rpm a 20 °C per 24 ore. Il pellet è stato solubilizzato in H20DEPC e
precipitato con 0,1 volumi di Sodio Acetato 3 M pH 6,4 e 2,2 volumi di Etanolo Assoluto per una notte a –20 °C. In seguito il campione è stato centrifugato a 13.000 rpm in centrifuga con rotore Kontron A8.24 per 30 min e il pellet lavato con Etanolo al 70% per eliminare i sali residui. L’RNA è stato quindi solubilizzato in un opportuno volume H20DEPC. I campioni sono
stati conservati a -80°C per le analisisuccessive.
SOLUZIONI UTILIZZATE: Tampone GITC guanidina tiocianato 4 M sodio citrato 25 mM pH 7,0 β-mercaptoetanolo 0,1 M N- lauril sarcosinato 0,5% (p/v) Cloruro di Cesio 5,7M cloruro di cesio 5,7 M
sodio acetato 25 mM pH 6,43.3 Isolamento dell’mRNA poliA
+Per l'isolamento dell’mRNA poliA+ è stato usato il PolyATtract®
mRNA Isolation Systems (Promega). Il sistema utilizza un primer oligo(dT)
biotinilato che ha la capacità di ibridare ad alta efficienza con la regione al 3' poliA+ presente nella maggior parte dei mRNA maturi eucariotici. Gli ibridi possono poi essere recuperati usando streptavidina (che ha la capacità di legarsi alla biotina) associata a particelle paramagnetiche (PMP), ed un supporto magnetico. Circa 1 mg di RNA totale in un volume di 500 μl di H2ODEPC è stato denaturato per 10 min a 65°C; sono stati aggiunti 3 μl di Biotinylated Oligo(dT) Probe e 13 μl di SSC 20x (NaCl 3 M; Na-citrato
0,3 M); il tutto è stato incubato a temperatura ambiente fino a raffreddamento per permettere la reazione di appaiamento. Contemporaneamente le particelle PMP sono state lavate e risospese in SSC 0,5x ed aggiunte poi alla reazione di appaiamento lasciando incubare a temperatura ambiente per circa 10 min per permettere il legame tra la biotina e la streptavidina. Catturate le particelle PMP e lavate con SSC 0,1x, sono stati in seguito eluiti gli mRNA risospendendo le particelle PMP in H2ODEPC e catturandole con il magnete, è
stata recuperata e trasferita la fase acquosa, contenente l’mRNA, in una nuova provetta priva di RNAsi. L’mRNA è stato poi precipitato e risospeso in 10 μl di H2O DEPC.
3.4 Controllo della qualità dell’mRNA poliA
+Gli mRNA poliA+ sono stati utilizzati per la costruzione delle librerie sottrattive di cDNA utilizzando la tecnica dell’ibridazione sottrattiva soppressiva (SSH). Al fine di rendere la sottrazione efficiente è necessario valutare il grado di contaminazione dell’mRNA da parte dell’rRNA (Sävenstrand et al., 2000).
Per valutare la contaminazione dell’mRNA è stata effettuata una retrotrascrizione reazione a catena della polimerasi (RT-PCR) su una piccola quantità di ciascun mRNA poliA+, utilizzando 2 set di primers specifici per l’rDNA di ratto (5,8 S; 18 S), le cui sequenze sono state trovate in banca dati (dati non mostrati).
3.5
Costruzione delle librerie sottrattive
Per ottenere sequenze di cDNA differenzialmente espresse è stato utilizzato il metodo della ibridazione soppressiva sottrattiva (suppression subtractive
hybridization, SSH), la cui attuazione è stata resa possibile dall’impiego del PCR-Select
™
cDNA Subtraction Kit (BD Biosciences). Questa metodologiapermette di ottenere librerie di cDNA arricchite di trascritti presenti soltanto in uno dei due campioni comparati (Diatchenko et al., 1996) e presenta la peculiarità di riuscire ad isolare anche sequenze poco espresse (Gurskaya et
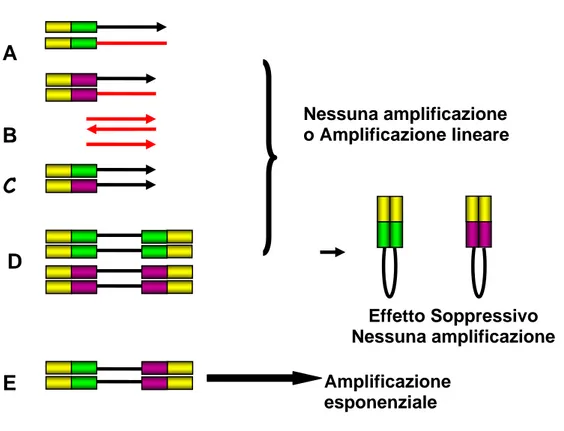
La tecnica della SSH comprende due ibridazioni sottrattive, seguite da due reazioni di PCR. Nella figura 3 è rappresentato in modo schematico il processo della SSH : ci riferiamo con il termine tester al cDNA ottenuto dall’mRNA isolato dal cervello di ratto trattato e con il termine driver all’cDNA ottenuto dall’mRNA del controllo.
a, b, c, d, + e Tester A Tester B Ligation con adattatore 1 Ligation con adattatore 2R I IBRIDAZIONE I IBRIDAZIONE Driver denaturato in eccesso II IBRIDAZIONE Driver in eccesso Tester a a b b c c d d e Amplificazioni utilizzando il primer (I PCR) Amplificazioni utilizzando i primers e (II PCR)
Figura 3: Rappresentazione schematica della tecnica della SSH.
Il cDNA è stato sintetizzato a partire da 2μg di mRNA di entrambi i campioni ed è stato digerito con l’enzima di restrizione Rsa I, che produce frammenti
blunt-end.Il cDNA del tester è stato suddiviso in due aliquote ed a ciascuna è
stato legato un diverso adattatore: 1 e 2R. Gli adattatori, mancando del gruppo fosfato al 5’, si legano solo all’estremità 5’ del cDNA.
Sono seguite due ibridazioni sottrattive successive. La prima è stata effettuata aggiungendo a ciascun tester denaturato, driver denaturato in eccesso. Durante questa fase la maggior parte dei trascritti del tester non differenziali, si lega al driver e la rimanente frazione del tester, che rimane a singolo
Effetto Soppressivo Nessuna amplificazione D A B C Nessuna amplificazione o Amplificazione lineare E Amplificazione esponenziale
filamento risulta arricchita di sequenze differenzialmente espresse. Inoltre in questo passaggio la concentrazione delle sequenze poco e molto espresse viene normalizzata, perché, essendo la cinetica di ibridazione di secondo ordine, la riassociazione delle molecole più concentrate è più veloce.
La seconda ibridazione è stata effettuata unendo, senza denaturare, i prodotti della prima ibridazione e aggiungendo driver denaturato in eccesso. Durante questo passaggio, oltre ad arricchirsi di sequenze differenzialmente espresse, la frazione a singolo filamento del tester 1 ibriderà con la stessa frazione nel
tester 2R, formando una popolazione di sequenze differenzialmente espresse a
doppio filamento caratterizzate dal fatto di essere asimmetricamente fiancheggiate dai due adattatori. Questo, insieme all’effetto soppressivo, rende possibile la loro amplificazione selettiva tramite due PCR successive utilizzando come primers sequenze complementari ai due adattatori.
Sequenze dei primers utilizzati
PCR primer 1 5’ –CTAATACGACTCACTATAGGGC- 3’
NESTED primer 1 5’ –TCGAGCGGCCGCCCGGCAGGT- 3’
Questa tecnica ha, quindi, permesso la costruzione di due librerie sottrattive:
Libreria Forward: ottenuta utilizzando come tester il cDNA trattato e come driver il cDNA controllo
Libreria Reverse: ottenuta utilizzando come tester il cDNA controllo e come driver il cDNA trattato
La libreria forward contiene i prodotti dei geni che sono attivati o modulati positivamente dal trattamento con ALC, mentre la libreria reverse contiene i prodotti dei geni inibiti o modulati negativamente dal trattamento con ALC. E’ stata valutata l’efficienza di sottrazione con:
a) Southern Blotting : gli amplificati delle librerie sottrattive sono stati ibridati con le sonde costituite dal cDNA delle librerie stesse (dati non mostrati).
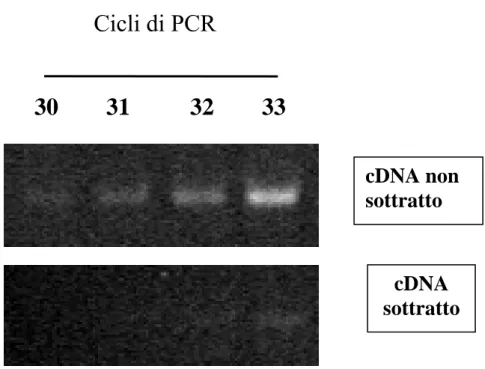
b) PCR con primer della gliceraldeide-3-fosfato deidrogenasi (G3PDH), gene costitutivo, come suggerito dal protocollo del PCR-Select™ cDNA Subtraction Kit (BD Biosciences) (fig 4).
c) Spot Blot : gli amplificati dei cDNA sottratti appartenenti alle librerie e dei cDNA non sottratti(vedi didascalia fig.4) sono stati ibridati con la sonda ottenuta marcando la G3PDH (dati non marcati).
Cicli di PCR
Gli amplificati ottenuti dalla seconda amplificazione sono stati inseriti nel vettore di clonaggio PCR® II-TOPO vector (TOPO TA Cloning® kit, Invitrogen)
I cloni sono stati recuperati in microtetraplates da 96 pozzetti contenenti 65 μl di LB A+, sono stati messi a crescere una notte in agitazione orizzontale a 37°C e, dopo l’aggiunta di 50 μl glicerolo, sono stati conservati a –20°C.
3.6 Screening primario delle librerie (Colony PCR screenin
g)3.6.1 Amplificazione delle sequenze differenziali
1μl di colonia è stato messo a crescere per 2 ore in 50 μl di LB A+ liquido a
37°C su agitatore orizzontale. La coltura è stata amplificata mediante PCR
30 31 32 33
cDNA non sottratto
cDNA sottratto
Figura 4 prova di sottrazione mediante PCR della libreria forward. CDNA
sottratto, cDNA costituente la libreria forward; cDNA non sottratto, cDNA del tester amplificato
utilizzando EuroTaq (EuroClone), in un volume di 25 μl usando le seguenti condizioni: 1 μl della coltura è stato utilizzato come stampo, poi sono stati aggiunti 2,5 μl di tampone 10x PCR EuroClone; 0,25 μl di dNTP Mix 10 mM; 0,25 μl di Primer M13F 10 μM; 0,25μl di Primer M13R 10 μM; 2,5 μl di
soluzione 25 mM di MgCl2; 0,25 μl di EuroTaq DNA Polymerase, 5U/μl;
H2O sterile a volume. La reazione così composta è stata posta nel
termociclizzatore (Applied Biosciences GeneAmp PCR System 2700) ed è stata iniziata l’amplificazione con la denaturazione dello stampo a 94°C per 10 min seguita da 30 cicli così composti: 94°C per 30 sec, 55°C per 30 sec, 72°C per 90 sec; al termine dei cicli è stata fatta seguire una estensione finale a 72°C per 10 min.
I prodotti di PCR sono stati analizzati su gel di agarosio al 2% (vedi fig.5 par 4.1).
Sequenze Primers utilizzati
M13 L 5’-GTTTTCCCAGTCACGAC-3’
M13 R 5’-CAGGAAACAGCTATGAC-3’
3.6.2 Spot Blot del prodotto di PCR
1 μl del prodotto di PCR di ciascun clone è stato denaturato (95oC per 1 min) e trasferito su membrana Hybond N+ (Amersham Biosciences). Le membrane
così ottenute sono state fissate mediante Hoefer™ UVC 500 cross-linker (Amersham Biosciences).
Lo screening primario, mediante la tecnica degli Spot Blot, ha permesso di eliminare i cloni falsi positivi, interpretando i risultati secondo il protocollo descritto nel PCR Select Differential Screening kit User Manual (BD
Biosciences). Dopo aver individuato i cloni candidati ad essere
differenzialmente espressi, si è passati all’isolamento del DNA plasmidico e quindi al sequenziamento, effettuato con sequenziatore automatico.
3.6.3 Marcatura delle sonde
Le membrane sono state ibridate utilizzando come sonde i cDNA che costituiscono le liberie forward (sonda T-C) e reverse (sonda C-T) marcate con marcatura non radioattiva, utilizzando DIG DNA Labelling Kit (ROCHE). Per la marcatura del cDNA circa 0,1-1μg del prodotto della seconda PCR sono stati digeriti con Rsa I per eliminare gli adattatori 1 e 2R da ogni sequenza di cDNA .
Per la marcatura del cDNA sono stati denaturati circa 0,1-1 μg di cDNA digerito in 15 μl di H2O (100°C) e dopo averli raffreddati in ghiaccio sono
2 μl di esanucleotidi, 2 μl di dNTP, 1 μl di Klenow. Dopo una incubazione per tutta la notte a 37°C, la reazione polimerasica è stata interrotta aggiungendo 2 μl di Na2EDTA 0,2 M (pH 8,0).
Il cDNA è stato precipitato con 2,5 μl di LiCl 4 M, 75 μl di Etanolo Assoluto freddo; questa reazione è stata lasciata per 1 ora a -80°C. Successivamente il cDNA è stato centrifugato a 12.000 g per 40 min a 4°C ed il pellet è stato lavato 2 volte con etanolo al 70%. Dopo i lavaggi il pellet, asciugato all'aria, è stato solubilizzato in 50 μl di H2O sterile.
3.6.4 Ibridazione su filtro
La membrana è stata preibridata per circa 3 ore nell'apposito termostato (20 ml di soluzione di preibridazione per circa 100 cm2 di membrana) alla temperatura di 68°C.
La sonda marcata è stata denaturata in H2O a 100°C per 10 minuti e poi posta
immediatamente in ghiaccio. Successivamente è stata preparata la soluzione di ibridazione aggiungendo la sonda marcata (25 ng/ml) a una nuova soluzione di preibridazione. Si è proceduto quindi con l’ibridazione.
Nella soluzione di preibridazione e in quella di ibridazione sono stati aggiunti i “competitori”, ovvero oligonucleotidi con sequenza complementare a quella degli adattatori utilizzati per la costruzione delle librerie sottrattive (vedi fig.3) ad una concentrazione finale pari a 0,1 μM.
Questo è stato necessario al fine di evitare ibridazioni tra eventuali adattatori rimasti nelle sonde T-C e C-T ed il cDNA in esame.
La sonda è stata recuperata e le membrane sono state sottoposte ad una serie di lavaggi a diversa stringenza, in dipendenza dell'omologia della sonda e della sua composizione (% in G+C).
La membrana è stata lavata due volte in SSC 2x/SDS 0,1% per 20 min a temperatura ambiente, in agitatore orizzontale.
Poi, aumentando la stringenza, è stata lavata due volte in SSC 0,1x/SDS 0,1% per 20 min a 68°C, in agitatore orizzontale. Quindi è stata equilibrata per 5 min con il tampone 1 e incubata per 1 ora in lenta agitazione con il tampone 2. Successivamente è stata posta per 30 min in lenta agitazione in una soluzione contenente l'anticorpo anti-digossigenina, diluito 1:10.000 nel tampone 2. In seguito sono stati effettuati, 3 lavaggi di 15 min nel tampone 1 a cui è stato aggiunto Tween 20 ad una concentrazione finale pari allo 0,3%; quindi la membrana è stata equilibrata per 5 min con il tampone 3.
Utilizzando il metodo della visualizzazione chemioluminescente si è proceduto in camera oscura cospargendo la membrana, adagiata in una pellicola trasparente, con Lumigen CSPD* (ROCHE) diluito 1:100 nel tampone 3 (circa 1 ml per membrane di 100 cm2).
Il tutto è stato lasciato al buio per 5 min a temperatura ambiente, quindi è stata tolta la soluzione in eccesso. Sulla membrana è stata posta una lastra
lasciata in esposizione per circa 2 ore. Successivamente la lastra radiografica è stata sviluppata e fissata con le soluzioni Kodak (seguendo le istruzioni fornite dalla ditta).
SOLUZIONI UTILIZZATE:
Tampone 1
NaCl 0,15M
Acido maleico 0,1M
Portare a pH 7,5 con NaOH e autoclavare
Blocking stock solution
Sciogliere il Blocking reagent fornito dalla ditta nel tampone 1 Conservare a –20°C Soluzione di preibridazione SSC 5x Blocking reagent 1% SLS 0,1% SDS 0,02% Tampone 2
Blocking Stock Solution, diluita 1:10 nel tampone 1
Tampone 3 Tris-HCl 100 mM, pH=9,5 NaCl 100 mM SSC 20x NaCl 3 M Na-Citrato 0,3
Portare a pH 7,0 con acido citrico e autoclavare
SDS 10%
3.7 Preparazione del DNA plasmidico su piccola scala
(MINIPREP) per sequenziamento
Il DNA plasmidico è stato estratto e purificato utilizzando High Pure Plasmid
Isolation Kit (Roche).
I cloni sono stati inoculati in bottigliette Macarty contenenti 10 ml di LB liquido e ampicillina alla concentrazione di 50 μg/ml e fatti crescere a 37°C su agitatore rotante per una notte. Per ogni clone, 6 ml di coltura suddivisi in tre aliquote da 2 ml ciascuna, sono stati centrifugati a 11.500 rpm per 1 min e 30 sec, in centrifuga da tavolo ALC 4226 in un rotore ALC 5531 (questo tipo di centrifuga viene utilizzato per tutta la preparazione). Successivamente le cellule sono state risospese delicatamente in 200 μl di Suspension buffer, fornito nel kit. I campioni provenienti dallo stesso clone, sono stati riuniti in un’unica provetta e a questa sono stati aggiunti 250 μl di Lising buffer, fornito nel kit; il campione è stato delicatamente mescolato per inversione, e lasciato a temperatura ambiente per 5 min. Sono stati aggiunti 350 μl di Binding
buffer, fornito nel kit, precedentemente raffreddato in ghiaccio. Il campione è
stato centrifugato a 12.000 g per 12 min. Il surnatante così ottenuto è stato posto delicatamente sulle colonnine fornite nel kit e centrifugato a 12.000 g per 1 min. Sono stati aggiunti 700 μl di Washing buffer, fornito nel kit, e centrifugato a 12.000 g per 1 min. Le colonnine sono state poste in provette sterili e si è proceduto all’eluizione del DNA con 50 μl di H2O sterile.
proceduto alla quantizzazione del materiale allo spettrofotometro e su gel di agarosio.
SOLUZIONI UTILIZZATE:
Ampicillina
soluzione stock 10mg/ml solubilizzata in Etanolo 70%
LB (Luria-Bertani) liquido
Triptone 1,0%
Yeast extract 0,5%
NaCl 1,0%
pH 7,0 con NaOH
3.8 ANALISI DELLE SEQUENZE
Le sequenze nucleotidiche dei cloni selezionati, ottenute con sequenziamento automatico, sono state analizzate mediante comparazione in banca dati (GenBank) con i programmi:
FASTA:(http://www.ebi.ac.uk/fasta33/)
BLAST: (http://www.ch.embnet.org/software/aBLAST.html) ADVANCED BLAST:
ExPASy Translate Tool : (http://www.expasy.org) Gli eventuali domini sono stati individuati con:
PROSITE : (http://www.expasy.org )
Pfam : (http://www.sanger.ac.uk/Software/Pfam)Tutti questi programmi sono disponibili in rete.
3.9 Analisi di espressione tramite RT-PCR relativa
La retrotrascrizione con la successiva PCR (RT-PCR) permette l’amplificazione di mRNA cellulare: vista l’elevata sensibilità, l’RT-PCR permette di valutare anche cambiamenti piccoli ma fisiologicamente rilevanti nell’espressione genica (Gause W.C. et al, 1995).
L’ RT-PCR relativa consente di comparare la quantità di un trascritto tra più campioni mediante la coamplificazione della sequenza di interesse e di un controllo interno al fine di normalizzare le differenze tra i vari campioni dovute alla qualità dell’ RNA totale, alla variabilità dell’efficienza dell’RT o ad una quantizzazione non accurata (Gause et al, 1995). Per questo tipo di PCR quantitativa è necessario che i primers per l’amplificato del gene di interesse (target) e quelli per il controllo interno siano compatibili e che i pesi molecolari del target e del controllo interno siano simili ma tali da permettere la distinzione degli amplificati su gel d’agarosio. Per questo tipo di analisi è
necessario che la reazione di PCR venga analizzata durante la fase lineare dell’amplificazione prima che entrambi i prodotti di amplificazione raggiungano la fase di plateau ossia di saturazione (Prediger, 2001)
La retrotrascrizione è stata eseguita con il kit SuperScript™ II RNase H -Reverse Transcriptase (Invitrogen) e con l’ Oligo(dT)12-18 Primer (Invitrogen) seguendo le istruzioni fornite dalla ditta, partendo da
RNA totale (4μg) isolato dal cervello di ratto trattato e dal controllo. Al fine di evitare errori di quantificazione dovuti ad una diversa concentrazione di RT o errori di pipettamento, è stato opportuno utilizzare un controllo interno, che viene coamplificato con il gene in esame. Come controllo interno è stata utilizzata la G3PDH che, essendo un gene costitutivo, presenta un’uguale espressione nel trattato e nel controllo. Per amplificare le sequenze dei cloni in esame sono stati utilizzati
primers gene-specifici; le sequenze dei primers sono riportate nella tabella
X. I primers sono stati scelti in modo da amplificare un frammento di peso molecolare di 452 pb per la G3PDH, di 216 pb per 5BG11, 147 pb per 5BE1 e di 150 pb per 5BF9.
Per ogni reazione di amplificazione da 30 μl sono stati utilizzati 0.6 μl di retrotrascritto come stampo; 3 μl del tampone di reazione 10x; 1.02 μl di soluzione di MgCl2 50mM; 0.6 μl di dNTP Mix 10mM; 0.6 μl di ogni primer
l’amplificazione con la denaturazione dello stampo a 94°C per 4 min seguito da cicli così composti: 94°C per 30 sec, 59°C per 30 sec, 72°C per 30 sec per un campione di cDNA trattato e un campione di cDNA controllo; al termine dei cicli è stata fatta seguire una estensione a 72°C per 7 min a tutti i campioni.
Per conoscere il punto di saturazione, ovvero il punto in cui l’aumento del prodotto di amplificazione non è più proporzionale alla quantità di stampo iniziale, è stata effettuata una PCR dalla quale sono state sottratte aliquote di campione al termine di cicli successivi; le aliquote sono state immediatamente trasferite in un termociclizzatore adiacente dove hanno subito la fase di estensione finale di 7 min a 72°C. In questo modo è stato possibile stabilire il numero di cicli appropriato per una valutazione quantitativa relativa mediante questa tecnica (nel nostro caso 24 cicli). Per ogni gene in analisi sono state fatte tre prove di RT-PCR relativa utilizzando come stampo cDNA ottenuti sia da tre retrotrascrizioni diverse sia da tre diverse estrazioni .
I prodotti di PCR sono stati analizzati su gel di agarosio 2% colorato con Etidio Bromuro. Gli amplificati sono stati quantizzati mediante scansione diretta del gel di agarosio al 2% con il densitometro UVP Image Store 5000 (Ultra Violet Product Ltd, Cambrige, England) equipaggiato con l’UVP GelBase-GelBlot TM Windows Software.
3.9.1 Analisi statistiche
Per normalizzare i campioni relativamente alla quantità di RNA totale utilizzato e alla efficienza della sintesi di cDNA, è stato fatto il rapporto tra le intensità delle bande di amplificazione dei vari campioni e la media dei prodotti di amplificazione della G3PDH. Questo rapporto è stato calcolato per 3-5 esperimenti indipendenti. E’ stata effettuata poi l’analisi statistica con il Mann-Whitney test.